
The Bright and the Dark Side of TGF-β Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications




Abstract
Simple Summary
Abstract
1. Introduction
2. Molecular Characteristics of Hepatocellular Carcinoma
3. Overview of the TGF-β Superfamily
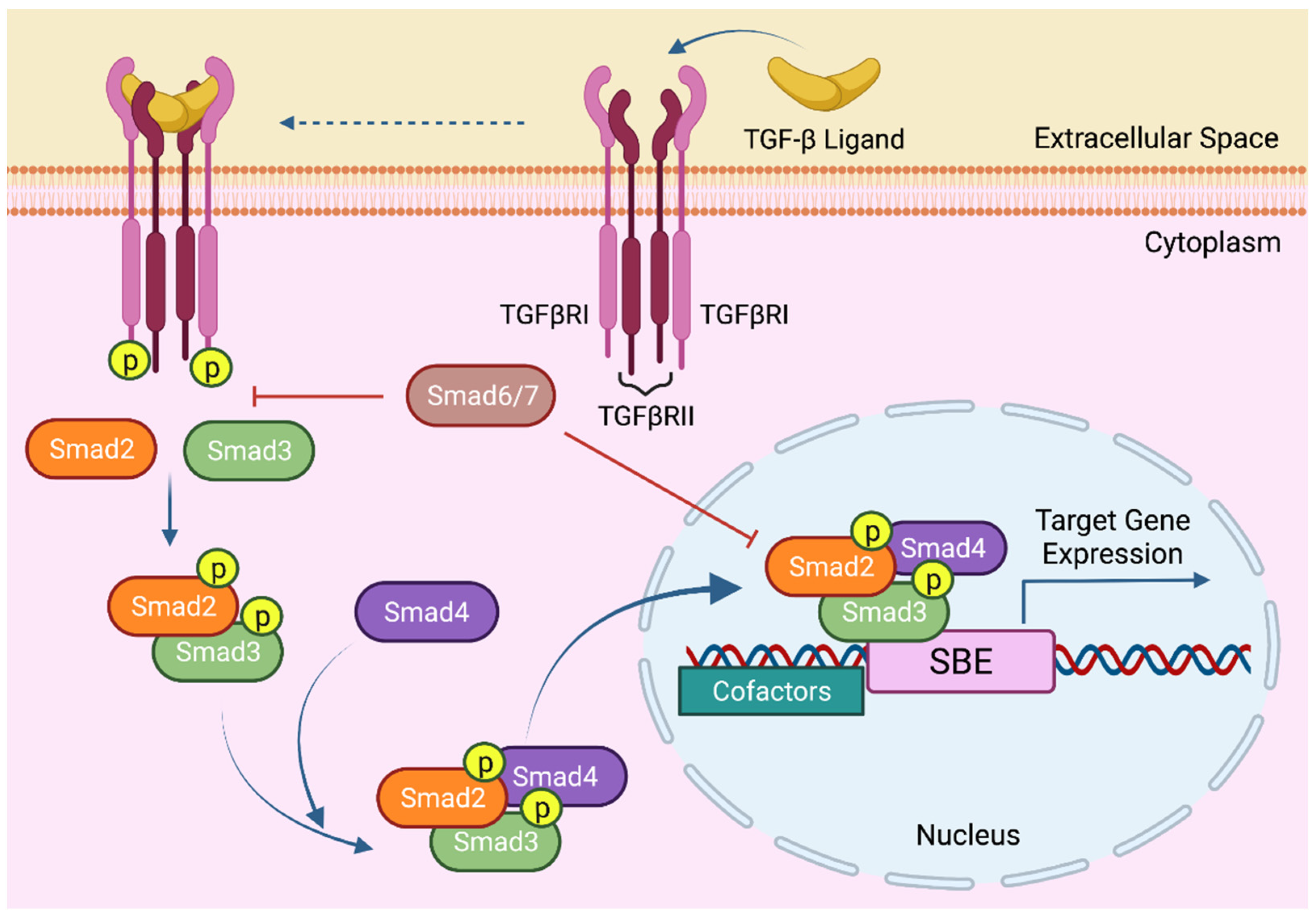
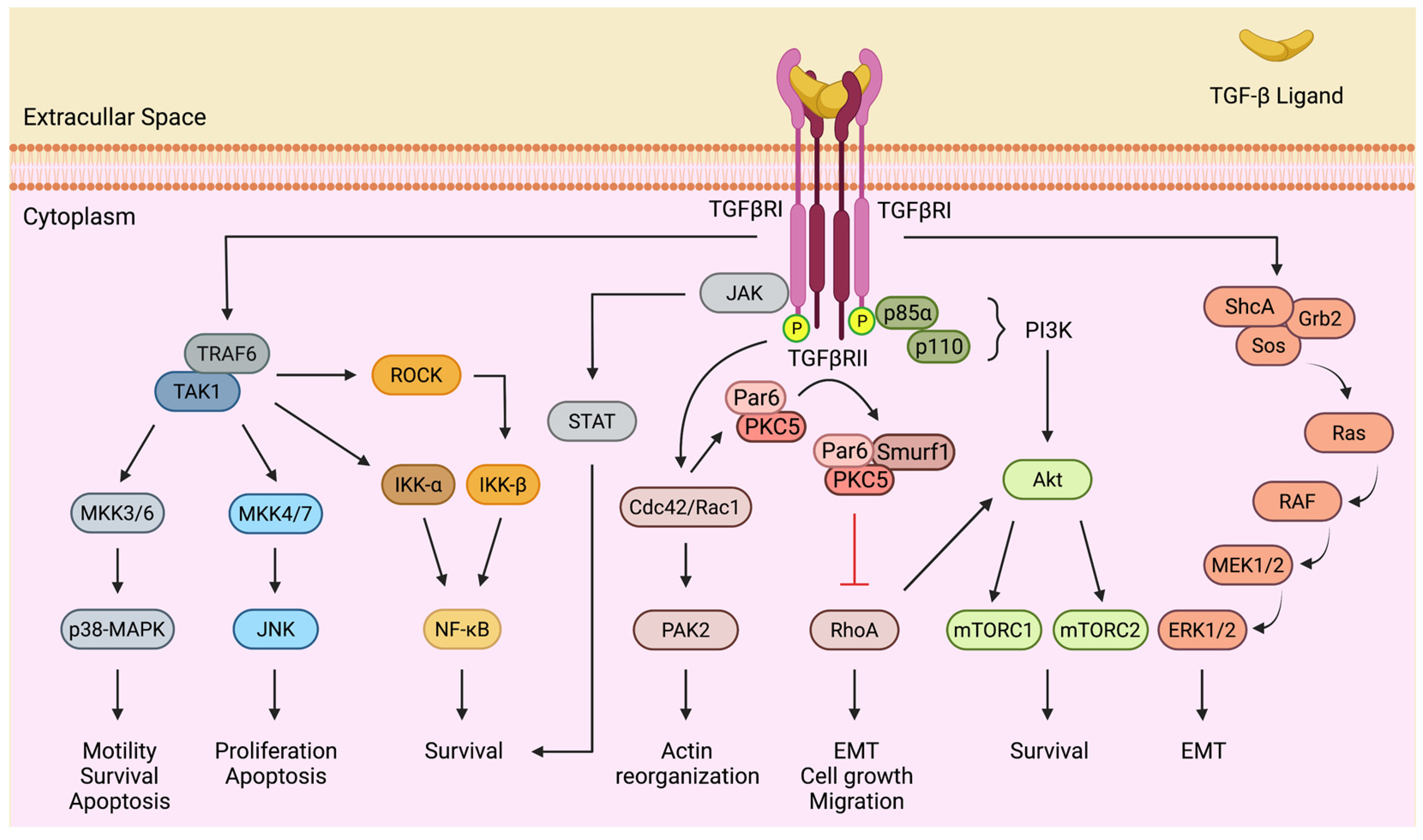
4. The Molecular Mechanisms of TGF-β Signaling
5. The Role of TGF-β Signaling in HCC
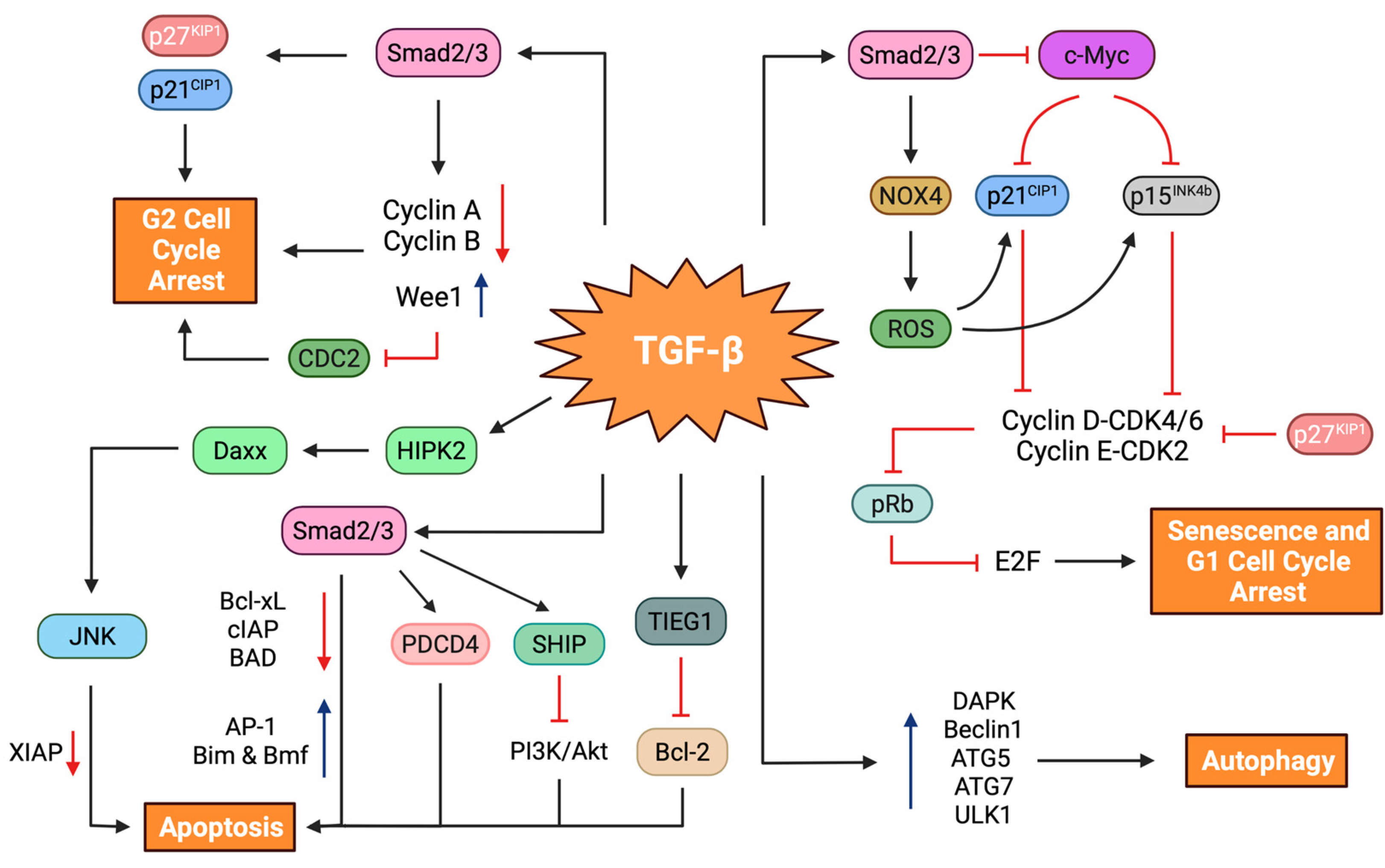
5.1. Tumor Suppressor Role
5.1.1. Cell Cycle Arrest
5.1.2. Cellular Senescence
5.1.3. Autophagy
5.1.4. Apoptosis
5.2. Dysregulation and Loss of the Tumor-Suppressive Functions of TGF-β Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mediator, Regulators, and Effectors | Functions | Dysregulations | Reference |
|---|---|---|---|
| TGFβRII | TGFβRII transmits signals from the cell surface into the cell and is inactivated by hypermethylation in HCC. | Reduced expression in HCC | [168] |
| Smad4 | Smad4 is the main effector in the Smadpathway and is inactivated by hypermethylation in HCC. | Reduced expression in HCC | [169] |
| Caveolin-1 | Caveolin-1 blocks the TGF-β growth-inhibitory responsiveness. | Increased expression in HCC | [170,171] |
| Smad7 | Smad7 interferes with R-Smad complex formation and antagonizes nuclear TGF-β signaling. | Increased expression in HCC | [174,175,176] |
| SPTBN1 | SPTBN1 encodes a Smad adaptor β-spectrin protein that modulates nuclear TGF-β/Smad signaling. | Reduced expression in HCC | [164,167,178] |
| cFLIP | cFLIP is an intracellular inhibitor of caspase-8 activation and a preeminent modulator of NF-κB signaling. | Increased expression in HCC | [183,184] |
| Bax | Bax is a key regulator of the intrinsic pathway of apoptosis and mediates permeabilization of the outer mitochondrial membrane. | Subcellular mislocalization or reduced expression in HCC | [185] |
| Bid | Bid induces permeabilization of the outer mitochondrial membrane. | Subcellular mislocalization or reduced expression in HCC | [186] |
| Zeb2 | Zeb2 is a Smad-interacting transcriptional corepressor. | Increased expression in HCC | [194] |
| Hey2 | Hey2 is a Smad-interacting transcriptional corepressor. | Increased expression in HCC | [189] |
| SRF | SRF is a Smad-interacting transcriptional corepressor. | Increased expression in HCC | [188] |
| SnoN | SnoN is a regulator of TGF-β signaling that binds to the N-terminus of R-Smads. | Abundant expression in HCC | [191] |
| Ski | Ski is a regulator of TGF-β signaling that binds to the N-terminus of R-Smads. | Abundant expression in HCC | [191] |
| EVI1 | EVI1 interacts with Smad3 and blocks TGF-β signaling. | Overexpressed in HCC | [192,193] |
| FHL | FHL proteins are tumor suppressor proteins that act as interaction partners of Smad molecules. | Epigenetically repressed in HCC | [195,196] |
| CXXC5 | CXXC5 enhances TGF-β signaling by forming a positive feedback loop. | Reduced expression in HCC | [77] |
| KLF17 | KLF17 enhances TGF-β signaling by forming a positive feedback loop | Reduced expression in HCC | [197] |
| Grk2 | GRK2 phosphorylates the Smad3 linker region and blocks TGF-β target gene expression. | Increased expression in HCC | [198] |
| Axl | Axl causes aberrant phosphorylation of the Smad3 linker region and enhances TGF-β resistance. | Increased expression in HCC | [199] |
| p15INK4b | p15INK4b plays a role as a cell growth regulator that impedes cell cycle G1 progression. | Deletion or promoter methylation in the INK4 locus | [72,200,201] |
| p16INK4a | p16INK4a plays a role as an inhibitor of CDK4 and CDK6. | Deletion or promoter methylation in the INK4 locus | [72,200,201] |
5.3. Functional Switch of TGF-β Signaling in HCC
5.4. Tumor Promoter Role of TGF-β Signaling in HCC
5.4.1. Cancer Cell Proliferation
5.4.2. RNA Modifications
5.4.3. Epithelial–Mesenchymal Transition and Tumor Microenvironment Remodeling
5.4.4. The Role of TGF-β in Immune Suppression
6. Therapeutic Implications of TGF-β Signaling
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primer 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Mejia, J.C.; Pasko, J. Primary Liver Cancers: Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma. Surg. Clin. 2020, 100, 535–549. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular Therapies and Precision Medicine for Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ Signalling in Context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-Dependent and Smad-Independent Pathways in TGF-β Family Signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-β as Multifaceted Orchestrator in HCC Progression: Signaling, EMT, Immune Microenvironment, and Novel Therapeutic Perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Sabbadini, F.; Bertolini, M.; De Matteis, S.; Mangiameli, D.; Contarelli, S.; Pietrobono, S.; Melisi, D. The Multifaceted Role of TGF-β in Gastrointestinal Tumors. Cancers 2021, 13, 3960. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Roberts, L.R. Hepatocellular Carcinoma: A Global View. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular Carcinoma Pathogenesis: From Genes to Environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Müller, M.; Bird, T.G.; Nault, J.-C. The Landscape of Gene Mutations in Cirrhosis and Hepatocellular Carcinoma. J. Hepatol. 2020, 72, 990–1002. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome Sequencing of Hepatocellular Carcinomas Identifies New Mutational Signatures and Potential Therapeutic Targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-Ancestry Mutational Landscape of Hepatocellular Carcinoma Genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Yang, X.; Guo, X.; Chen, Y.; Chen, G.; Ma, Y.; Huang, K.; Zhang, Y.; Zhao, Q.; Winkler, C.A.; An, P.; et al. Telomerase Reverse Transcriptase Promoter Mutations in Hepatitis B Virus-Associated Hepatocellular Carcinoma. Oncotarget 2016, 7, 27838–27847. [Google Scholar] [CrossRef]
- Heidenreich, B.; Rachakonda, P.S.; Hemminki, K.; Kumar, R. TERT Promoter Mutations in Cancer Development. Curr. Opin. Genet. Dev. 2014, 24, 30–37. [Google Scholar] [CrossRef]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour Evolution in Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-Specific Pten Deficiency Results in Steatohepatitis and Hepatocellular Carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, L.; Guan, X.-Y. The Genetic and Epigenetic Alterations in Human Hepatocellular Carcinoma: A Recent Update. Protein Cell 2014, 5, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-M.; Kim-Ha, J.; Choi, W.-Y.; Lee, J.; Kim, D.; Lee, J.; Choi, E.; Kim, Y.-J. Interplay of Genetic and Epigenetic Alterations in Hepatocellular Carcinoma. Epigenomics 2016, 8, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Korkut, A.; Zaidi, S.; Kanchi, R.S.; Rao, S.; Gough, N.R.; Schultz, A.; Li, X.; Lorenzi, P.L.; Berger, A.C.; Robertson, G.; et al. A Pan-Cancer Analysis Reveals High-Frequency Genetic Alterations in Mediators of Signaling by the TGF-β Superfamily. Cell Syst. 2018, 7, 422–437.e7. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef]
- Wrighton, K.H.; Lin, X.; Feng, X.-H. Phospho-Control of TGF-β Superfamily Signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef]
- Söderberg, S.S.; Karlsson, G.; Karlsson, S. Complex and Context Dependent Regulation of Hematopoiesis by TGF-β Superfamily Signaling. Ann. N. Y. Acad. Sci. 2009, 1176, 55–69. [Google Scholar] [CrossRef]
- Ten Dijke, P.; Hill, C.S. New Insights into TGF-β-Smad Signalling. Trends Biochem. Sci. 2004, 29, 265–273. [Google Scholar] [CrossRef]
- Yan, X.; Liu, Z.; Chen, Y. Regulation of TGF-β Signaling by Smad7. Acta Biochim. Biophys. Sin. 2009, 41, 263–272. [Google Scholar] [CrossRef]
- Brown, N.F.; Marshall, J.F. Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers 2019, 11, 1221. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Ota, M.; Rifkin, D.B. Matrix Control of Transforming Growth Factor-β Function. J. Biochem. 2012, 152, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Sheppard, D. Cross Talk among TGF-β Signaling Pathways, Integrins, and the Extracellular Matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Q.; Zhao, M.; Walton, K.; Harrison, C.; Nie, G. Multiple Soluble TGF-β Receptors in Addition to Soluble Endoglin Are Elevated in Preeclamptic Serum and They Synergistically Inhibit TGF-β Signaling. J. Clin. Endocrinol. Metab. 2017, 102, 3065–3074. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β Receptors: In and beyond TGF-β Signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The Role of TGF-β/SMAD4 Signaling in Cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Kamato, D.; Do, B.H.; Osman, N.; Ross, B.P.; Mohamed, R.; Xu, S.; Little, P.J. Smad Linker Region Phosphorylation Is a Signalling Pathway in Its Own Right and Not Only a Modulator of Canonical TGF-β Signalling. Cell. Mol. Life Sci. CMLS 2020, 77, 243–251. [Google Scholar] [CrossRef]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming Growth Factor-β Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
- Imamura, T.; Takase, M.; Nishihara, A.; Oeda, E.; Hanai, J.; Kawabata, M.; Miyazono, K. Smad6 Inhibits Signalling by the TGF-β Superfamily. Nature 1997, 389, 622–626. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFβ-Inducible Antagonist of TGF-β Signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Lin, X.; Feng, X.-H. Posttranslational Regulation of Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022087. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Pathways in TGF-β Signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Tu, S.; Huang, W.; Huang, C.; Luo, Z.; Yan, X. Contextual Regulation of TGF-β Signaling in Liver Cancer. Cells 2019, 8, 1235. [Google Scholar] [CrossRef]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.-H.; Landström, M. The Type I TGF-β Receptor Engages TRAF6 to Activate TAK1 in a Receptor Kinase-Independent Manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling Interplay between Transforming Growth Factor-β Receptor and PI3K/AKT Pathways in Cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.-F. Signaling Cross-Talk between TGF-β/BMP and Other Pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.-H.; Landström, M. TGF-β Promotes PI3K-AKT Signaling and Prostate Cancer Cell Migration through the TRAF6-Mediated Ubiquitylation of P85α. Sci. Signal. 2017, 10, eaal4186. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-L.; Wang, J.; Chan, C.-H.; Lee, S.-W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 Ligase TRAF6 Regulates Akt Ubiquitination and Activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Lehnert, H. The Role of Small GTPases of the Rho/Rac Family in TGF-β-Induced EMT and Cell Motility in Cancer. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2018, 247, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.-R.; Zhang, Y.; Wrana, J.L. Regulation of the Polarity Protein Par6 by TGFβ Receptors Controls Epithelial Cell Plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef]
- Tang, L.-Y.; Heller, M.; Meng, Z.; Yu, L.-R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-β (TGF-β) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef]
- Yamamoto, T.; Matsuda, T.; Muraguchi, A.; Miyazono, K.; Kawabata, M. Cross-Talk between IL-6 and TGF-β Signaling in Hepatoma Cells. FEBS Lett. 2001, 492, 247–253. [Google Scholar] [CrossRef]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-β IL-6 Axis Mediates Selective and Adaptive Mechanisms of Resistance to Molecular Targeted Therapy in Lung Cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef]
- Yu, Y.; Feng, X.-H. TGF-β Signaling in Cell Fate Control and Cancer. Curr. Opin. Cell Biol. 2019, 61, 56–63. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad Signaling Pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Padua, D.; Massagué, J. Roles of TGFβ in Metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of Function between Tumor Prevention and Carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell Cycle Control in Cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming Growth Factor-β Induces Senescence in Hepatocellular Carcinoma Cells and Inhibits Tumor Growth. Hepatology 2010, 52, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, Y.; Wang, F.; Guo, J.; Fu, W.; Li, M.; Zheng, Q.; Liu, Y.; Fan, L.; Li, L.; et al. Bmi1 Drives Hepatocarcinogenesis by Repressing the TGFβ2/SMAD Signalling Axis. Oncogene 2020, 39, 1063–1079. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFβ Signaling in Growth Control, Cancer, and Heritable Disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Gomis, R.R.; Alarcón, C.; He, W.; Wang, Q.; Seoane, J.; Lash, A.; Massagué, J. A FoxO-Smad Synexpression Group in Human Keratinocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 12747–12752. [Google Scholar] [CrossRef]
- Seoane, J.; Le, H.-V.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and Forkhead Pathways in the Control of Neuroepithelial and Glioblastoma Cell Proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Claassen, G.F.; Hann, S.R. A Role for Transcriptional Repression of P21CIP1 by C-Myc in Overcoming Transforming Growth Factor β -Induced Cell-Cycle Arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 9498–9503. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A.M. Control of Cell Cycle Transcription during G1 and S Phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.-F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [PubMed]
- Gungor, M.Z.; Uysal, M.; Ozturk, M.; Senturk, S. Systematic Analysis of Cytostatic TGF-β Response in Mesenchymal-Like Hepatocellular Carcinoma Cell Lines. J. Gastrointest. Cancer 2021, 52, 1320–1335. [Google Scholar] [CrossRef] [PubMed]
- Kitisin, K.; Ganesan, N.; Tang, Y.; Jogunoori, W.; Volpe, E.A.; Kim, S.S.; Katuri, V.; Kallakury, B.; Pishvaian, M.; Albanese, C.; et al. Disruption of Transforming Growth Factor-β Signaling through β-Spectrin ELF Leads to Hepatocellular Cancer through Cyclin D1 Activation. Oncogene 2007, 26, 7103–7110. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, O.; Ueno, T.; Kimura, R.; Ohtsubo, M.; Nakamura, T.; Koga, H.; Torimura, T.; Uchida, S.; Yamashita, K.; Sata, M. Inhibition of Proteasome-Dependent Degradation of Wee1 in G2-Arrested Hep3B Cells by TGF β 1. Mol. Carcinog. 2003, 36, 171–182. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, Q.; Li, Y.; Yang, Z.; Yang, L.; Zong, Z.; Wang, B.; Meng, X.; Li, Q.; Liu, J.; et al. Transforming Growth Factor-β1 Suppresses Hepatocellular Carcinoma Proliferation via Activation of Hippo Signaling. Oncotarget 2017, 8, 29785–29794. [Google Scholar] [CrossRef]
- Kim, J.B.; Lee, S.; Kim, H.R.; Park, S.-Y.; Lee, M.; Yoon, J.-H.; Kim, Y.J. Transforming Growth Factor-β Decreases Side Population Cells in Hepatocellular Carcinoma in Vitro. Oncol. Lett. 2018, 15, 8723–8728. [Google Scholar] [CrossRef]
- Yan, X.; Wu, J.; Jiang, Q.; Cheng, H.; Han, J.-D.J.; Chen, Y.-G. CXXC5 Suppresses Hepatocellular Carcinoma by Promoting TGF-β-Induced Cell Cycle Arrest and Apoptosis. J. Mol. Cell Biol. 2018, 10, 48–59. [Google Scholar] [CrossRef]
- Demirci, D.; Dayanc, B.; Mazi, F.A.; Senturk, S. The Jekyll and Hyde of Cellular Senescence in Cancer. Cells 2021, 10, 208. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence-Stemness Alliance—A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the Unique Role of the Retinoblastoma Tumor Suppressor during Cellular Senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Huda, N.; Liu, G.; Hong, H.; Yan, S.; Khambu, B.; Yin, X.-M. Hepatic Senescence, the Good and the Bad. World J. Gastroenterol. 2019, 25, 5069–5081. [Google Scholar] [CrossRef]
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in Chronic Liver Disease: Is the Future in Aging? J. Hepatol. 2016, 65, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and Tumour Clearance Is Triggered by P53 Restoration in Murine Liver Carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence Surveillance of Pre-Malignant Hepatocytes Limits Liver Cancer Development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of P53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Romero-Gallo, J.; Sozmen, E.G.; Chytil, A.; Russell, W.E.; Whitehead, R.; Parks, W.T.; Holdren, M.S.; Her, M.F.; Gautam, S.; Magnuson, M.; et al. Inactivation of TGF-β Signaling in Hepatocytes Results in an Increased Proliferative Response after Partial Hepatectomy. Oncogene 2005, 24, 3028–3041. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P. IT-LIVER Consortium TGF-β Signalling and Liver Disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, M.; Arslan-Ergul, A.; Bagislar, S.; Senturk, S.; Yuzugullu, H. Senescence and Immortality in Hepatocellular Carcinoma. Cancer Lett. 2009, 286, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Kim, H.-J.; Yoon, Y.-S.; Cho, H.; Lim, I.K.; Lee, J.-H. Iron Chelation-Induced Senescence-like Growth Arrest in Hepatocyte Cell Lines: Association of Transforming Growth Factor β1 (TGF-β1)-Mediated P27Kip1 Expression. Biochem. J. 2002, 366, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.K.; Chua, M.-S.; He, J.; So, S.K. Suppression of Glypican 3 Inhibits Growth of Hepatocellular Carcinoma Cells through Up-Regulation of TGF-β2. Neoplasia 2011, 13, 735–747. [Google Scholar] [CrossRef]
- Karabicici, M.; Alptekin, S.; Fırtına Karagonlar, Z.; Erdal, E. Doxorubicin-Induced Senescence Promotes Stemness and Tumorigenicity in EpCAM-/CD133- Nonstem Cell Population in Hepatocellular Carcinoma Cell Line, HuH-7. Mol. Oncol. 2021, 15, 2185–2202. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Wiemann, S.U.; Satyanarayana, A.; Tsahuridu, M.; Tillmann, H.L.; Zender, L.; Klempnauer, J.; Flemming, P.; Franco, S.; Blasco, M.A.; Manns, M.P.; et al. Hepatocyte Telomere Shortening and Senescence Are General Markers of Human Liver Cirrhosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 935–942. [Google Scholar] [CrossRef]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef]
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular Senescence: Immunosurveillance and Future Senescence-Induced Therapy in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 589908. [Google Scholar] [CrossRef]
- Nault, J.-C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The Role of Telomeres and Telomerase in Cirrhosis and Liver Cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Lin, S.Y.; Elledge, S.J. Multiple Tumor Suppressor Pathways Negatively Regulate Telomerase. Cell 2003, 113, 881–889. [Google Scholar] [CrossRef]
- Li, H.; Xu, D.; Li, J.; Berndt, M.C.; Liu, J.-P. Transforming Growth Factor β Suppresses Human Telomerase Reverse Transcriptase (HTERT) by Smad3 Interactions with c-Myc and the HTERT Gene. J. Biol. Chem. 2006, 281, 25588–25600. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. TGF-β in Progression of Liver Disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Kim, J.-S.; Nitta, T.; Mohuczy, D.; O’Malley, K.A.; Moldawer, L.L.; Dunn, W.A.; Behrns, K.E. Impaired Autophagy: A Mechanism of Mitochondrial Dysfunction in Anoxic Rat Hepatocytes. Hepatology 2008, 47, 1725–1736. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the Liver: Functions in Health and Disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Teckman, J.H.; Perlmutter, D.H. Retention of Mutant α1-Antitrypsin Z in Endoplasmic Reticulum Is Associated with an Autophagic Response. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G961–G974. [Google Scholar] [CrossRef]
- Ke, P.-Y. Diverse Functions of Autophagy in Liver Physiology and Liver Diseases. Int. J. Mol. Sci. 2019, 20, 300. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis through Elimination of P62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-Deficient Mice Develop Multiple Liver Tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.T.; Soh, N.J.H.; Chang, H.-C.; Yu, V.C. P62/SQSTM1 in Liver Diseases: The Usual Suspect with Multifarious Identities. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an Autophagy Gene Essential for Early Embryonic Development, Is a Haploinsufficient Tumor Suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of Autophagy and Inhibition of Tumorigenesis by Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- White, E. The Role for Autophagy in Cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and Signaling: Their Role in Cell Survival and Cell Death. Cell Death Differ. 2005, 12 (Suppl 2), 1509–1518. [Google Scholar] [CrossRef]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic Programmed Cell Death by Selective Catalase Degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef]
- Ding, Y.; Kim, S.I.; Lee, S.-Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy Regulates TGF-β Expression and Suppresses Kidney Fibrosis Induced by Unilateral Ureteral Obstruction. J. Am. Soc. Nephrol. JASN 2014, 25, 2835–2846. [Google Scholar] [CrossRef]
- Ding, Y.; Choi, M.E. Regulation of Autophagy by TGF-β: Emerging Role in Kidney Fibrosis. Semin. Nephrol. 2014, 34, 62–71. [Google Scholar] [CrossRef]
- Ghavami, S.; Cunnington, R.H.; Gupta, S.; Yeganeh, B.; Filomeno, K.L.; Freed, D.H.; Chen, S.; Klonisch, T.; Halayko, A.J.; Ambrose, E.; et al. Autophagy Is a Regulator of TGF-β1-Induced Fibrogenesis in Primary Human Atrial Myofibroblasts. Cell Death Dis. 2015, 6, e1696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, N.; Ping, J.; Xu, L. TGF-β1-induced Autophagy Activates Hepatic Stellate Cells via the ERK and JNK Signaling Pathways. Int. J. Mol. Med. 2021, 47, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Kiyono, K.; Miyazono, K. Regulation of Autophagy by Transforming Growth Factor-β (TGF-β) Signaling. Autophagy 2010, 6, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Trelford, C.B.; Di Guglielmo, G.M. Assessing Methods to Quantitatively Validate TGFβ-Dependent Autophagy. Biol. Open 2020, 9, bio055103. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy Is Activated by TGF-β and Potentiates TGF-β-Mediated Growth Inhibition in Human Hepatocellular Carcinoma Cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef] [PubMed]
- Trelford, C.B.; Di Guglielmo, G.M. Canonical and Non-Canonical TGFβ Signaling Activate Autophagy in an ULK1-Dependent Manner. Front. Cell Dev. Biol. 2021, 9, 712124. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy Promotes Tumor Cell Survival and Restricts Necrosis, Inflammation, and Tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy Promotes Hepatocellular Carcinoma Cell Invasion through Activation of Epithelial-Mesenchymal Transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef]
- Hardy, S.D.; Shinde, A.; Wang, W.-H.; Wendt, M.K.; Geahlen, R.L. Regulation of Epithelial-Mesenchymal Transition and Metastasis by TGF-β, P-Bodies, and Autophagy. Oncotarget 2017, 8, 103302–103314. [Google Scholar] [CrossRef]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy Modulates Transforming Growth Factor β 1 Induced Epithelial to Mesenchymal Transition in Non-Small Cell Lung Cancer Cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef] [PubMed]
- Dünker, N.; Schmitt, K.; Krieglstein, K. TGF-β Is Required for Programmed Cell Death in Interdigital Webs of the Developing Mouse Limb. Mech. Dev. 2002, 113, 111–120. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Pollard, J.W. Transforming Growth Factor β3 Induces Cell Death during the First Stage of Mammary Gland Involution. Development 2000, 127, 3107–3118. [Google Scholar] [CrossRef] [PubMed]
- Levéen, P.; Larsson, J.; Ehinger, M.; Cilio, C.M.; Sundler, M.; Sjöstrand, L.J.; Holmdahl, R.; Karlsson, S. Induced Disruption of the Transforming Growth Factor β Type II Receptor Gene in Mice Causes a Lethal Inflammatory Disorder That Is Transplantable. Blood 2002, 100, 560–568. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming Growth Factor-β Regulation of Immune Responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Oberhammer, F.; Bursch, W.; Tiefenbacher, R.; Fröschl, G.; Pavelka, M.; Purchio, T.; Schulte-Hermann, R. Apoptosis Is Induced by Transforming Growth Factor-β 1 within 5 Hours in Regressing Liver without Significant Fragmentation of the DNA. Hepatology 1993, 18, 1238–1246. [Google Scholar] [CrossRef]
- Oberhammer, F.A.; Pavelka, M.; Sharma, S.; Tiefenbacher, R.; Purchio, A.F.; Bursch, W.; Schulte-Hermann, R. Induction of Apoptosis in Cultured Hepatocytes and in Regressing Liver by Transforming Growth Factor β 1. Proc. Natl. Acad. Sci. USA 1992, 89, 5408–5412. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernández, M.; Fabregat, I. Upregulation of the NADPH Oxidase NOX4 by TGF-β in Hepatocytes Is Required for Its pro-Apoptotic Activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef]
- Kumar, S. Caspase Function in Programmed Cell Death. Cell Death Differ. 2007, 14, 32–43. [Google Scholar] [CrossRef]
- Kim, B.-C.; Mamura, M.; Choi, K.S.; Calabretta, B.; Kim, S.-J. Transforming Growth Factor β1 Induces Apoptosis through Cleavage of BAD in a Smad3-Dependent Mechanism in FaO Hepatoma Cells. Mol. Cell. Biol. 2002, 22, 1369–1378. [Google Scholar] [CrossRef]
- Shima, Y.; Nakao, K.; Nakashima, T.; Kawakami, A.; Nakata, K.; Hamasaki, K.; Kato, Y.; Eguchi, K.; Ishii, N. Activation of Caspase-8 in Transforming Growth Factor-β-Induced Apoptosis of Human Hepatoma Cells. Hepatology 1999, 30, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.; Fernández, M.; Benito, M.; Fabregat, I. CIAP-1, but Not XIAP, Is Cleaved by Caspases during the Apoptosis Induced by TGF-β in Fetal Rat Hepatocytes. FEBS Lett. 2002, 520, 93–96. [Google Scholar] [CrossRef]
- Tachibana, I.; Imoto, M.; Adjei, P.N.; Gores, G.J.; Subramaniam, M.; Spelsberg, T.C.; Urrutia, R. Overexpression of the TGFβ-Regulated Zinc Finger Encoding Gene, TIEG, Induces Apoptosis in Pancreatic Epithelial Cells. J. Clin. Investig. 1997, 99, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Yang, Y.-A.; Zhang, G.-M.; Feigenbaum, L.; Zhang, Y.E. Smad3 Reduces Susceptibility to Hepatocarcinoma by Sensitizing Hepatocytes to Apoptosis through Downregulation of Bcl-2. Cancer Cell 2006, 9, 445–457. [Google Scholar] [CrossRef]
- Ramjaun, A.R.; Tomlinson, S.; Eddaoudi, A.; Downward, J. Upregulation of Two BH3-Only Proteins, Bmf and Bim, during TGF β-Induced Apoptosis. Oncogene 2007, 26, 970–981. [Google Scholar] [CrossRef]
- Ramesh, S.; Qi, X.-J.; Wildey, G.M.; Robinson, J.; Molkentin, J.; Letterio, J.; Howe, P.H. TGF β-Mediated BIM Expression and Apoptosis Are Regulated through SMAD3-Dependent Expression of the MAPK Phosphatase MKP2. EMBO Rep. 2008, 9, 990–997. [Google Scholar] [CrossRef]
- Jang, C.-W.; Chen, C.-H.; Chen, C.-C.; Chen, J.; Su, Y.-H.; Chen, R.-H. TGF-β Induces Apoptosis through Smad-Mediated Expression of DAP-Kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef]
- Fabregat, I.; Roncero, C.; Fernández, M. Survival and Apoptosis: A Dysregulated Balance in Liver Cancer. Liver Int. Off. J. Int. Assoc. Study Liver 2007, 27, 155–162. [Google Scholar] [CrossRef]
- Yoo, J.; Ghiassi, M.; Jirmanova, L.; Balliet, A.G.; Hoffman, B.; Fornace, A.J.; Liebermann, D.A.; Bottinger, E.P.; Roberts, A.B. Transforming Growth Factor-β-Induced Apoptosis Is Mediated by Smad-Dependent Expression of GADD45b through P38 Activation. J. Biol. Chem. 2003, 278, 43001–43007. [Google Scholar] [CrossRef]
- Perlman, R.; Schiemann, W.P.; Brooks, M.W.; Lodish, H.F.; Weinberg, R.A. TGF-β-Induced Apoptosis Is Mediated by the Adapter Protein Daxx That Facilitates JNK Activation. Nat. Cell Biol. 2001, 3, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.G.; Stollberg, N.; Schmitz, M.L.; Will, H. HIPK2 Regulates Transforming Growth Factor-β-Induced c-Jun NH2-Terminal Kinase Activation and Apoptosis in Human Hepatoma Cells. Cancer Res. 2003, 63, 8271–8277. [Google Scholar] [PubMed]
- Matsuhashi, S.; Manirujjaman, M.; Hamajima, H.; Ozaki, I. Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions. Int. J. Mol. Sci. 2019, 20, 2304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ozaki, I.; Mizuta, T.; Hamajima, H.; Yasutake, T.; Eguchi, Y.; Ideguchi, H.; Yamamoto, K.; Matsuhashi, S. Involvement of Programmed Cell Death 4 in Transforming Growth Factor-β1-Induced Apoptosis in Human Hepatocellular Carcinoma. Oncogene 2006, 25, 6101–6112. [Google Scholar] [CrossRef] [PubMed]
- Valderrama-Carvajal, H.; Cocolakis, E.; Lacerte, A.; Lee, E.-H.; Krystal, G.; Ali, S.; Lebrun, J.-J. Activin/TGF-β Induce Apoptosis through Smad-Dependent Expression of the Lipid Phosphatase SHIP. Nat. Cell Biol. 2002, 4, 963–969. [Google Scholar] [CrossRef]
- Kaur, S.; Wang, F.; Venkatraman, M.; Arsura, M. X-Linked Inhibitor of Apoptosis (XIAP) Inhibits c-Jun N-Terminal Kinase 1 (JNK1) Activation by Transforming Growth Factor β1 (TGF-β1) through Ubiquitin-Mediated Proteosomal Degradation of the TGF-β1-Activated Kinase 1 (TAK1). J. Biol. Chem. 2005, 280, 38599–38608. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 Mediates Smad-Independent Activation of JNK and P38 by TGF-β. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef]
- Park, S.S.; Eom, Y.-W.; Kim, E.H.; Lee, J.H.; Min, D.S.; Kim, S.; Kim, S.-J.; Choi, K.S. Involvement of C-Src Kinase in the Regulation of TGF-β1-Induced Apoptosis. Oncogene 2004, 23, 6272–6281. [Google Scholar] [CrossRef][Green Version]
- Black, D.; Lyman, S.; Qian, T.; Lemasters, J.J.; Rippe, R.A.; Nitta, T.; Kim, J.-S.; Behrns, K.E. Transforming Growth Factor β Mediates Hepatocyte Apoptosis through Smad3 Generation of Reactive Oxygen Species. Biochimie 2007, 89, 1464–1473. [Google Scholar] [CrossRef][Green Version]
- Herrera, B.; Murillo, M.M.; Alvarez-Barrientos, A.; Beltrán, J.; Fernández, M.; Fabregat, I. Source of Early Reactive Oxygen Species in the Apoptosis Induced by Transforming Growth Factor-β in Fetal Rat Hepatocytes. Free Radic. Biol. Med. 2004, 36, 16–26. [Google Scholar] [CrossRef]
- Herrera, B.; Fernández, M.; Alvarez, A.M.; Roncero, C.; Benito, M.; Gil, J.; Fabregat, I. Activation of Caspases Occurs Downstream from Radical Oxygen Species Production, Bcl-XL down-Regulation, and Early Cytochrome C Release in Apoptosis Induced by Transforming Growth Factor β in Rat Fetal Hepatocytes. Hepatology 2001, 34, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Herzer, K.; Grosse-Wilde, A.; Krammer, P.H.; Galle, P.R.; Kanzler, S. Transforming Growth Factor-β-Mediated Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Expression and Apoptosis in Hepatoma Cells Requires Functional Cooperation between Smad Proteins and Activator Protein-1. Mol. Cancer Res. MCR 2008, 6, 1169–1177. [Google Scholar] [CrossRef] [PubMed][Green Version]
- David, C.J.; Massagué, J. Contextual Determinants of TGFβ Action in Development, Immunity and Cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zaidi, S.; Rao, S.; Chen, J.-S.; Phan, L.; Farci, P.; Su, X.; Shetty, K.; White, J.; Zamboni, F.; et al. Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-β Pathway. Gastroenterology 2018, 154, 195–210. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [CrossRef]
- Chen, J.; Gingold, J.A.; Su, X. Immunomodulatory TGF-β Signaling in Hepatocellular Carcinoma. Trends Mol. Med. 2019, 25, 1010–1023. [Google Scholar] [CrossRef]
- Mamiya, T.; Yamazaki, K.; Masugi, Y.; Mori, T.; Effendi, K.; Du, W.; Hibi, T.; Tanabe, M.; Ueda, M.; Takayama, T.; et al. Reduced Transforming Growth Factor-β Receptor II Expression in Hepatocellular Carcinoma Correlates with Intrahepatic Metastasis. Lab. Investig. J. Tech. Methods Pathol. 2010, 90, 1339–1345. [Google Scholar] [CrossRef]
- Mu, X.; Lin, S.; Yang, J.; Chen, C.; Chen, Y.; Herzig, M.C.; Washburn, K.; Halff, G.A.; Walter, C.A.; Sun, B.; et al. TGF-β Signaling Is Often Attenuated during Hepatotumorigenesis, but Is Retained for the Malignancy of Hepatocellular Carcinoma Cells. PLoS ONE 2013, 8, e63436. [Google Scholar] [CrossRef]
- Meyer, C.; Liu, Y.; Kaul, A.; Peipe, I.; Dooley, S. Caveolin-1 Abrogates TGF-β Mediated Hepatocyte Apoptosis. Cell Death Dis. 2013, 4, e466. [Google Scholar] [CrossRef]
- Moreno-Càceres, J.; Caballero-Díaz, D.; Nwosu, Z.C.; Meyer, C.; López-Luque, J.; Malfettone, A.; Lastra, R.; Serrano, T.; Ramos, E.; Dooley, S.; et al. The Level of Caveolin-1 Expression Determines Response to TGF-β as a Tumour Suppressor in Hepatocellular Carcinoma Cells. Cell Death Dis. 2017, 8, e3098. [Google Scholar] [CrossRef] [PubMed]
- Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Transforming Growth Factor-β Gene Expression Signature in Mouse Hepatocytes Predicts Clinical Outcome in Human Cancer. Hepatology 2008, 47, 2059–2067. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Mikulits, W.; Dooley, S.; Fabregat, I.; Moustakas, A.; ten Dijke, P.; Portincasa, P.; Winter, P.; Janssen, R.; Leporatti, S.; et al. The Rationale for Targeting TGF-β in Chronic Liver Diseases. Eur. J. Clin. Investig. 2016, 46, 349–361. [Google Scholar] [CrossRef]
- Wang, H.; Song, X.; Liao, H.; Wang, P.; Zhang, Y.; Che, L.; Zhang, J.; Zhou, Y.; Cigliano, A.; Ament, C.; et al. Overexpression of Mothers Against Decapentaplegic Homolog 7 Activates the Yes-Associated Protein/NOTCH Cascade and Promotes Liver Carcinogenesis in Mice and Humans. Hepatology 2021, 74, 248–263. [Google Scholar] [CrossRef]
- Zhang, S.; Fei, T.; Zhang, L.; Zhang, R.; Chen, F.; Ning, Y.; Han, Y.; Feng, X.-H.; Meng, A.; Chen, Y.-G. Smad7 Antagonizes Transforming Growth Factor β Signaling in the Nucleus by Interfering with Functional Smad-DNA Complex Formation. Mol. Cell. Biol. 2007, 27, 4488–4499. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef]
- Baek, H.J.; Pishvaian, M.J.; Tang, Y.; Kim, T.H.; Yang, S.; Zouhairi, M.E.; Mendelson, J.; Shetty, K.; Kallakury, B.; Berry, D.L.; et al. Transforming Growth Factor-β Adaptor, β2-Spectrin, Modulates Cyclin Dependent Kinase 4 to Reduce Development of Hepatocellular Cancer. Hepatology 2011, 53, 1676–1684. [Google Scholar] [CrossRef]
- Yao, Z.-X.; Jogunoori, W.; Choufani, S.; Rashid, A.; Blake, T.; Yao, W.; Kreishman, P.; Amin, R.; Sidawy, A.A.; Evans, S.R.T.; et al. Epigenetic Silencing of β-Spectrin, a TGF-β Signaling/Scaffolding Protein in a Human Cancer Stem Cell Disorder: Beckwith-Wiedemann Syndrome. J. Biol. Chem. 2010, 285, 36112–36120. [Google Scholar] [CrossRef]
- Tang, Y.; Katuri, V.; Dillner, A.; Mishra, B.; Deng, C.-X.; Mishra, L. Disruption of Transforming Growth Factor-β Signaling in ELF β-Spectrin-Deficient Mice. Science 2003, 299, 574–577. [Google Scholar] [CrossRef]
- Fabregat, I. Dysregulation of Apoptosis in Hepatocellular Carcinoma Cells. World J. Gastroenterol. 2009, 15, 513–520. [Google Scholar] [CrossRef]
- Karin, M. NF-KappaB as a Critical Link between Inflammation and Cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, Death, and Autophagy in Cancer: NF-ΚB Turns up Everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Luedde, T. Apoptosis and Necroptosis in the Liver: A Matter of Life and Death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Shiraki, K.; Inoue, H.; Kawakita, T.; Yamanaka, T.; Deguchi, M.; Sugimoto, K.; Sakai, T.; Ohmori, S.; Fujikawa, K.; et al. Cellular FLICE/Caspase-8-Inhibitory Protein as a Principal Regulator of Cell Death and Survival in Human Hepatocellular Carcinoma. Lab. Investig. J. Tech. Methods Pathol. 2003, 83, 1033–1043. [Google Scholar] [CrossRef]
- Funk, K.; Czauderna, C.; Klesse, R.; Becker, D.; Hajduk, J.; Oelgeklaus, A.; Reichenbach, F.; Fimm-Todt, F.; Lauterwasser, J.; Galle, P.R.; et al. BAX Redistribution Induces Apoptosis Resistance and Selective Stress Sensitivity in Human HCC. Cancers 2020, 12, 1437. [Google Scholar] [CrossRef]
- Beerheide, W.; Tan, Y.J.; Teng, E.; Ting, A.E.; Jedpiyawongse, A.; Srivatanakul, P. Downregulation of Proapoptotic Proteins Bax and Bcl-X(S) in P53 Overexpressing Hepatocellular Carcinomas. Biochem. Biophys. Res. Commun. 2000, 273, 54–61. [Google Scholar] [CrossRef]
- Tang, J.; Gifford, C.C.; Samarakoon, R.; Higgins, P.J. Deregulation of Negative Controls on TGF-β1 Signaling in Tumor Progression. Cancers 2018, 10, 159. [Google Scholar] [CrossRef]
- Lee, H.-J.; Yun, C.-H.; Lim, S.H.; Kim, B.-C.; Baik, K.G.; Kim, J.-M.; Kim, W.-H.; Kim, S.-J. SRF Is a Nuclear Repressor of Smad3-Mediated TGF-β Signaling. Oncogene 2007, 26, 173–185. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Zhu, B.; Zhang, Y.; Saiyin, H.; Wumaier, R.; Yu, L.; Sun, L.; Xiao, Q. HEY2 Acting as a Co-Repressor with Smad3 and Smad4 Interferes with the Response of TGF-β in Hepatocellular Carcinoma. Am. J. Transl. Res. 2019, 11, 4367–4381. [Google Scholar]
- Safaee, S.; Fardi, M.; Hemmat, N.; Khosravi, N.; Derakhshani, A.; Silvestris, N.; Baradaran, B. Silencing ZEB2 Induces Apoptosis and Reduces Viability in Glioblastoma Cell Lines. Molecules 2021, 26, 901. [Google Scholar] [CrossRef]
- He, J.; Tegen, S.B.; Krawitz, A.R.; Martin, G.S.; Luo, K. The Transforming Activity of Ski and SnoN Is Dependent on Their Ability to Repress the Activity of Smad Proteins. J. Biol. Chem. 2003, 278, 30540–30547. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Konishi, C.; Gen, Y.; Endo, M.; Dohi, O.; Tomie, A.; Kitaichi, T.; Yamada, N.; Iwai, N.; Nishikawa, T.; et al. EVI1, a Target Gene for Amplification at 3q26, Antagonizes Transforming Growth Factor-β-Mediated Growth Inhibition in Hepatocellular Carcinoma. Cancer Sci. 2015, 106, 929–937. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kurokawa, M.; Mitani, K.; Irie, K.; Matsuyama, T.; Takahashi, T.; Chiba, S.; Yazaki, Y.; Matsumoto, K.; Hirai, H. The Oncoprotein Evi-1 Represses TGF-β Signalling by Inhibiting Smad3. Nature 1998, 394, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-B.; Gao, F.-Z.; Chen, X.-F. MiRNA-1179 Suppresses the Metastasis of Hepatocellular Carcinoma by Interacting with ZEB2. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5149–5157. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, Z.; Yan, J.; Yang, X.; Liu, A.; Qiu, W.; Zhu, J.; Han, J.; Zhang, H.; Lin, J.; et al. Human Four-and-a-Half LIM Family Members Suppress Tumor Cell Growth through a TGF-β-like Signaling Pathway. J. Clin. Investig. 2009, 119, 349–361. [Google Scholar] [CrossRef]
- Wang, J.; Huang, F.; Huang, J.; Kong, J.; Liu, S.; Jin, J. Epigenetic Analysis of FHL1 Tumor Suppressor Gene in Human Liver Cancer. Oncol. Lett. 2017, 14, 6109–6116. [Google Scholar] [CrossRef]
- Ali, A.; Zhang, P.; Liangfang, Y.; Wenshe, S.; Wang, H.; Lin, X.; Dai, Y.; Feng, X.-H.; Moses, R.; Wang, D.; et al. KLF17 Empowers TGF-β/Smad Signaling by Targeting Smad3-Dependent Pathway to Suppress Tumor Growth and Metastasis during Cancer Progression. Cell Death Dis. 2015, 6, e1681. [Google Scholar] [CrossRef]
- Ho, J.; Cocolakis, E.; Dumas, V.M.; Posner, B.I.; Laporte, S.A.; Lebrun, J.-J. The G Protein-Coupled Receptor Kinase-2 Is a TGFβ-Inducible Antagonist of TGFβ Signal Transduction. EMBO J. 2005, 24, 3247–3258. [Google Scholar] [CrossRef]
- Reichl, P.; Dengler, M.; van Zijl, F.; Huber, H.; Führlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl Activates Autocrine Transforming Growth Factor-β Signaling in Hepatocellular Carcinoma. Hepatology 2015, 61, 930–941. [Google Scholar] [CrossRef]
- Yang, B.; Guo, M.; Herman, J.G.; Clark, D.P. Aberrant Promoter Methylation Profiles of Tumor Suppressor Genes in Hepatocellular Carcinoma. Am. J. Pathol. 2003, 163, 1101–1107. [Google Scholar] [CrossRef]
- Roussel, M.F. The INK4 Family of Cell Cycle Inhibitors in Cancer. Oncogene 1999, 18, 5311–5317. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Pang, L.; Dai, W.; Wu, S.; Ren, T.; Duan, Y.; Zheng, Y.; Bi, S.; Zhang, X.; Kong, J. Role of Forkhead Box O Proteins in Hepatocellular Carcinoma Biology and Progression (Review). Front. Oncol. 2021, 11, 667730. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.N.M.; Fuchs, E.; Mikula, M.; Huber, H.; Beug, H.; Mikulits, W. PDGF Essentially Links TGF-β Signaling to Nuclear β-Catenin Accumulation in Hepatocellular Carcinoma Progression. Oncogene 2007, 26, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Murillo, M.M.; del Castillo, G.; Sánchez, A.; Fernández, M.; Fabregat, I. Involvement of EGF Receptor and C-Src in the Survival Signals Induced by TGF-β1 in Hepatocytes. Oncogene 2005, 24, 4580–4587. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Bertran, E.; Campbell, J.; Fausto, N.; Fabregat, I. The Transforming Growth Factor-β (TGF-β) Mediates Acquisition of a Mesenchymal Stem Cell-like Phenotype in Human Liver Cells. J. Cell. Physiol. 2011, 226, 1214–1223. [Google Scholar] [CrossRef]
- Caballero-Díaz, D.; Bertran, E.; Peñuelas-Haro, I.; Moreno-Càceres, J.; Malfettone, A.; López-Luque, J.; Addante, A.; Herrera, B.; Sánchez, A.; Alay, A.; et al. Clathrin Switches Transforming Growth Factor-β Role to pro-Tumorigenic in Liver Cancer. J. Hepatol. 2020, 72, 125–134. [Google Scholar] [CrossRef]
- Dolicka, D.; Sobolewski, C.; Gjorgjieva, M.; Correia de Sousa, M.; Berthou, F.; De Vito, C.; Colin, D.J.; Bejuy, O.; Fournier, M.; Maeder, C.; et al. Tristetraprolin Promotes Hepatic Inflammation and Tumor Initiation but Restrains Cancer Progression to Malignancy. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 597–621. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, K.; Yan, W.; Hu, L.; Yuan, J.; Dong, Y.; Li, Y.; Jing, K.; Yang, Y.; Guo, M. Epigenetic Silencing of DACH1 Induces Loss of Transforming Growth Factor-β1 Antiproliferative Response in Human Hepatocellular Carcinoma. Hepatology 2013, 58, 2012–2022. [Google Scholar] [CrossRef]
- Bévant, K.; Desoteux, M.; Abdel Wahab, A.H.A.; Abdel Wahab, S.A.; Metwally, A.M.; Coulouarn, C. DNA Methylation of TGFβ Target Genes: Epigenetic Control of TGFβ Functional Duality in Liver Cancer. Cells 2021, 10, 2207. [Google Scholar] [CrossRef]
- Ooshima, A.; Park, J.; Kim, S.-J. Phosphorylation Status at Smad3 Linker Region Modulates Transforming Growth Factor-β-Induced Epithelial-Mesenchymal Transition and Cancer Progression. Cancer Sci. 2019, 110, 481–488. [Google Scholar] [CrossRef]
- Matsuzaki, K. Smad Phosphoisoform Signaling Specificity: The Right Place at the Right Time. Carcinogenesis 2011, 32, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K.; Murata, M.; Yamaguchi, T.; Suwa, K.; Okazaki, K. Clinico-Pathological Importance of TGF-β/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis. Cancers 2018, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Matsuzaki, K.; Yoshida, K.; Sekimoto, G.; Tahashi, Y.; Mori, S.; Uemura, Y.; Sakaida, N.; Fujisawa, J.; Seki, T.; et al. Hepatitis B Virus X Protein Shifts Human Hepatic Transforming Growth Factor (TGF)-β Signaling from Tumor Suppression to Oncogenesis in Early Chronic Hepatitis B. Hepatology 2009, 49, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, G.; Yuan, H.; Wang, J.; Guo, Y.; Chen, T.; Zhai, R.; Shao, D.; Ni, W.; Tai, G. Mucin1 Shifts Smad3 Signaling from the Tumor-Suppressive PSmad3C/P21(WAF1) Pathway to the Oncogenic PSmad3L/c-Myc Pathway by Activating JNK in Human Hepatocellular Carcinoma Cells. Oncotarget 2015, 6, 4253–4265. [Google Scholar] [CrossRef]
- Nagata, H.; Hatano, E.; Tada, M.; Murata, M.; Kitamura, K.; Asechi, H.; Narita, M.; Yanagida, A.; Tamaki, N.; Yagi, S.; et al. Inhibition of C-Jun NH2-Terminal Kinase Switches Smad3 Signaling from Oncogenesis to Tumor- Suppression in Rat Hepatocellular Carcinoma. Hepatology 2009, 49, 1944–1953. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic Inflammation Associated with Hepatitis C Virus Infection Perturbs Hepatic Transforming Growth Factor β Signaling, Promoting Cirrhosis and Hepatocellular Carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef]
- Liu, R.; Tang, C.; Shen, A.; Luo, H.; Wei, X.; Zheng, D.; Sun, C.; Li, Z.; Zhu, D.; Li, T.; et al. IL-37 Suppresses Hepatocellular Carcinoma Growth by Converting PSmad3 Signaling from JNK/PSmad3L/c-Myc Oncogenic Signaling to PSmad3C/P21 Tumor-Suppressive Signaling. Oncotarget 2016, 7, 85079–85096. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, S.K.; Lim, C.R.; Park, H.W.; Liu, F.; Kim, S.-J.; Kim, B.-C. Heme Oxygenase-1/Carbon Monoxide Axis Suppresses Transforming Growth Factor-β1-Induced Growth Inhibition by Increasing ERK1/2-Mediated Phosphorylation of Smad3 at Thr-179 in Human Hepatocellular Carcinoma Cell Lines. Biochem. Biophys. Res. Commun. 2018, 498, 609–615. [Google Scholar] [CrossRef]
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming Growth Factor-β and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatology 2019, 69, 222–236. [Google Scholar] [CrossRef]
- Alarcón, C.; Zaromytidou, A.-I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs Drive Smad Transcriptional Activation and Turnover in BMP and TGF-β Pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in Cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Ostman, A. Hallmarks of Cancer: Interactions with the Tumor Stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.-J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef] [PubMed]
- Costanza, B.; Umelo, I.A.; Bellier, J.; Castronovo, V.; Turtoi, A. Stromal Modulators of TGF-β in Cancer. J. Clin. Med. 2017, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, M.; Luo, Z.; Wen, Z.; Yan, X. The Dichotomous Role of TGF-β in Controlling Liver Cancer Cell Survival and Proliferation. J. Genet. Genomics Yi Chuan Xue Bao 2020, 47, 497–512. [Google Scholar] [CrossRef]
- Qu, N.; Bo, X.; Li, B.; Ma, L.; Wang, F.; Zheng, Q.; Xiao, X.; Huang, F.; Shi, Y.; Zhang, X. Role of N6-Methyladenosine (M6A) Methylation Regulators in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 755206. [Google Scholar] [CrossRef]
- Zhang, L.; Hou, C.; Chen, C.; Guo, Y.; Yuan, W.; Yin, D.; Liu, J.; Sun, Z. The Role of N6-Methyladenosine (M6A) Modification in the Regulation of CircRNAs. Mol. Cancer 2020, 19, 105. [Google Scholar] [CrossRef]
- Sivasudhan, E.; Blake, N.; Lu, Z.-L.; Meng, J.; Rong, R. Dynamics of M6A RNA Methylome on the Hallmarks of Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2021, 9, 642443. [Google Scholar] [CrossRef]
- Chen, J.; Fang, X.; Zhong, P.; Song, Z.; Hu, X. N6-Methyladenosine Modifications: Interactions with Novel RNA-Binding Proteins and Roles in Signal Transduction. RNA Biol. 2019, 16, 991–1000. [Google Scholar] [CrossRef]
- Liu, J.; Sun, G.; Pan, S.; Qin, M.; Ouyang, R.; Li, Z.; Huang, J. The Cancer Genome Atlas (TCGA) Based M6A Methylation-Related Genes Predict Prognosis in Hepatocellular Carcinoma. Bioengineered 2020, 11, 759–768. [Google Scholar] [CrossRef]
- Liu, X.; Qin, J.; Gao, T.; Li, C.; Chen, X.; Zeng, K.; Xu, M.; He, B.; Pan, B.; Xu, X.; et al. Analysis of METTL3 and METTL14 in Hepatocellular Carcinoma. Aging 2020, 12, 21638–21659. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chai, G.; Wu, Y.; Li, J.; Chen, F.; Liu, J.; Luo, G.; Tauler, J.; Du, J.; Lin, S.; et al. RNA M6A Methylation Regulates the Epithelial Mesenchymal Transition of Cancer Cells and Translation of Snail. Nat. Commun. 2019, 10, 2065. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Zheng, H.; Kang, Y. Multilayer Control of the EMT Master Regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef]
- Pinzani, M. Epithelial-Mesenchymal Transition in Chronic Liver Disease: Fibrogenesis or Escape from Death? J. Hepatol. 2011, 55, 459–465. [Google Scholar] [CrossRef]
- Jou, J.; Diehl, A.M. Epithelial-Mesenchymal Transitions and Hepatocarcinogenesis. J. Clin. Investig. 2010, 120, 1031–1034. [Google Scholar] [CrossRef]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of Epithelial to Mesenchymal Transition in Hepatocellular Carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling Pathway Cooperation in TGF-β-Induced Epithelial-Mesenchymal Transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef]
- Fu, H.; He, Y.; Qi, L.; Chen, L.; Luo, Y.; Chen, L.; Li, Y.; Zhang, N.; Guo, H. CPLA2α Activates PI3K/AKT and Inhibits Smad2/3 during Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma Cells. Cancer Lett. 2017, 403, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.C. Cytosolic Phospholipase A2: Physiological Function and Role in Disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Ciarrocchi, A. Long Noncoding RNA and Epithelial Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 1924. [Google Scholar] [CrossRef]
- Zhou, J.-N.; Zeng, Q.; Wang, H.-Y.; Zhang, B.; Li, S.-T.; Nan, X.; Cao, N.; Fu, C.-J.; Yan, X.-L.; Jia, Y.-L.; et al. MicroRNA-125b Attenuates Epithelial-Mesenchymal Transitions and Targets Stem-like Liver Cancer Cells through Small Mothers against Decapentaplegic 2 and 4. Hepatology 2015, 62, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ooi, L.L.P.J.; Hui, K.M. MicroRNA-216a/217-Induced Epithelial-Mesenchymal Transition Targets PTEN and SMAD7 to Promote Drug Resistance and Recurrence of Liver Cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.-H.; Wan, J.-L.; Zeng, L.-Y.; Xie, L.; Sun, H.-C.; Qin, L.-X.; Wang, L.; Zhou, J.; Ren, Z.-G.; Li, Y.-X.; et al. MiR-612 Suppresses the Invasive-Metastatic Cascade in Hepatocellular Carcinoma. J. Exp. Med. 2013, 210, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-Y.; Zhang, K.; Chen, D.-Q.; Chen, J.; Feng, B.; Song, H.; Chen, Y.; Zhu, Z.; Lu, L.; De, W.; et al. MicroRNA-451: Epithelial-Mesenchymal Transition Inhibitor and Prognostic Biomarker of Hepatocelluar Carcinoma. Oncotarget 2015, 6, 18613–18630. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The MiR-200 Family and MiR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.-Y.; et al. An Autocrine TGF-β/ZEB/MiR-200 Signaling Network Regulates Establishment and Maintenance of Epithelial-Mesenchymal Transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef]
- Suzuki, H.I. MicroRNA Control of TGF-β Signaling. Int. J. Mol. Sci. 2018, 19, 1901. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Tsui, Y.-M.; Shi, C.; Wang, Y.; Zhang, X.; Yan, Q.; Chen, M.; Jiang, C.; Yuan, Y.-F.; et al. RALYL Increases Hepatocellular Carcinoma Stemness by Sustaining the MRNA Stability of TGF-β2. Nat. Commun. 2021, 12, 1518. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadée, C.; et al. The SMAD2/3 Interactome Reveals That TGFβ Controls M6A MRNA Methylation in Pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, H.; Tao, K.; Li, R.; Song, Z.; Zhao, Q.; Zhang, F.; Dou, K. Expression and Clinical Significance of the Stem Cell Marker CD133 in Hepatocellular Carcinoma. Int. J. Clin. Pract. 2008, 62, 1212–1218. [Google Scholar] [CrossRef]
- Miraglia, S.; Godfrey, W.; Yin, A.H.; Atkins, K.; Warnke, R.; Holden, J.T.; Bray, R.A.; Waller, E.K.; Buck, D.W. A Novel Five-Transmembrane Hematopoietic Stem Cell Antigen: Isolation, Characterization, and Molecular Cloning. Blood 1997, 90, 5013–5021. [Google Scholar] [CrossRef]
- You, H.; Ding, W.; Rountree, C.B. Epigenetic Regulation of Cancer Stem Cell Marker CD133 by Transforming Growth Factor-β. Hepatology 2010, 51, 1635–1644. [Google Scholar] [CrossRef]
- Malfettone, A.; Soukupova, J.; Bertran, E.; Crosas-Molist, E.; Lastra, R.; Fernando, J.; Koudelkova, P.; Rani, B.; Fabra, Á.; Serrano, T.; et al. Transforming Growth Factor-β-Induced Plasticity Causes a Migratory Stemness Phenotype in Hepatocellular Carcinoma. Cancer Lett. 2017, 392, 39–50. [Google Scholar] [CrossRef]
- Pardali, K.; Moustakas, A. Actions of TGF-β as Tumor Suppressor and pro-Metastatic Factor in Human Cancer. Biochim. Biophys. Acta 2007, 1775, 21–62. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Y.; Wang, L. A Feedback Inhibition between MiRNA-127 and TGFβ/c-Jun Cascade in HCC Cell Migration via MMP13. PLoS ONE 2013, 8, e65256. [Google Scholar] [CrossRef]
- Qin, G.; Luo, M.; Chen, J.; Dang, Y.; Chen, G.; Li, L.; Zeng, J.; Lu, Y.; Yang, J. Reciprocal Activation between MMP-8 and TGF-β1 Stimulates EMT and Malignant Progression of Hepatocellular Carcinoma. Cancer Lett. 2016, 374, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hsu, S.-H.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. TGFβ-Mediated Upregulation of Hepatic MiR-181b Promotes Hepatocarcinogenesis by Targeting TIMP3. Oncogene 2010, 29, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; He, X.; Ding, J.; Liang, L.; Zhao, Y.; Zhang, Z.; Yao, X.; Pan, Z.; Zhang, P.; Li, J.; et al. Upregulation of MiR-23a Approximately 27a Approximately 24 Decreases Transforming Growth Factor-β-Induced Tumor-Suppressive Activities in Human Hepatocellular Carcinoma Cells. Int. J. Cancer 2008, 123, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Ke, A.-W.; Shi, G.-M.; Zhou, J.; Huang, X.-Y.; Shi, Y.-H.; Ding, Z.-B.; Wang, X.-Y.; Devbhandari, R.P.; Fan, J. CD151 Amplifies Signaling by Integrin A6β1 to PI3K and Induces the Epithelial-Mesenchymal Transition in HCC Cells. Gastroenterology 2011, 140, 1629–1641.e15. [Google Scholar] [CrossRef]
- Fransvea, E.; Mazzocca, A.; Antonaci, S.; Giannelli, G. Targeting Transforming Growth Factor (TGF)-βRI Inhibits Activation of β1 Integrin and Blocks Vascular Invasion in Hepatocellular Carcinoma. Hepatology 2009, 49, 839–850. [Google Scholar] [CrossRef]
- Giannelli, G.; Bergamini, C.; Fransvea, E.; Sgarra, C.; Antonaci, S. Laminin-5 with Transforming Growth Factor-β1 Induces Epithelial to Mesenchymal Transition in Hepatocellular Carcinoma. Gastroenterology 2005, 129, 1375–1383. [Google Scholar] [CrossRef]
- Miyazaki, K. Laminin-5 (Laminin-332): Unique Biological Activity and Role in Tumor Growth and Invasion. Cancer Sci. 2006, 97, 91–98. [Google Scholar] [CrossRef]
- Luo, Y.; Huang, K.; Zheng, J.; Zhang, J.; Zhang, L. TGF-β1 Promotes Cell Migration in Hepatocellular Carcinoma by Suppressing Reelin Expression. Gene 2019, 688, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Qiu, M.; Wan, L.; Wang, G.; Huang, T.; Chen, Z.; Jiang, S.; Li, X.; Xie, L.; Cai, L. TGF-β1 Promotes Hepatocellular Carcinoma Invasion and Metastasis via ERK Pathway-Mediated FGFR4 Expression. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 45, 1690–1699. [Google Scholar] [CrossRef]
- De Jong, O.G.; van der Waals, L.M.; Kools, F.R.W.; Verhaar, M.C.; van Balkom, B.W.M. Lysyl Oxidase-like 2 Is a Regulator of Angiogenesis through Modulation of Endothelial-to-Mesenchymal Transition. J. Cell. Physiol. 2019, 234, 10260–10269. [Google Scholar] [CrossRef]
- Shao, B.; Zhao, X.; Liu, T.; Zhang, Y.; Sun, R.; Dong, X.; Liu, F.; Zhao, N.; Zhang, D.; Wu, L.; et al. LOXL2 Promotes Vasculogenic Mimicry and Tumour Aggressiveness in Hepatocellular Carcinoma. J. Cell. Mol. Med. 2019, 23, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Nakamura, I.; Roberts, L.R. The Tumor Microenvironment in Hepatocellular Carcinoma: Current Status and Therapeutic Targets. Semin. Cancer Biol. 2011, 21, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-Outschoorn, U.; et al. Metabolic Reprogramming of Cancer-Associated Fibroblasts by TGF-β Drives Tumor Growth: Connecting TGF-β Signaling with “Warburg-like” Cancer Metabolism and L-Lactate Production. Cell Cycle Georget. Tex 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lu, Y.; Lin, Y.-Y.; Zheng, Z.-Y.; Fang, J.-H.; He, S.; Zhuang, S.-M. Vascular Mimicry Formation Is Promoted by Paracrine TGF-β and SDF1 of Cancer-Associated Fibroblasts and Inhibited by MiR-101 in Hepatocellular Carcinoma. Cancer Lett. 2016, 383, 18–27. [Google Scholar] [CrossRef]
- Thompson, A.I.; Conroy, K.P.; Henderson, N.C. Hepatic Stellate Cells: Central Modulators of Hepatic Carcinogenesis. BMC Gastroenterol. 2015, 15, 63. [Google Scholar] [CrossRef]
- Le Pabic, H.; Bonnier, D.; Wewer, U.M.; Coutand, A.; Musso, O.; Baffet, G.; Clément, B.; Théret, N. ADAM12 in Human Liver Cancers: TGF-β-Regulated Expression in Stellate Cells Is Associated with Matrix Remodeling. Hepatology 2003, 37, 1056–1066. [Google Scholar] [CrossRef]
- Park, S.-A.; Kim, M.-J.; Park, S.-Y.; Kim, J.-S.; Lim, W.; Nam, J.-S.; Yhong Sheen, Y. TIMP-1 Mediates TGF-β-Dependent Crosstalk between Hepatic Stellate and Cancer Cells via FAK Signaling. Sci. Rep. 2015, 5, 16492. [Google Scholar] [CrossRef]
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef]
- Amann, T.; Bataille, F.; Spruss, T.; Mühlbauer, M.; Gäbele, E.; Schölmerich, J.; Kiefer, P.; Bosserhoff, A.-K.; Hellerbrand, C. Activated Hepatic Stellate Cells Promote Tumorigenicity of Hepatocellular Carcinoma. Cancer Sci. 2009, 100, 646–653. [Google Scholar] [CrossRef]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming Growth Factor-β 1 (TGF-β1) Induces Angiogenesis through Vascular Endothelial Growth Factor (VEGF)-Mediated Apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Tumor Angiogenesis: MMP-Mediated Induction of Intravasation- and Metastasis-Sustaining Neovasculature. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 44–46, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current Perspectives on the Immunosuppressive Tumor Microenvironment in Hepatocellular Carcinoma: Challenges and Opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Qiu, X.; Li, J.; Zheng, S.; Li, L.; Zhao, H. TGF-β Secreted by Tumor-Associated Macrophages Promotes Proliferation and Invasion of Colorectal Cancer via MiR-34a-VEGF Axis. Cell Cycle Georget. Tex 2018, 17, 2766–2778. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Gerken, G. Local Control of the Immune Response in the Liver. Immunol. Rev. 2000, 174, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Prieto, J.; Melero, I.; Sangro, B. Immunological Landscape and Immunotherapy of Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681–700. [Google Scholar] [CrossRef]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 Fosters HCC Development by Enhancing TGF-β-Mediated Alternative Activation of Macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef]
- Hong, S.; Lim, S.; Li, A.G.; Lee, C.; Lee, Y.S.; Lee, E.-K.; Park, S.H.; Wang, X.-J.; Kim, S.-J. Smad7 Binds to the Adaptors TAB2 and TAB3 to Block Recruitment of the Kinase TAK1 to the Adaptor TRAF2. Nat. Immunol. 2007, 8, 504–513. [Google Scholar] [CrossRef]
- Chen, K.-J.; Lin, S.-Z.; Zhou, L.; Xie, H.-Y.; Zhou, W.-H.; Taki-Eldin, A.; Zheng, S.-S. Selective Recruitment of Regulatory T Cell through CCR6-CCL20 in Hepatocellular Carcinoma Fosters Tumor Progression and Predicts Poor Prognosis. PLoS ONE 2011, 6, e24671. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 Is Essential for TGF-β to Convert Naive CD4+CD25− Cells to CD25+Foxp3+ Regulatory T Cells and for Expansion of These Cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25− Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Genestier, L.; Kasibhatla, S.; Brunner, T.; Green, D.R. Transforming Growth Factor β1 Inhibits Fas Ligand Expression and Subsequent Activation-Induced Cell Death in T Cells via Downregulation of c-Myc. J. Exp. Med. 1999, 189, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Wolfraim, L.A.; Walz, T.M.; James, Z.; Fernandez, T.; Letterio, J.J. P21Cip1 and P27Kip1 Act in Synergy to Alter the Sensitivity of Naive T Cells to TGF-β-Mediated G1 Arrest through Modulation of IL-2 Responsiveness. J. Immunol. 2004, 173, 3093–3102. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-β 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef]
- Walker, L.S.K. Treg and CTLA-4: Two Intertwining Pathways to Immune Tolerance. J. Autoimmun. 2013, 45, 49–57. [Google Scholar] [CrossRef]
- Zheng, S.G.; Wang, J.H.; Stohl, W.; Kim, K.S.; Gray, J.D.; Horwitz, D.A. TGF-β Requires CTLA-4 Early after T Cell Activation to Induce FoxP3 and Generate Adaptive CD4+CD25+ Regulatory Cells. J. Immunol. 2006, 176, 3321–3329. [Google Scholar] [CrossRef]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebemberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.-J.; et al. TGFβ1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef]
- Faivre, S.; Rimassa, L.; Finn, R.S. Molecular Therapies for HCC: Looking Outside the Box. J. Hepatol. 2020, 72, 342–352. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.J.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. SEARCH: A Phase III, Randomized, Double-Blind, Placebo-Controlled Trial of Sorafenib plus Erlotinib in Patients with Advanced Hepatocellular Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K.; Minoshima, Y.; Iwata, M.; Funahashi, Y. Antitumor Activity of Lenvatinib (E7080): An Angiogenesis Inhibitor That Targets Multiple Receptor Tyrosine Kinases in Preclinical Human Thyroid Cancer Models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Seufferlein, T. Regorafenib. Recent Results Cancer Res. Fortschritte Krebsforsch. Progres Dans Rech. Sur Cancer 2018, 211, 45–56. [Google Scholar] [CrossRef]
- Tovoli, F.; Granito, A.; De Lorenzo, S.; Bolondi, L. Regorafenib for the Treatment of Hepatocellular Carcinoma. Drugs Today Barc. Spain 1998 2018, 54, 5–13. [Google Scholar] [CrossRef]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.B.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib Suppresses Tumor Growth and Metastasis in Hepatocellular Carcinoma by a Dual Blockade of VEGFR2 and MET. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef]
- Amicone, L.; Marchetti, A. Microenvironment and Tumor Cells: Two Targets for New Molecular Therapies of Hepatocellular Carcinoma. Transl. Gastroenterol. Hepatol. 2018, 3, 24. [Google Scholar] [CrossRef]
- Kim, B.-G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel Therapies Emerging in Oncology to Target the TGF-β Pathway. J. Hematol. Oncol. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, Cell Plasticity and Metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Cheng, J.-W.; Hu, J.-W.; Li, H.; Ma, X.-L.; Tang, W.-G.; Sun, Y.-F.; Guo, W.; Huang, A.; Zhou, K.-Q.; et al. KPNA3 Confers Sorafenib Resistance to Advanced Hepatocellular Carcinoma via TWIST Regulated Epithelial-Mesenchymal Transition. J. Cancer 2019, 10, 3914–3925. [Google Scholar] [CrossRef] [PubMed]
- Fernando, J.; Malfettone, A.; Cepeda, E.B.; Vilarrasa-Blasi, R.; Bertran, E.; Raimondi, G.; Fabra, À.; Alvarez-Barrientos, A.; Fernández-Salguero, P.; Fernández-Rodríguez, C.M.; et al. A Mesenchymal-like Phenotype and Expression of CD44 Predict Lack of Apoptotic Response to Sorafenib in Liver Tumor Cells. Int. J. Cancer 2015, 136, E161–E172. [Google Scholar] [CrossRef] [PubMed]
- Ungerleider, N.; Han, C.; Zhang, J.; Yao, L.; Wu, T. TGFβ Signaling Confers Sorafenib Resistance via Induction of Multiple RTKs in Hepatocellular Carcinoma Cells. Mol. Carcinog. 2017, 56, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Badawi, M.; Kim, J.; Dauki, A.; Sutaria, D.; Motiwala, T.; Reyes, R.; Wani, N.; Kolli, S.; Jiang, J.; Coss, C.C.; et al. CD44 Positive and Sorafenib Insensitive Hepatocellular Carcinomas Respond to the ATP-Competitive MTOR Inhibitor INK128. Oncotarget 2018, 9, 26032–26045. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The Great Escape: Tumour Cell Plasticity in Resistance to Targeted Therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Weng, C.-H.; Chen, L.-Y.; Lin, Y.-C.; Shih, J.-Y.; Lin, Y.-C.; Tseng, R.-Y.; Chiu, A.-C.; Yeh, Y.-H.; Liu, C.; Lin, Y.-T.; et al. Epithelial-Mesenchymal Transition (EMT) beyond EGFR Mutations per Se Is a Common Mechanism for Acquired Resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Iyer, S.; Wang, Z.-G.; Akhtari, M.; Zhao, W.; Seth, P. Targeting TGFβ Signaling for Cancer Therapy. Cancer Biol. Ther. 2005, 4, 261–266. [Google Scholar] [CrossRef][Green Version]
- Liu, S.; Ren, J.; Ten Dijke, P. Targeting TGFβ Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 8. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, E.; Vaquero, J.; Férnandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF-β Pathway: A Pharmacological Target in Hepatocellular Carcinoma? Cancers 2021, 13, 3248. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ Pathway with Galunisertib, a TGFβRI Small Molecule Inhibitor, Promotes Anti-Tumor Immunity Leading to Durable, Complete Responses, as Monotherapy and in Combination with Checkpoint Blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Pasche, B. Transforming Growth Factor β as a Therapeutic Target in Systemic Sclerosis. Nat. Rev. Rheumatol. 2009, 5, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Chung, C.-L.; Hu, T.-H.; Chen, J.-J.; Liu, P.-F.; Chen, C.-L. Recent Progress in TGF-β Inhibitors for Cancer Therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ Pathway for Cancer Therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a Novel Transforming Growth Factor β Receptor Type I and Type II Dual Inhibitor, as a Therapeutic Approach to Suppressing Pancreatic Cancer Metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef]
- Doi, T.; Fujiwara, Y.; Koyama, T.; Ikeda, M.; Helwig, C.; Watanabe, M.; Vugmeyster, Y.; Kudo, M. Phase I Study of the Bifunctional Fusion Protein Bintrafusp Alfa in Asian Patients with Advanced Solid Tumors, Including a Hepatocellular Carcinoma Safety-Assessment Cohort. Oncologist 2020, 25, e1292–e1302. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical Development of Therapies Targeting TGFβ: Current Knowledge and Future Perspectives. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]




| Bright Side | Dark Side |
|---|---|
| Cellular Senescence | Cancer Cell Proliferation |
| G1 Cell Cycle Arrest | RNA Modifications |
| G2 Cell Cycle Arrest | Epithelial–Mesenchymal Transition (EMT) |
| Autophagy | Invasion and Angiogenesis |
| Apoptosis | ECM Remodeling |
| Mode of Action | Target | Name | Type | References |
|---|---|---|---|---|
| TGF-β ligand inhibitors | TGF-β1, TGF-β2, and TGF-β3 | Fresolimumab | Neutralizing antibody | [318] |
| TGF-β1, TGF-β2, and TGF-β3 | SAR439459 | Neutralizing antibody | [320] | |
| TGF-β1, TGF-β2, and TGF-β3 | NIS793 | Neutralizing antibody | [320] | |
| TGF-β1 and TGF-β3 | AVID200 | Ligand trap | [308] | |
| TGF-β1 | Metelimumab | Neutralizing antibody | [324] | |
| TGF-β2 | Trabedersen | Antisense oligonucleotide | [308] | |
| TGF-β2 | Lucanix | Vaccine | [321] | |
| TGF-β1 | LY2382770 | Neutralizing antibody | [325] | |
| TGF-β1 | SRK-181 | Neutralizing antibody | [321] | |
| TGF-β2 | Lerdelimumab | Monoclonal antibody | [326] | |
| TGF-β1 | Disitertide | Peptide | [326] | |
| TGF-β1 and TGF-β2 | FANG Vaccine | Vaccine | [326] | |
| TGF-β1 | ISTH0036 | Antisense oligonucleotide | [325] | |
| TGF-β receptor inhibitors | TGFβRI | Galunisertib | Small molecule inhibitor | [318] |
| TGFβRI and TGFβRII | LY2109761 | Small molecule inhibitor | [327] | |
| TGFβRI | PF-03446962 | Monoclonal antibody | [318] | |
| TGFβRI | IMC-TR1 | Monoclonal antibody | [320] | |
| TGFβRI | Vactosertib | Small molecule inhibitor | [318] | |
| TGFβRI and TGFβRII | P144 | Ligand trap | [325] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gungor, M.Z.; Uysal, M.; Senturk, S. The Bright and the Dark Side of TGF-β Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications. Cancers 2022, 14, 940. https://doi.org/10.3390/cancers14040940
Gungor MZ, Uysal M, Senturk S. The Bright and the Dark Side of TGF-β Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications. Cancers. 2022; 14(4):940. https://doi.org/10.3390/cancers14040940
Chicago/Turabian StyleGungor, Medine Zeynep, Merve Uysal, and Serif Senturk. 2022. "The Bright and the Dark Side of TGF-β Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications" Cancers 14, no. 4: 940. https://doi.org/10.3390/cancers14040940
APA StyleGungor, M. Z., Uysal, M., & Senturk, S. (2022). The Bright and the Dark Side of TGF-β Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications. Cancers, 14(4), 940. https://doi.org/10.3390/cancers14040940