Peptides against Low Density Lipoprotein (LDL) Aggregation Inhibit Intracellular Cholesteryl Ester Loading and Proliferation of Pancreatic Tumor Cells

 , , and
, , and 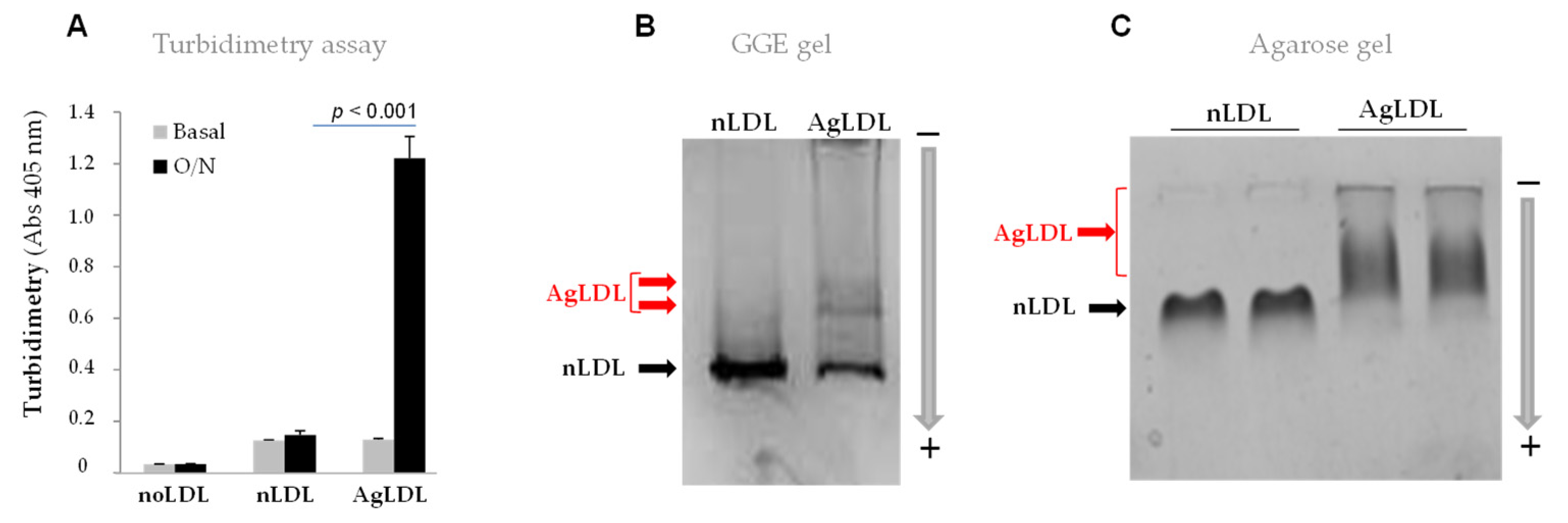{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Peptide Sequence and Synthesis
2.2. LDL Isolation and Purification
2.3. LDL Modification by Sphingomyelinase (SMase) in the Presence and Absence of Peptides
2.4. Characterization of SMase-Induced LDL Aggregation by Turbidimetry
2.5. Cell Culture of Pancreatic Tumor Cell Lines
2.6. Monitorization of the Efficacy of Peptides to Inhibit LDL-Induced Intracellular Cholesteryl Ester Accumulation
2.7. Determination of Intracellular Cholesteryl Ester/Free Cholesterol Ratio
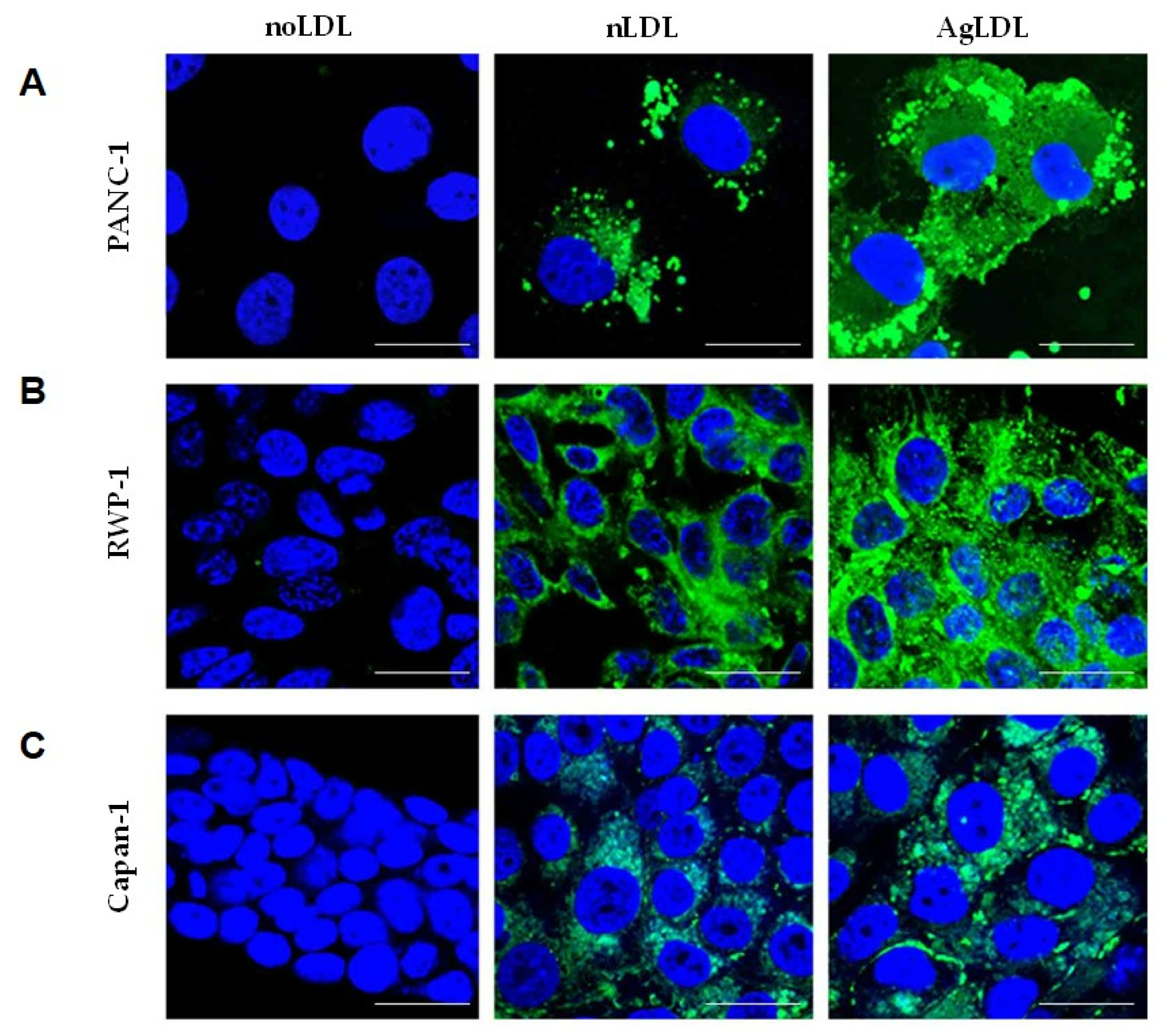
2.8. Confocal Microscopy Analysis
2.9. Flow Cytometry Analysis
2.10. Statistical Analysis
3. Results
3.1. Effect of SMase on Human LDL Aggregation
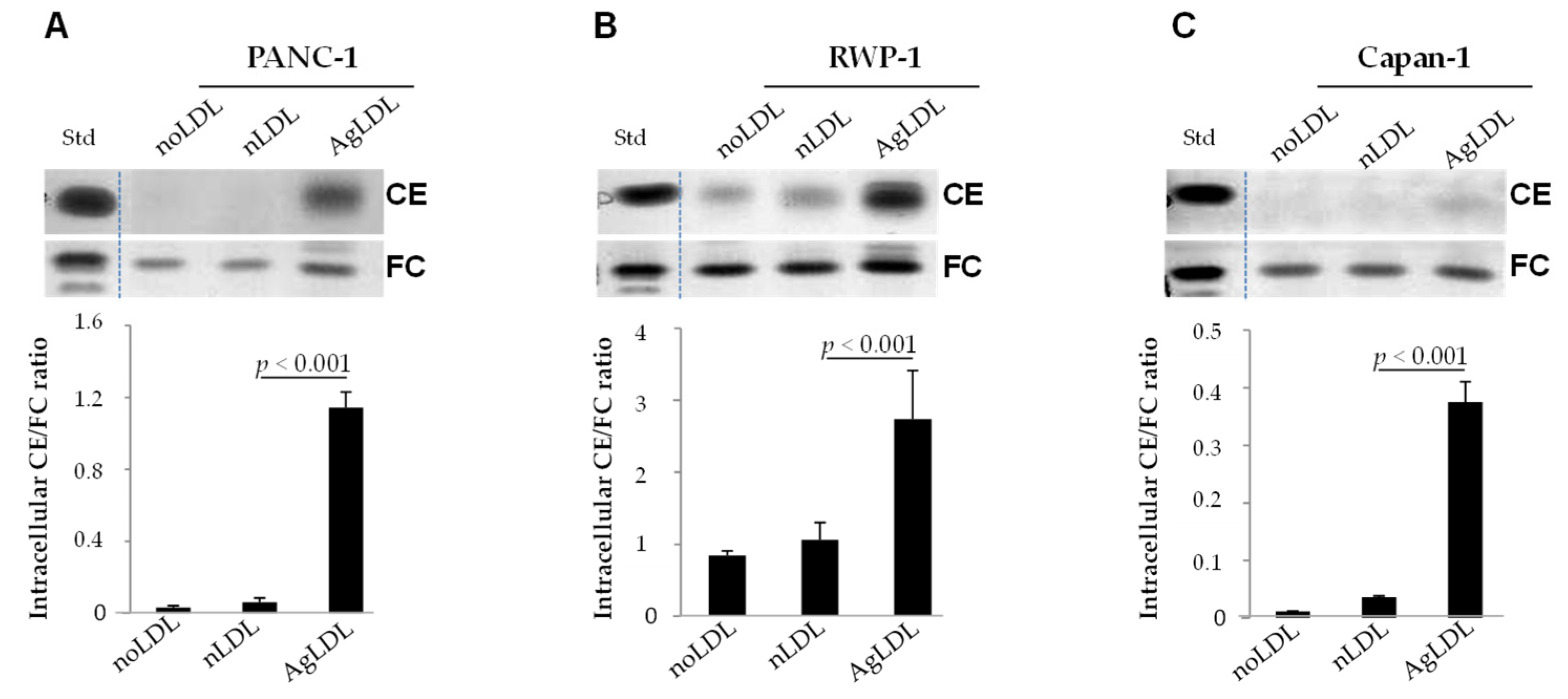
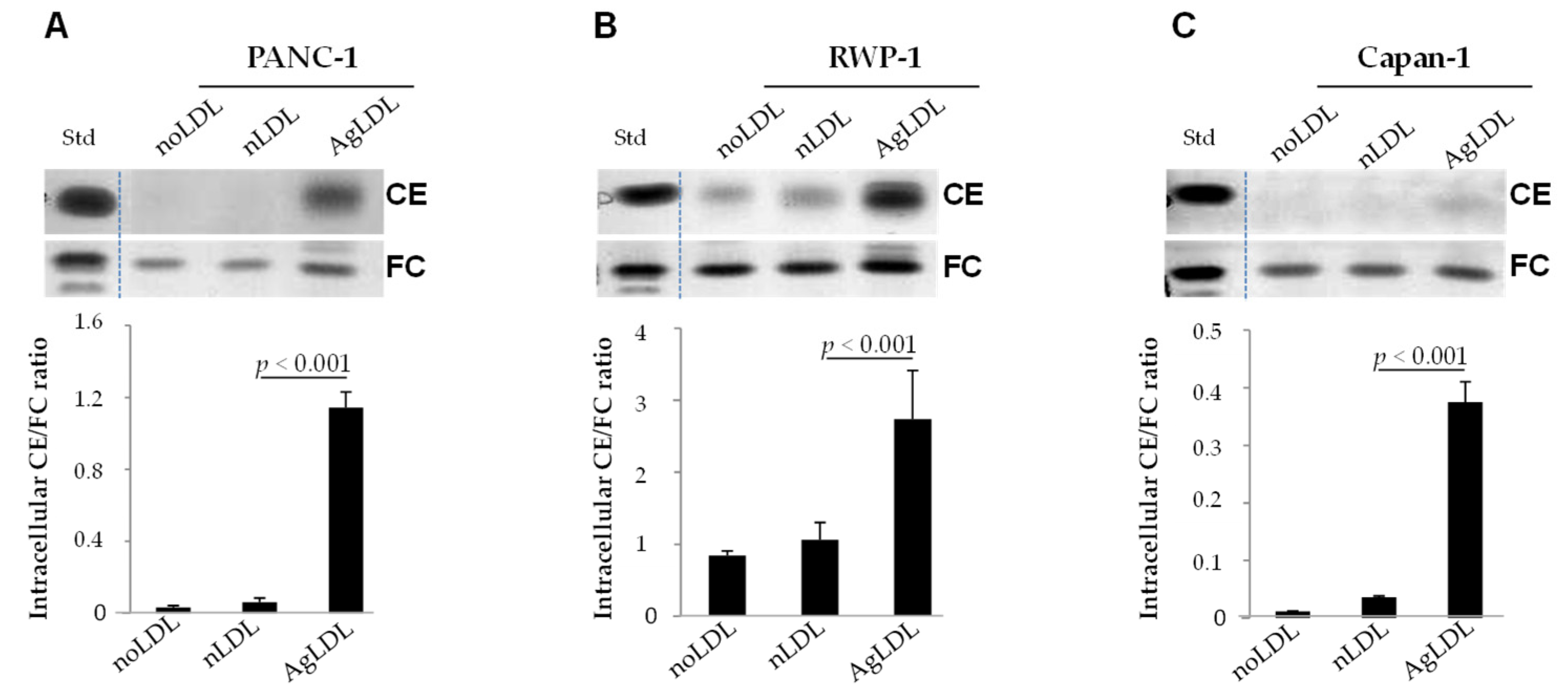
3.2. Effect of nLDLs and AgLDLs on Intracellular CE/FC Ratio in PANC-1, RWP-1 and Capan-1 Cells
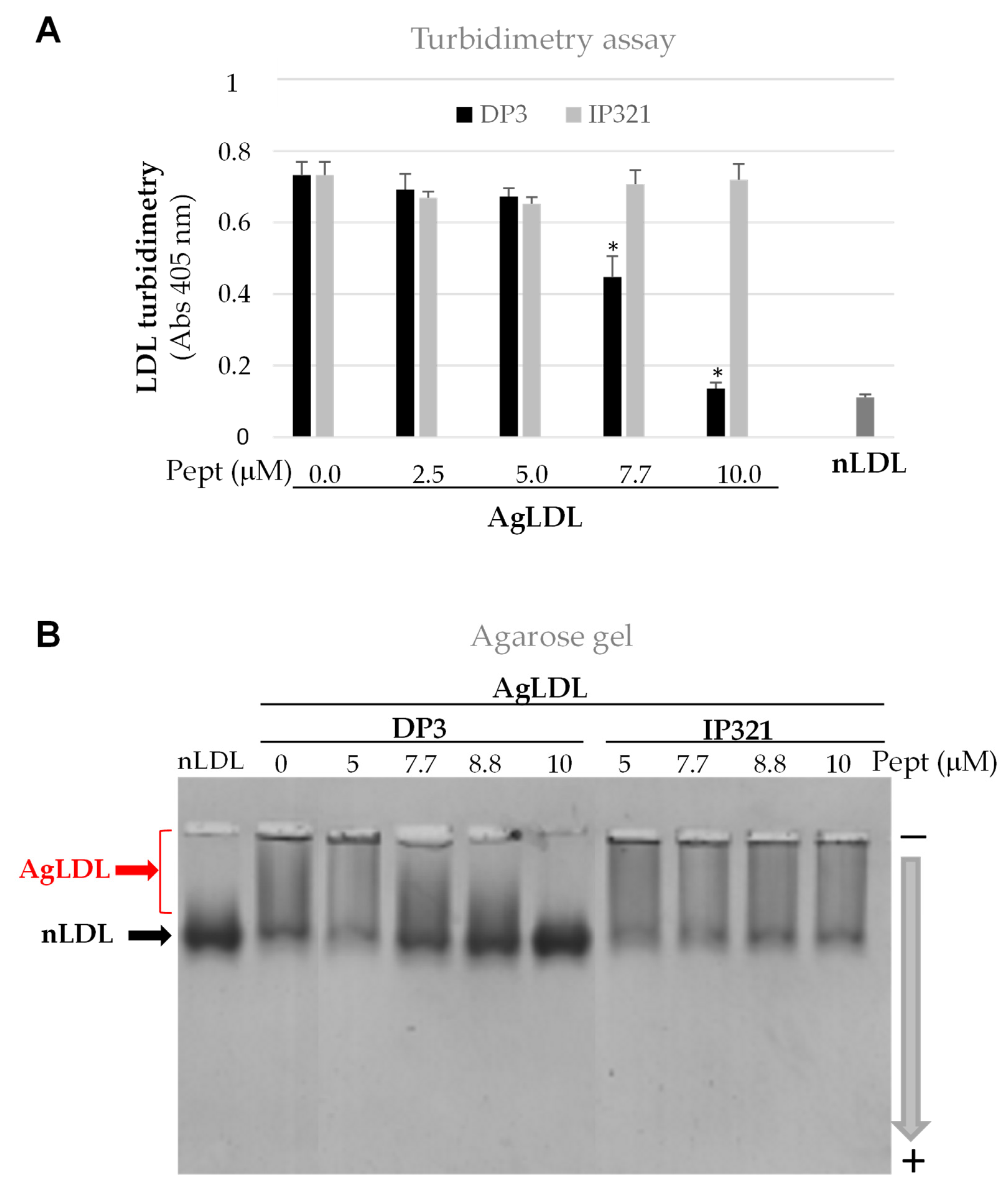
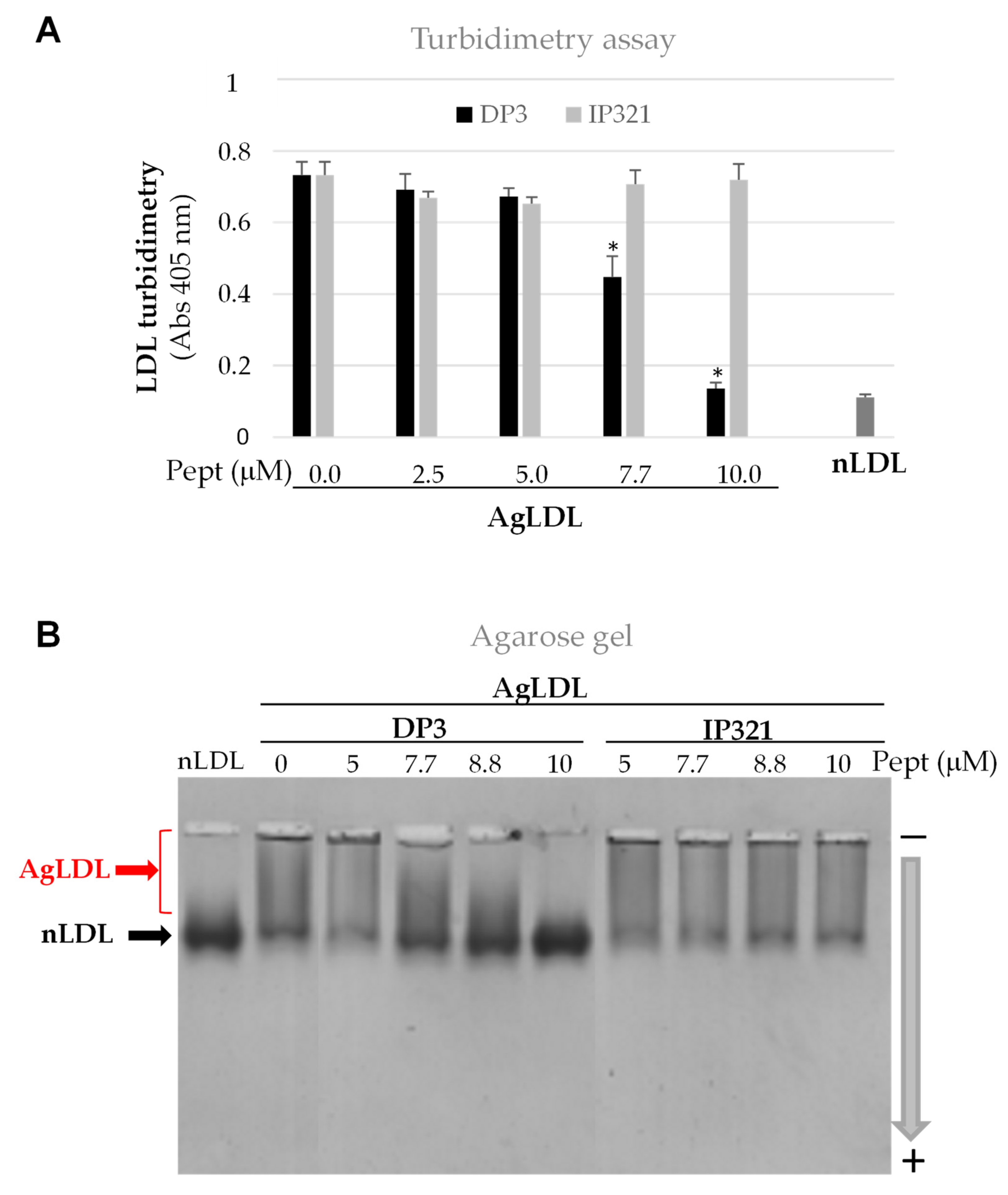
3.3. Effect of Peptide DP3 on SMase-Induced LDL Modification
3.4. Effect of DP3 on Intracellular CE/FC Ratio Induced by AgLDL in PANC-1, RWP-1, and Capan-1 Pancreatic Tumor Cells
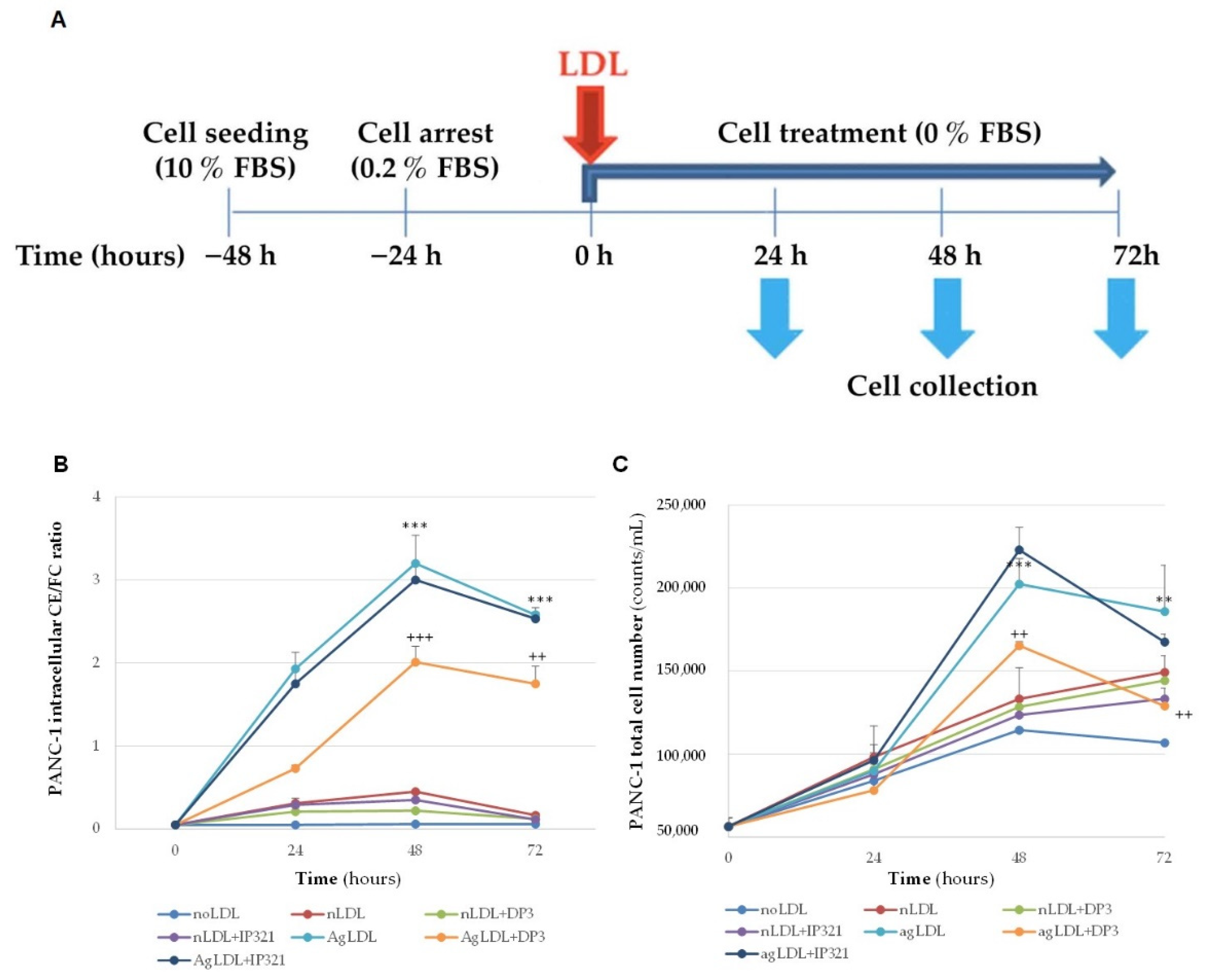
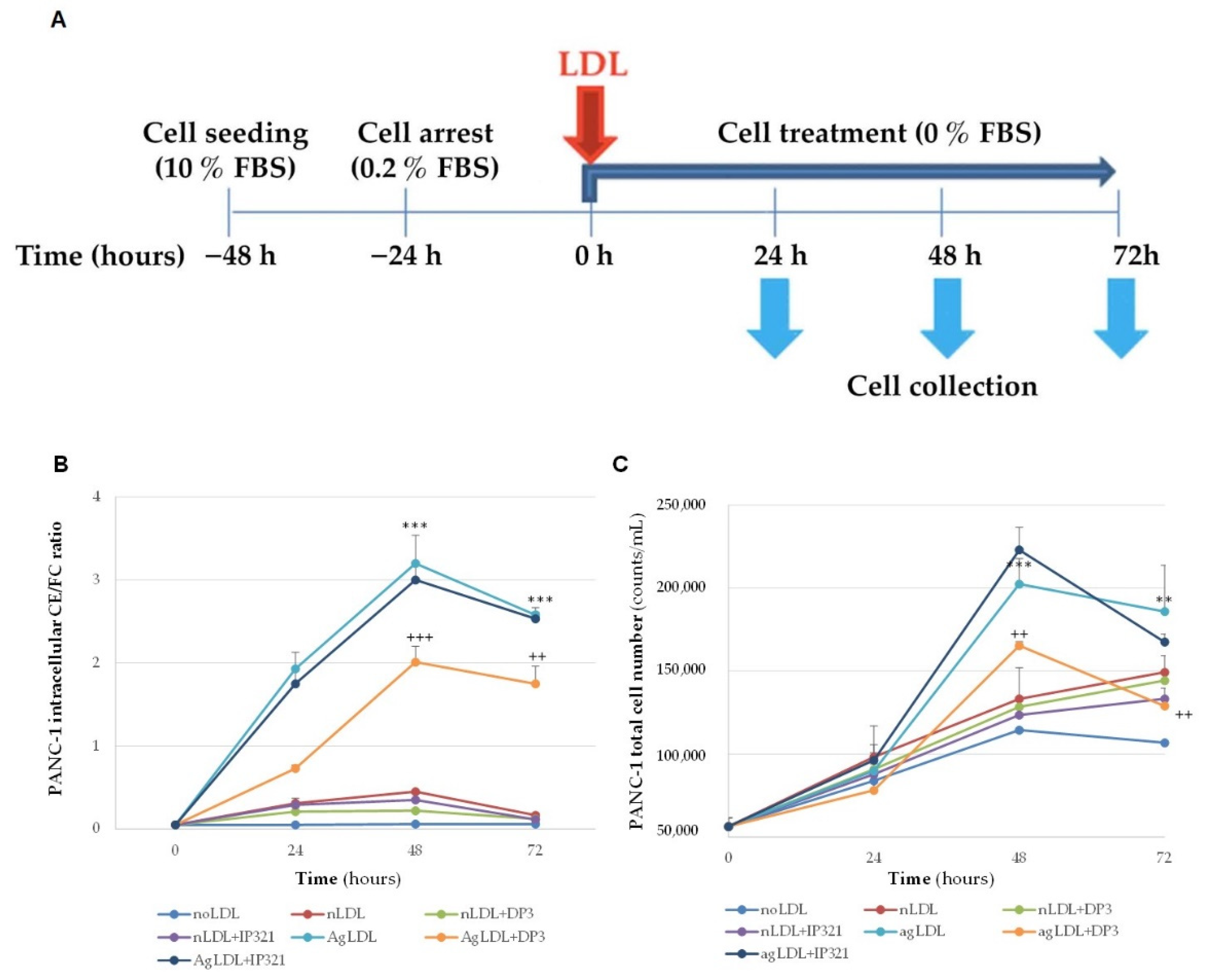
3.5. Time-Course Effect of nLDLs and AgLDLs on PANC-1 Cholesteryl Ester Loading and Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, 102655. [Google Scholar] [CrossRef]
- Jain, T.; Dudeja, V. The war against pancreatic cancer in 2020-advances on all fronts. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 99–100. [Google Scholar] [CrossRef]
- Park, S.K.; Oh, C.-M.; Kim, M.-H.; Ha, E.; Choi, Y.-S.; Ryoo, J.-H. Metabolic syndrome, metabolic components, and their relation to the risk of pancreatic cancer. Cancer 2020, 126, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.J.; Zhai, L.; Zhang, D.F. Association of cholesterol with risk of pancreatic cancer: A meta-analysis. World J. Gastroenterol. 2015, 21, 3711–3719. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qin, S.; Wang, M.; Zhang, T.; Zhang, S. Association between cholesterol intake and pancreatic cancer risk: Evidence from a meta-analysis. Sci. Rep. 2015, 5, 8243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, P.A.; Nikolic, D.; Rizzo, M. Small, dense LDL: An update. Curr. Opin. Cardiol. 2017, 32, 454–459. [Google Scholar] [CrossRef] [Green Version]
- Camejo, G.; Olsson, U.; Hurt-Camejo, E.; Baharamian, N.; Bondjers, G. The extracellular matrix on atherogenesis and diabetes-associated vascular disease. Atheroscler. Suppl. 2002, 3, 3–9. [Google Scholar] [CrossRef]
- Davidsson, P.; Hulthe, J.; Fagerberg, B.; Olsson, B.M.; Hallberg, C.; Dahllöf, B.; Camejo, G. A proteomic study of the apolipoproteins in LDL subclasses in patients with the metabolic syndrome and type 2 diabetes. J. Lipid Res. 2005, 46, 1999–2006. [Google Scholar] [CrossRef] [Green Version]
- Borén, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef]
- Bång-Rudenstam, A.; Cerezo-Magaña, M.; Belting, M. Pro-metastatic functions of lipoproteins and extracellular vesicles in the acidic tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 79–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Orozco, C.A.; Martinez-Bosch, N.; Guerrero, P.E.; Vinaixa, J.; Dalotto-Moreno, T.; Iglesias, M.; Moreno, M.; Djurec, M.; Poirier, F.; Gabius, H.-J.; et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc. Natl. Acad. Sci. USA 2018, 115, E3769–E3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruuth, M.; Nguyen, S.D.; Vihervaara, T.; Hilvo, M.; Laajala, T.D.; Kondadi, P.K.; Gisterå, A.; Lähteenmäki, H.; Kittilä, T.; Huusko, J.; et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur. Heart J. 2018, 39, 2562–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marathe, S.; Kuriakose, G.; Williams, K.J.; Tabas, I. Sphingomyelinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Ozawa, S.; Tsukuda, M.; Kubota, E.; Miyazaki, K.; St-Pierre, Y.; Hata, R.-I. Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J. 2007, 274, 3171–3183. [Google Scholar] [CrossRef]
- Ferrara, B.; Pignatelli, C.; Cossutta, M.; Citro, A.; Courty, J.; Piemonti, L. The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options. Cancers 2021, 3, 4442. [Google Scholar] [CrossRef]
- Llorente-Cortés, V.; Otero-Viñas, M.; Sánchez, S.; Rodríguez, C.; Badimon, L. Low-density lipoprotein upregulates low-density lipoprotein receptor-related protein expression in vascular smooth muscle cells: Possible involvement of sterol regulatory element binding protein-2-dependent mechanism. Circulation 2002, 106, 3104–3110. [Google Scholar] [CrossRef]
- Llorente-Cortés, V.; Otero-Viñas, M.; Camino-López, S.; Llampayas, O.; Badimon, L. Aggregated low-density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells. Circulation 2004, 110, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Castellano, J.; Aledo, R.; Sendra, J.; Costales, P.; Juan-Babot, O.; Badimon, L.; Llorente-Cortés, V. Hypoxia stimulates low-density lipoprotein receptor-related protein-1 expression through hypoxia-inducible factor-1α in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Yang, E.J.; Shim, J.S. Cholesterol Trafficking: An Emerging Therapeutic Target for Angiogenesis and Cancer. Cells 2019, 8, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogachkov, Y.Y.; Chen, L.; Le Master, E.; Fancher, I.S.; Zhao, Y.; Aguilar, V.; Oh, M.-J.; Wary, K.K.; DiPietro, L.A.; Levitan, I. LDL induces cholesterol loading and inhibits endothelial proliferation and angiogenesis in Matrigels: Correlation with impaired angiogenesis during wound healing. Am. J. Physiol. Cell Physiol. 2020, 318, C762–C776. [Google Scholar] [CrossRef]
- Solimando, A.G.; De Summa, S.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef] [PubMed]
- Rozeveld, C.N.; Johnson, K.M.; Zhang, L.; Razidlo, G.L. KRAS Controls Pancreatic Cancer Cell Lipid Metabolism and Invasive Potential through the Lipase HSL. Cancer Res. 2020, 80, 4932–4945. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Guillaumond, F. LDL Receptor: An open route to feed pancreatic tumor cells. Mol. Cell. Oncol. 2016, 3, e1033586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gu, D.; Lee, S.S.-Y.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costales, P.; Fuentes-Prior, P.; Castellano, J.; Revuelta-Lopez, E.; Corral-Rodríguez, M.Á.; Nasarre, L.; Badimon, L.; Llorente-Cortes, V. K domain CR9 of low density lipoprotein (LDL) receptor-related protein 1 (LRP1) is critical for aggregated LDL-induced foam cell formation from human vascular smooth muscle cells. J. Biol. Chem. 2015, 290, 14852–14865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benitez-Amaro, A.; Pallara, C.; Nasarre, L.; Rivas-Urbina, A.; Benitez, S.; Vea, A.; Bornachea, O.; de Gonzalo-Calvo, D.; Serra-Mir, G.; Villegas, S.; et al. Molecular basis for the protective effects of low-density lipoprotein receptor-related protein 1 (LRP1)-derived peptides against LDL aggregation. Biochim. Biophys. Acta-Biomembr. 2019, 1861, 1302–1316. [Google Scholar] [CrossRef]
- Benitez-Amaro, A.; Pallara, C.; Nasarre, L.; Ferreira, R.; de Gonzalo-Calvo, D.; Prades, R.; Tarragó, T.; Llorente-Cortés, V. Development of Innovative Antiatherosclerotic Peptides through the Combination of Molecular Modeling and a Dual (Biochemical-Cellular) Screening System. Adv. Ther. 2020, 3, 2000037. [Google Scholar] [CrossRef]
- Ditiatkovski, M.; Palsson, J.; Chin-Dusting, J.; Remaley, A.T.; Sviridov, D. Apolipoprotein A-I Mimetic Peptides: Discordance Between In Vitro and In Vivo Properties-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1301–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas-Urbina, A.; Rull, A.; Aldana-Ramos, J.; Santos, D.; Puig, N.; Farre-Cabrerizo, N.; Benitez, S.; Perez, A.; de Gonzalo-Calvo, D.; Escola-Gil, J.C.; et al. Subcutaneous Administration of Apolipoprotein J-Derived Mimetic Peptide d-[113-122]apoJ Improves LDL and HDL Function and Prevents Atherosclerosis in LDLR-KO Mice. Biomolecules 2020, 10, 829. [Google Scholar] [CrossRef] [PubMed]
- Wool, G.D.; Cabana, V.G.; Lukens, J.; Shaw, P.X.; Binder, C.J.; Witztum, J.L.; Reardon, C.A.; Getz, G.S. 4F Peptide reduces nascent atherosclerosis and induces natural antibody production in apolipoprotein E-null mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 290–300. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, P.; Fredrikson, G.N.; Samnegard, A.; Ericsson, C.-G.; Öhrvik, J.; Fisher, R.M.; Nilsson, J.; Hamsten, A. High plasma concentrations of autoantibodies against native peptide 210 of apoB-100 are related to less coronary atherosclerosis and lower risk of myocardial infarction. Eur. Heart J. 2008, 29, 2218–2226. [Google Scholar] [CrossRef] [PubMed]
- Bornachea, O.; Benitez-Amaro, A.; Vea, A.; Nasarre, L.; de Gonzalo-Calvo, D.; Escola-Gil, J.C.; Cedo, L.; Iborra, A.; Martinez-Martinez, L.; Juarez, C.; et al. Immunization with the Gly1127-Cys1140 amino acid sequence of the LRP1 receptor reduces atherosclerosis in rabbits. Molecular, immunohistochemical and nuclear imaging studies. Theranostics 2020, 10, 3263. [Google Scholar] [CrossRef]
- Lieber, M.; Mazzetta, J.; Nelson-Rees, W.; Kaplan, M.; Todaro, G. Establishment of a continuous tumor-cell line (PANC-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 1975, 15, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.L.; Matook, G.M.; Meitner, P.A.; Turner, M.D.; Calabresl, P.; Bogaar, H.A.; Jolly, G.A. Establishment and Characterization of Two Human Pancreatic Cancer Cell Lines Tumorigenic in Athymic Mice. Cancer Res. 1982, 42, 2705–2714. [Google Scholar]
- Kyriazis, A.P.; Kyriazis, A.A.; Scarpelli, D.G.; Fogh, J.; Rao, M.S.; Lepera, R. Human pancreatic adenocarcinoma line Capan-1 in tissue culture and the nude mouse. Morphologic, biologic, and biochemical characteristics. Am. J. Pathol. 1982, 106, 250–260. [Google Scholar]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangère, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; de Barros, J.-P.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Menard, J.A.; Christianson, H.C.; Kucharzewska, P.; Bourseau-Guilmain, E.; Svensson, K.J.; Lindqvist, E.; Chandran, V.I.; Kjellén, L.; Welinder, C.; Bengzon, J.; et al. Metastasis Stimulation by Hypoxia and Acidosis-Induced Extracellular Lipid Uptake Is Mediated by Proteoglycan-Dependent Endocytosis. Cancer Res. 2016, 76, 4828–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Cheng, J.-X. Cholesterol Esterification Inhibition Suppresses Prostate Cancer Metastasis by Impairing the Wnt/β-Catenin Pathway. Mol. Cancer Res. 2018, 16, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-W.; Lo, Y.-H.; Chen, C.-H.; Lin, C.-Y.; Tsai, C.-H.; Chen, P.-J.; Yang, Y.-F.; Wang, C.-H.; Tan, C.-H.; Hou, M.-F.; et al. VLDL and LDL, but not HDL, promote breast cancer cell proliferation, metastasis and angiogenesis. Cancer Lett. 2017, 388, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.Y.; Ko, J.-H.; Um, J.-Y.; Chinnathambi, A.; Alharbi, S.A.; Sethi, G.; Ahn, K.S. LDL cholesterol promotes the proliferation of prostate and pancreatic cancer cells by activating the STAT3 pathway. J. Cell. Physiol. 2021, 236, 5253–5264. [Google Scholar] [CrossRef] [PubMed]
- González-Ortiz, A.; Galindo-Hernández, O.; Hernández-Acevedo, G.N.; Hurtado-Ureta, G.; García-González, V. Impact of cholesterol-pathways on breast cancer development, a metabolic landscape. J. Cancer 2021, 12, 4307–4321. [Google Scholar] [CrossRef]
- Ghahremanfard, F.; Mirmohammadkhani, M.; Shahnazari, B.; Gholami, G.; Mehdizadeh, J. The Valuable Role of Measuring Serum Lipid Profile in Cancer Progression. Oman Med. J. 2015, 30, 353–357. [Google Scholar] [CrossRef]
- Wang, C.; Li, P.; Xuan, J.; Zhu, C.; Liu, J.; Shan, L.; Du, Q.; Ren, Y.; Ye, J. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 729–742. [Google Scholar] [CrossRef]
- de Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortés, V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef] [Green Version]
- Mulas, M.F.; Abete, C.; Pulisci, D.; Pani, A.; Massidda, B.; Dessì, S.; Mandas, A. Cholesterol esters as growth regulators of lymphocytic leukaemia cells. Cell Prolif. 2011, 44, 360–371. [Google Scholar] [CrossRef]
- Bemlih, S.; Poirier, M.-D.; El Andaloussi, A. Acyl-coenzyme A: Cholesterol acyltransferase inhibitor Avasimibe affect survival and proliferation of glioma tumor cell lines. Cancer Biol. Ther. 2010, 9, 1025–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Song, B.-L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.S.-Y.; Li, J.; Tai, J.N.; Ratliff, T.L.; Park, K.; Cheng, J.-X. Avasimibe Encapsulated in Human Serum Albumin Blocks Cholesterol Esterification for Selective Cancer Treatment. ACS Nano 2015, 9, 2420–2432. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benitez-Amaro, A.; Martínez-Bosch, N.; Manero-Rupérez, N.; Claudi, L.; La Chica Lhoëst, M.T.; Soler, M.; Ros-Blanco, L.; Navarro, P.; Llorente-Cortés, V. Peptides against Low Density Lipoprotein (LDL) Aggregation Inhibit Intracellular Cholesteryl Ester Loading and Proliferation of Pancreatic Tumor Cells. Cancers 2022, 14, 890. https://doi.org/10.3390/cancers14040890
Benitez-Amaro A, Martínez-Bosch N, Manero-Rupérez N, Claudi L, La Chica Lhoëst MT, Soler M, Ros-Blanco L, Navarro P, Llorente-Cortés V. Peptides against Low Density Lipoprotein (LDL) Aggregation Inhibit Intracellular Cholesteryl Ester Loading and Proliferation of Pancreatic Tumor Cells. Cancers. 2022; 14(4):890. https://doi.org/10.3390/cancers14040890
Chicago/Turabian StyleBenitez-Amaro, Aleyda, Neus Martínez-Bosch, Noemí Manero-Rupérez, Lene Claudi, Maria Teresa La Chica Lhoëst, Marta Soler, Lia Ros-Blanco, Pilar Navarro, and Vicenta Llorente-Cortés. 2022. "Peptides against Low Density Lipoprotein (LDL) Aggregation Inhibit Intracellular Cholesteryl Ester Loading and Proliferation of Pancreatic Tumor Cells" Cancers 14, no. 4: 890. https://doi.org/10.3390/cancers14040890
APA StyleBenitez-Amaro, A., Martínez-Bosch, N., Manero-Rupérez, N., Claudi, L., La Chica Lhoëst, M. T., Soler, M., Ros-Blanco, L., Navarro, P., & Llorente-Cortés, V. (2022). Peptides against Low Density Lipoprotein (LDL) Aggregation Inhibit Intracellular Cholesteryl Ester Loading and Proliferation of Pancreatic Tumor Cells. Cancers, 14(4), 890. https://doi.org/10.3390/cancers14040890