
Tumor Cell-Autonomous Pro-Metastatic Activities of PD-L1 in Human Breast Cancer Are Mediated by PD-L1-S283 and Chemokine Axes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
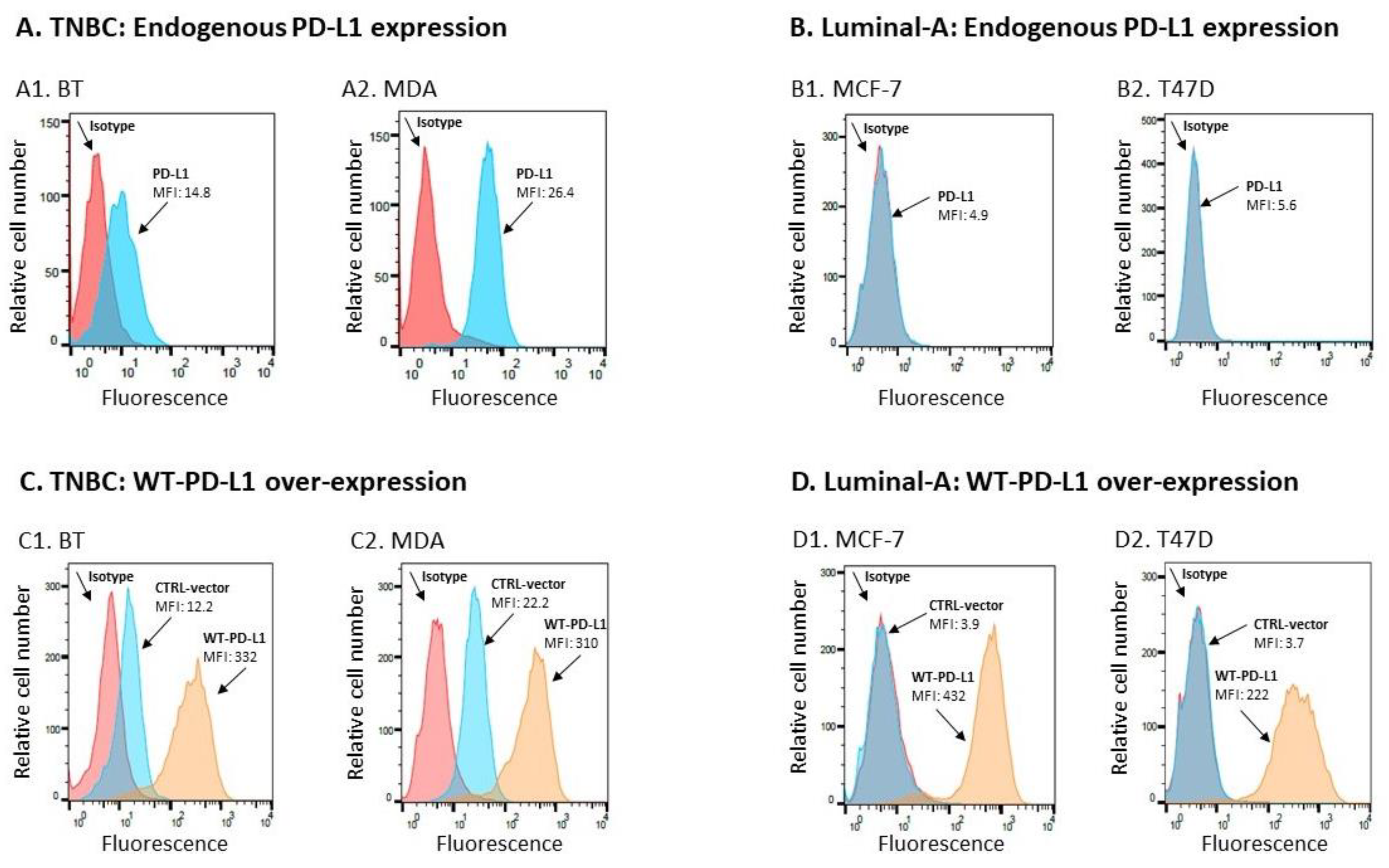
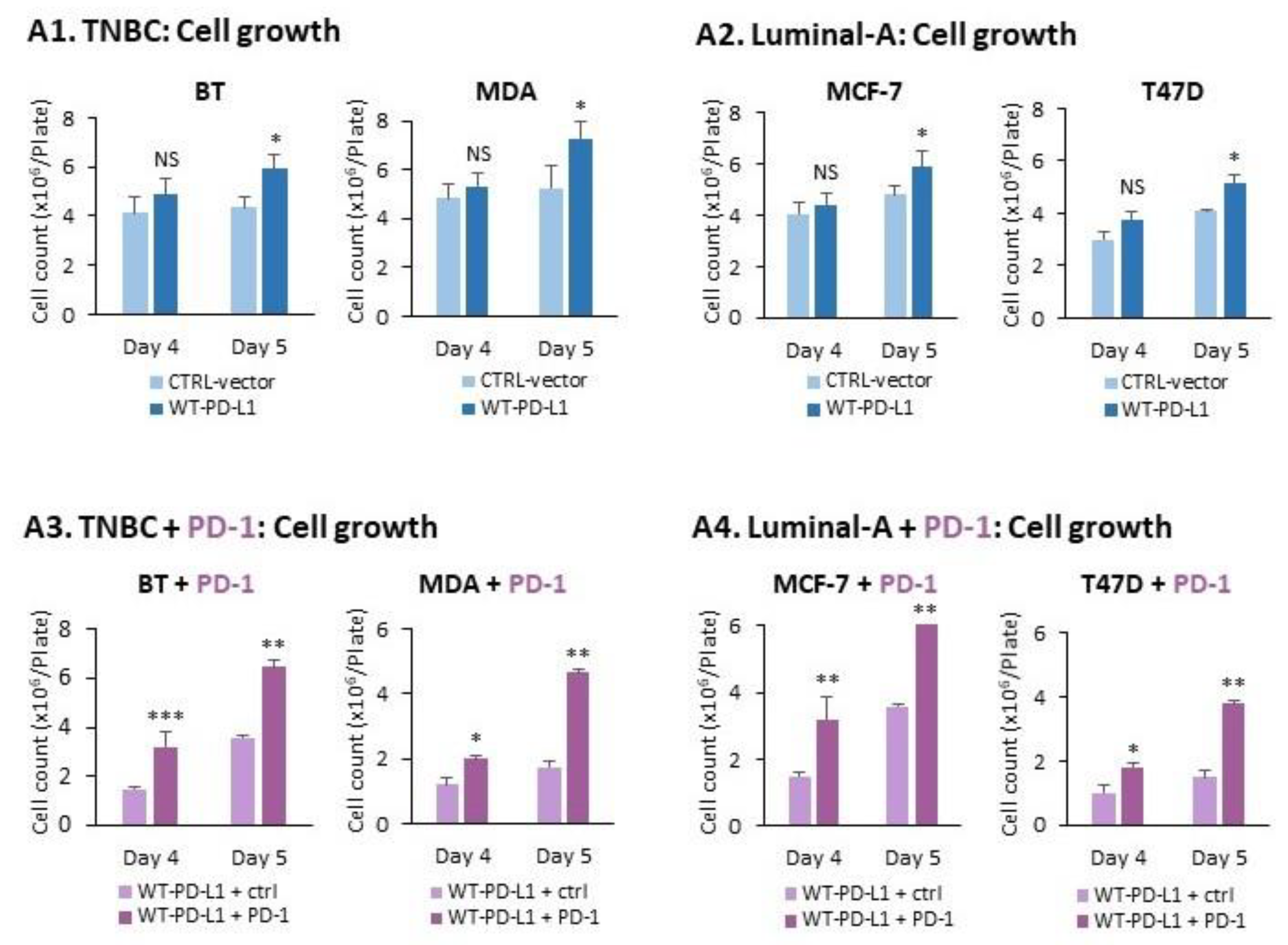
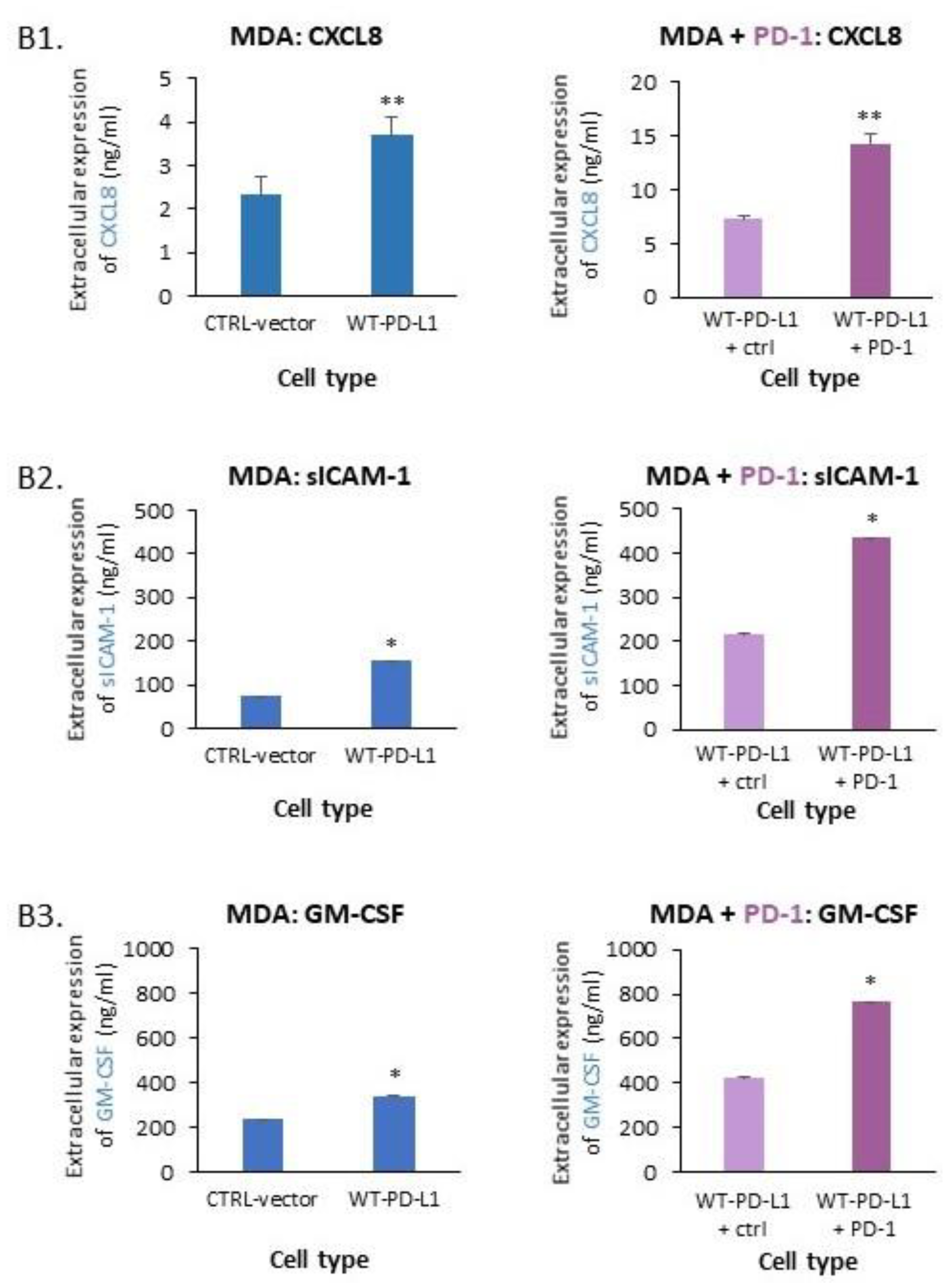
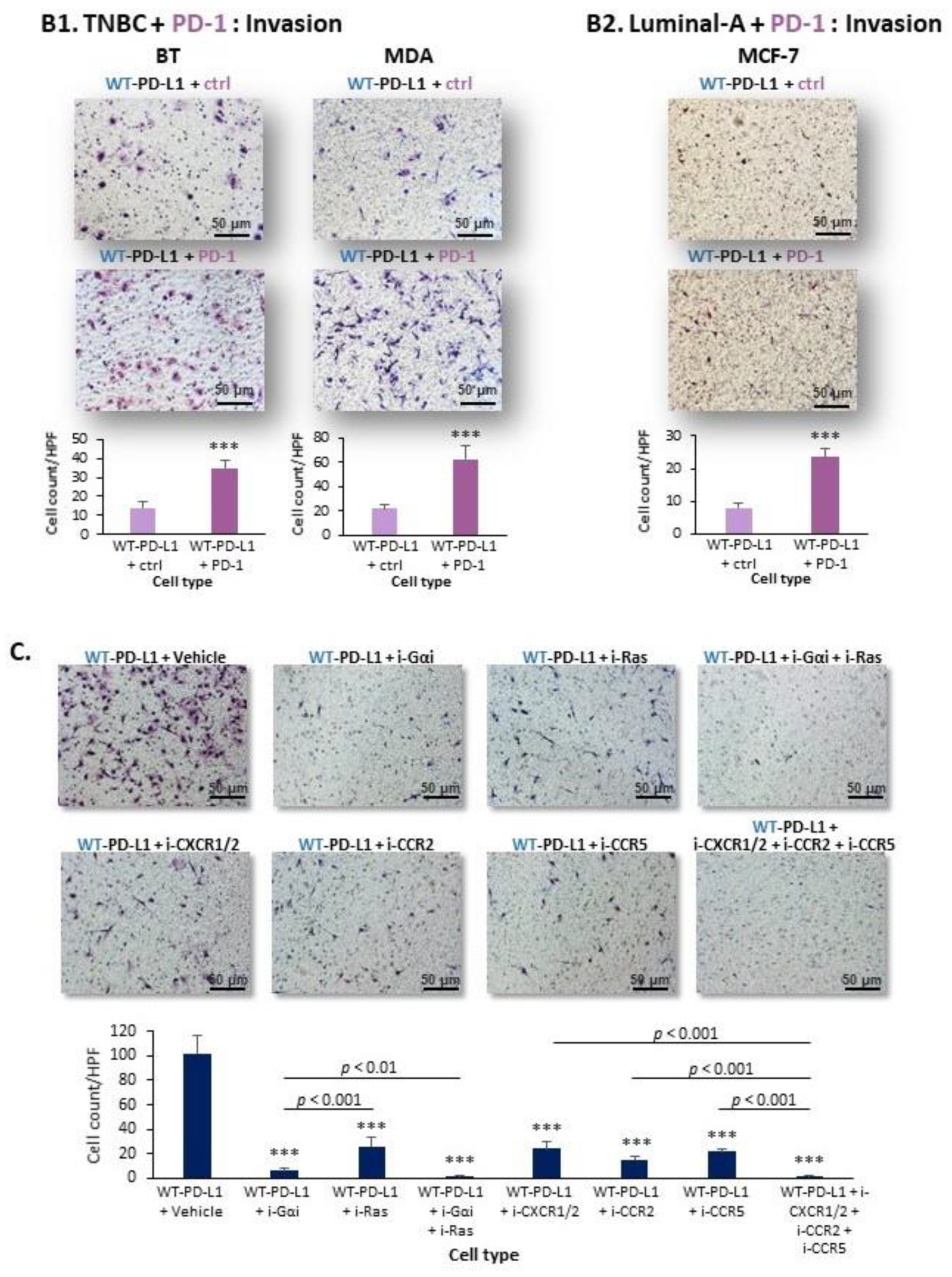
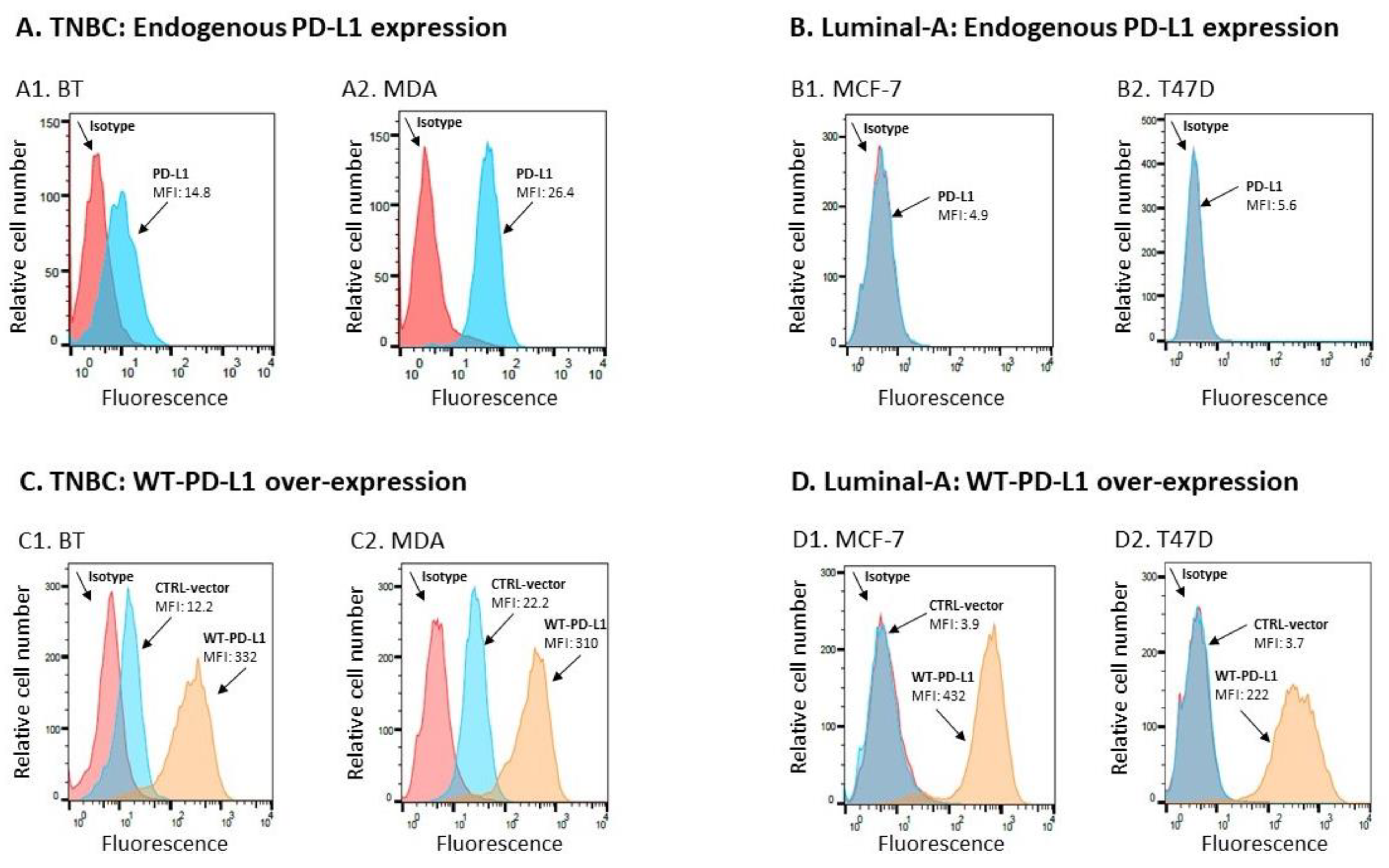
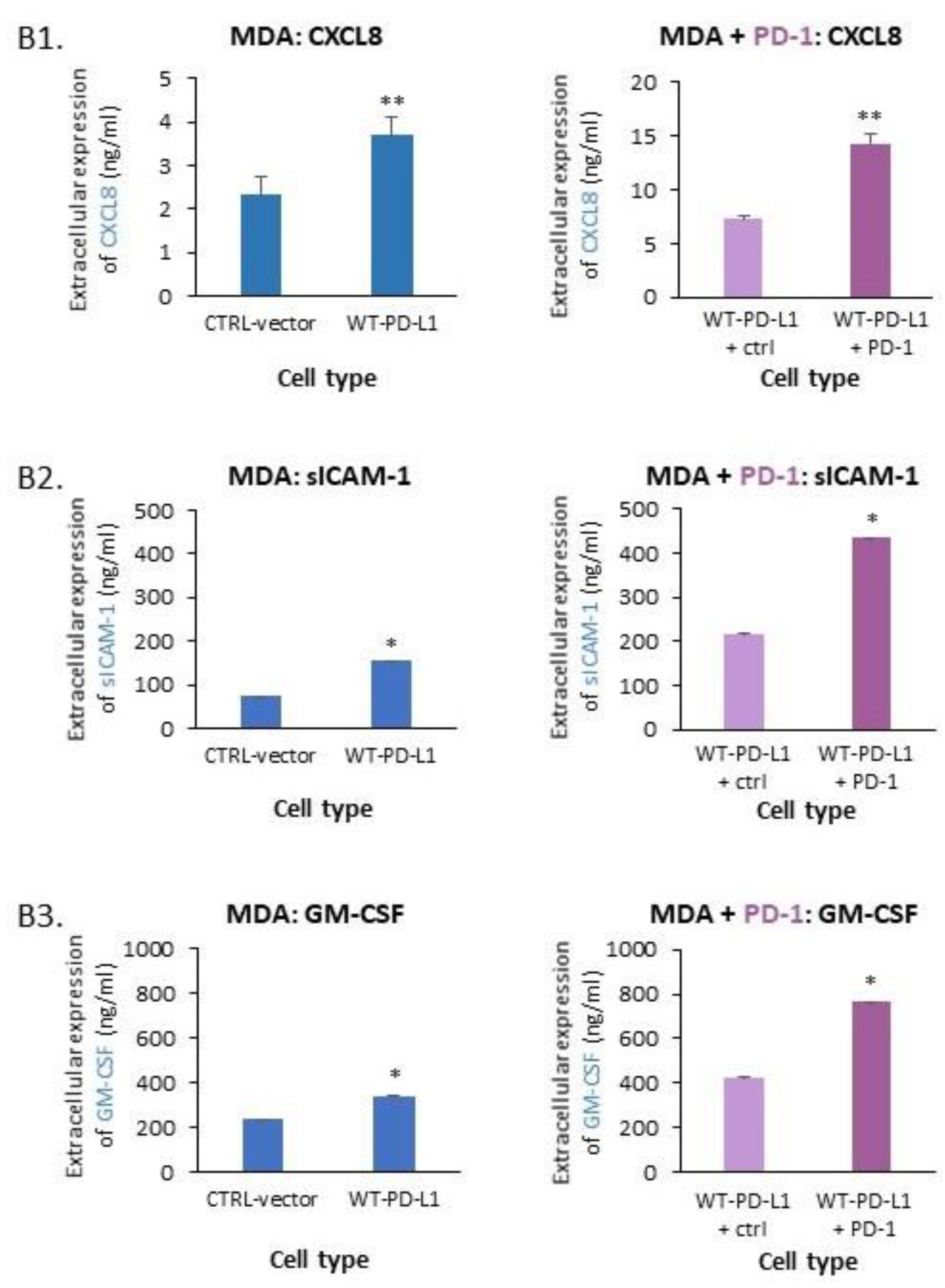
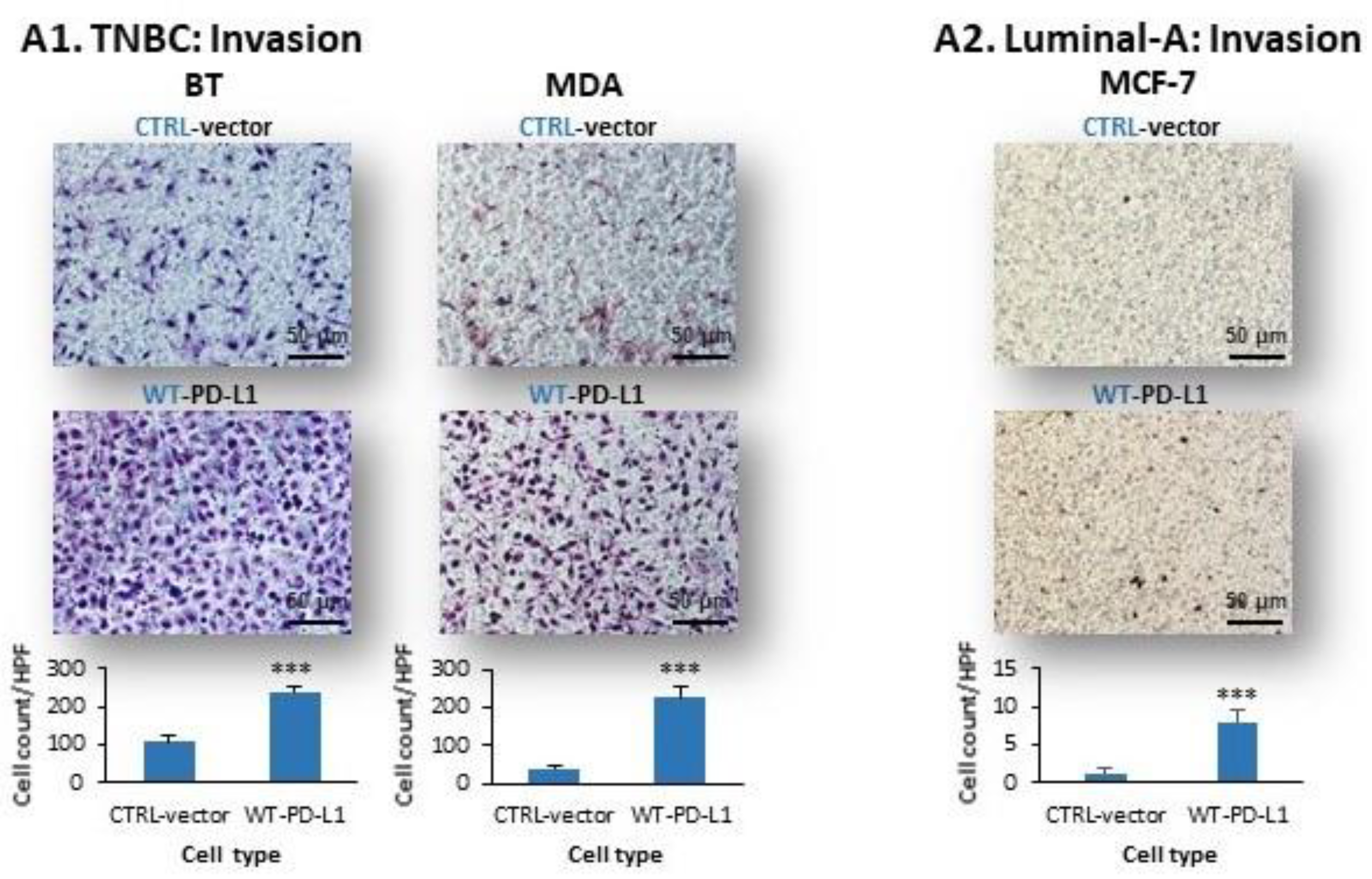
2.1. PD-L1 Exerts Cell-Autonomous Metastasis-Supporting Activities That Are Increased by Exposure of Breast Tumor Cells to PD-1
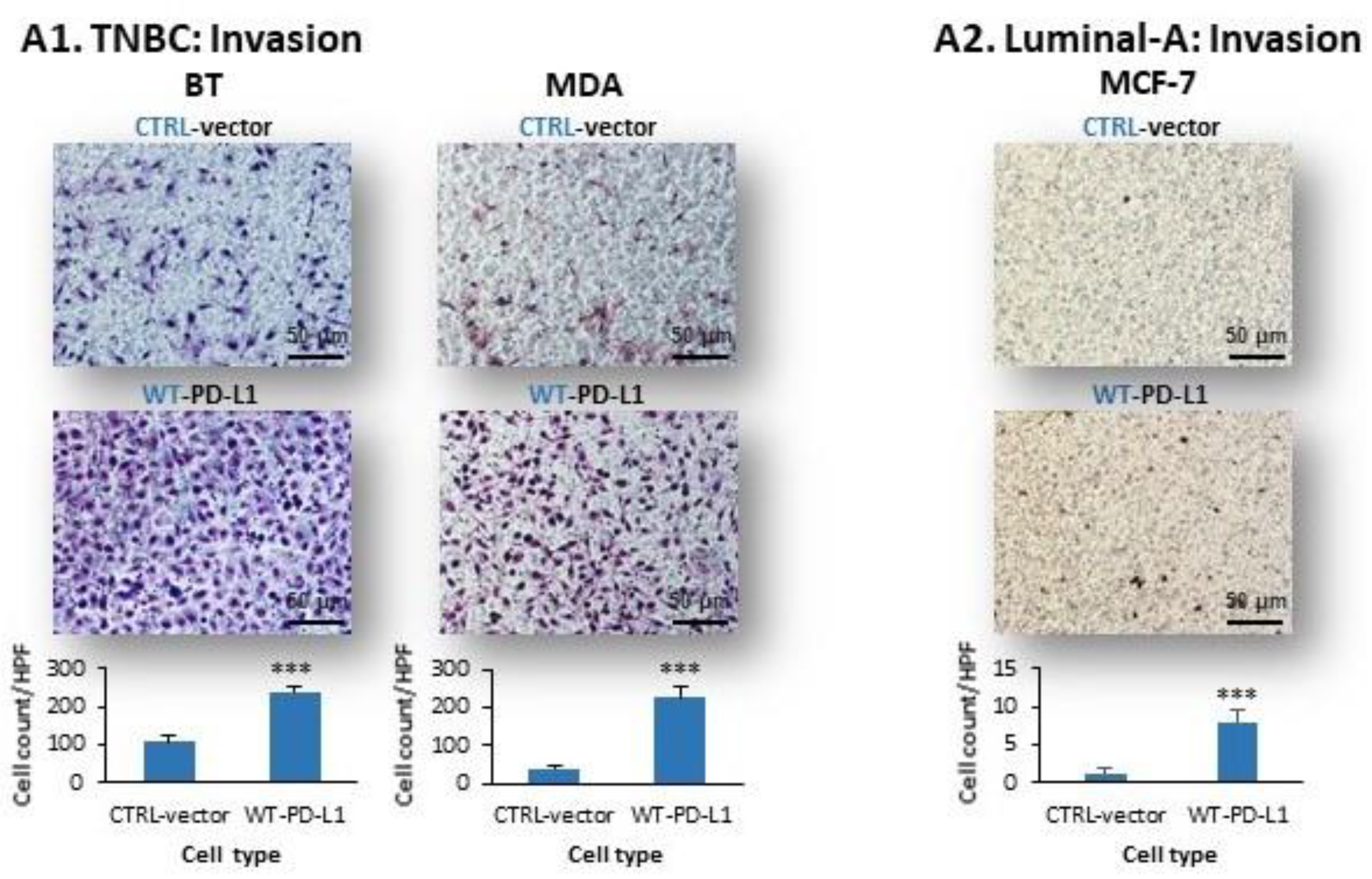
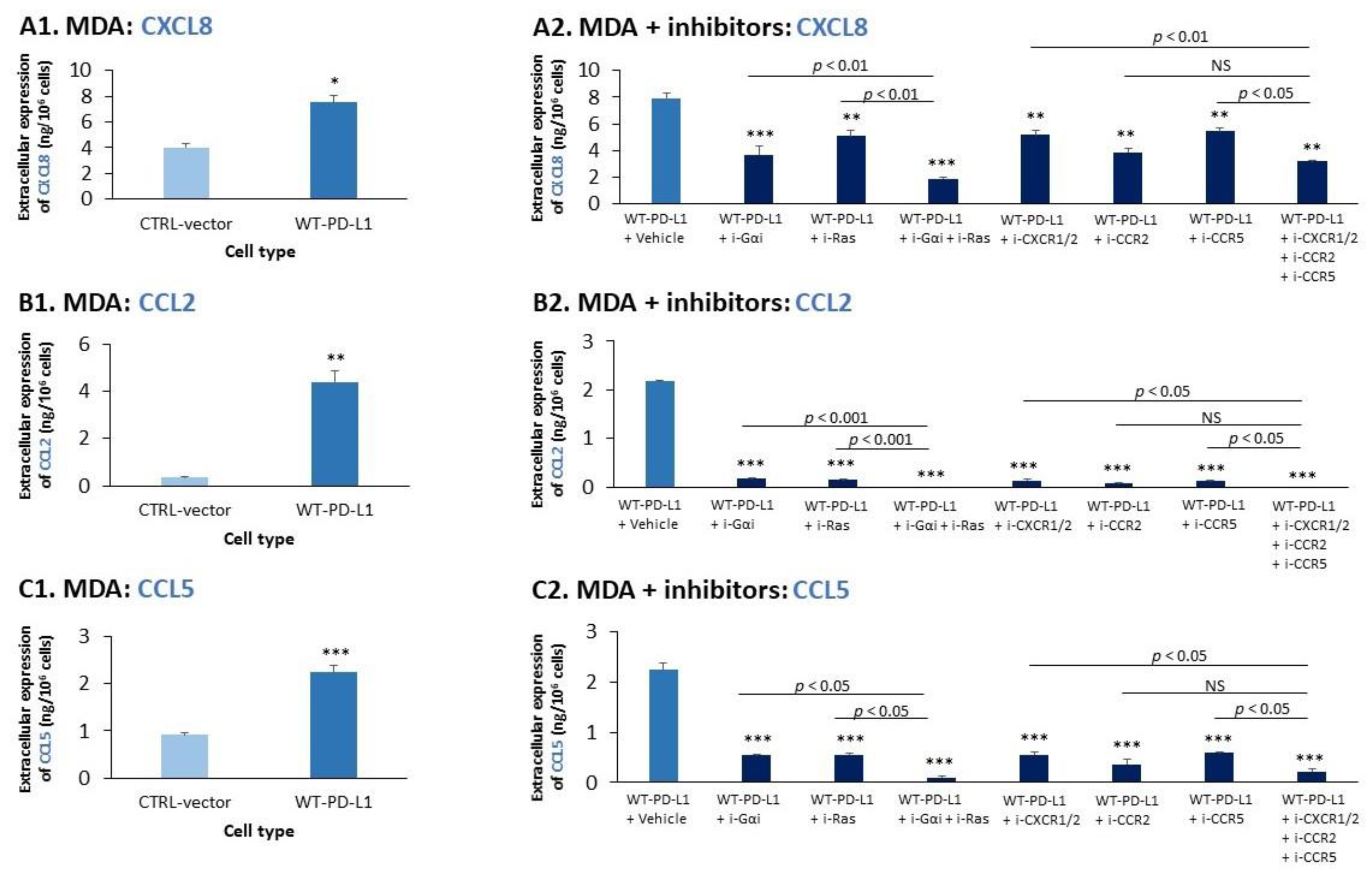
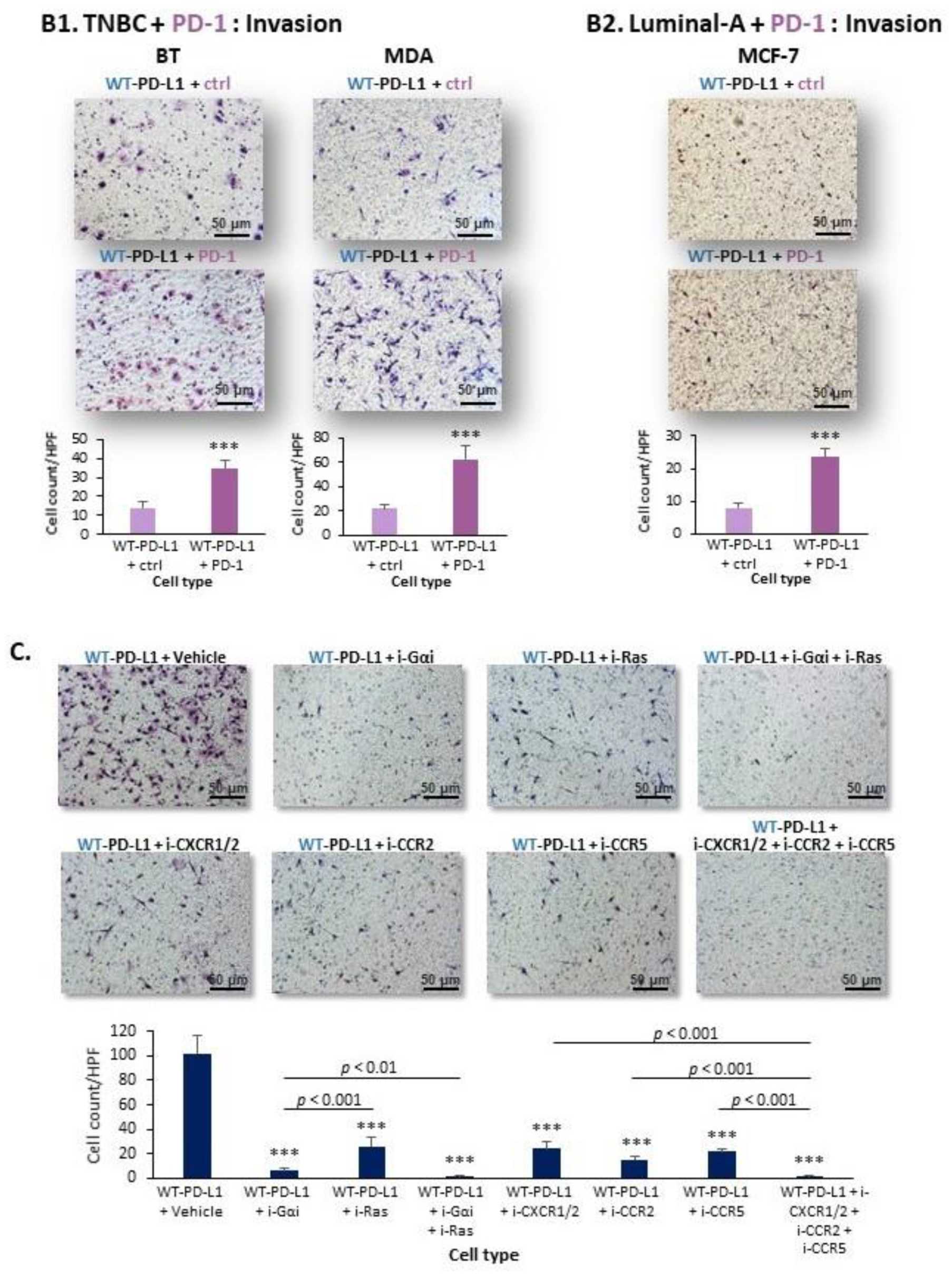
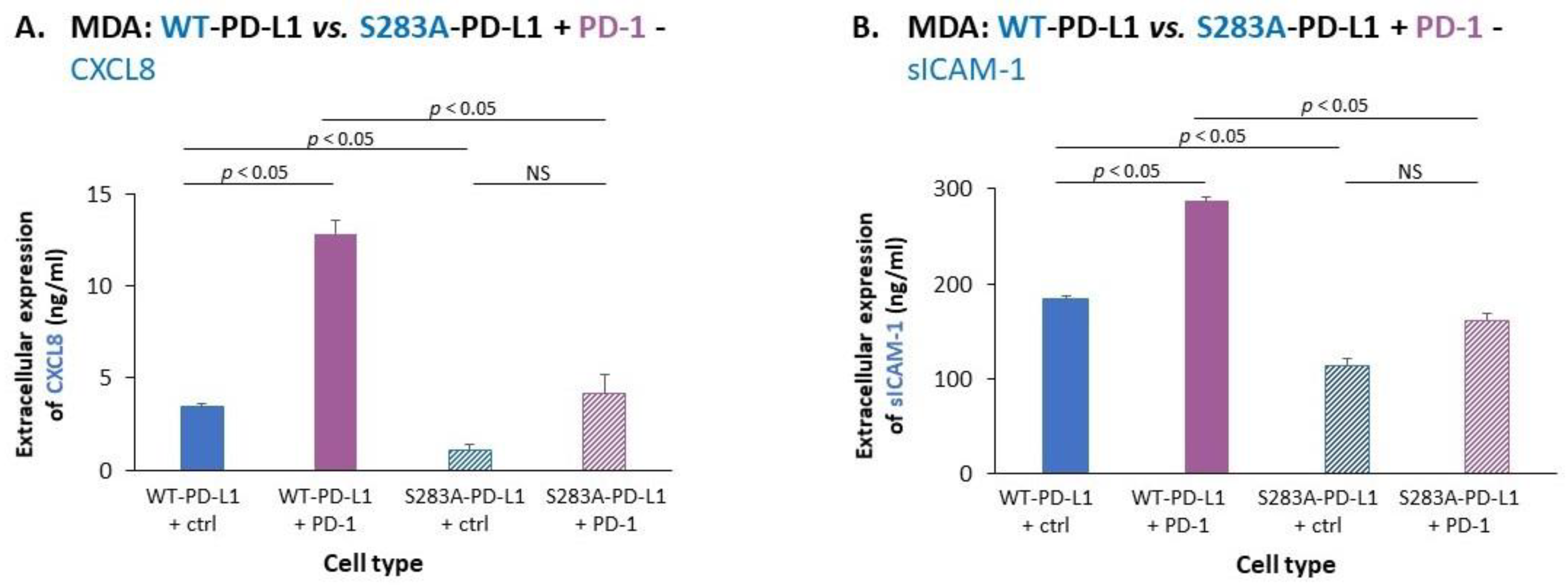
2.2. Cell-Autonomous PD-L1-Induced Invasion of TNBC Cells Is Promoted by Chemokine Axes
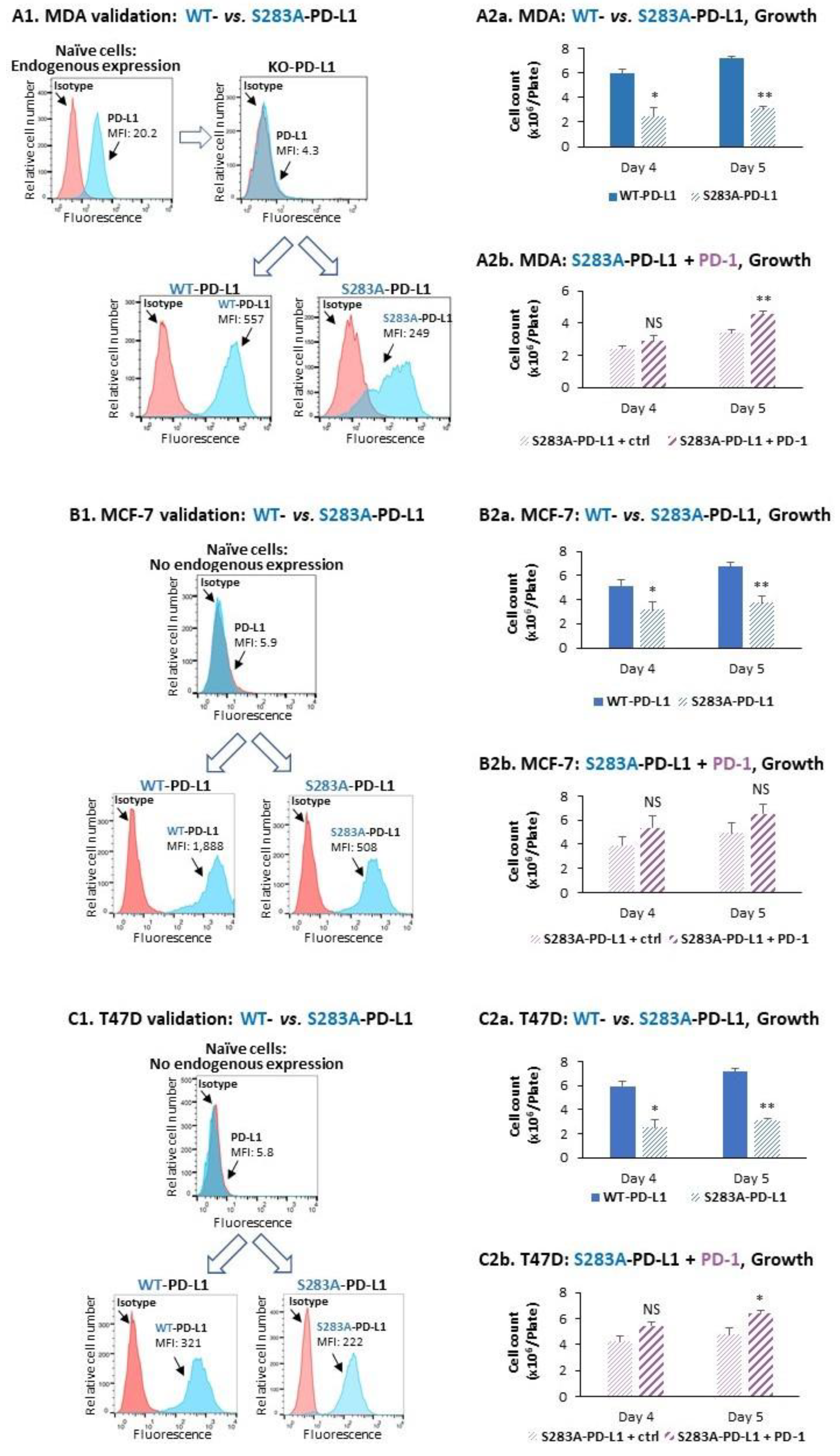
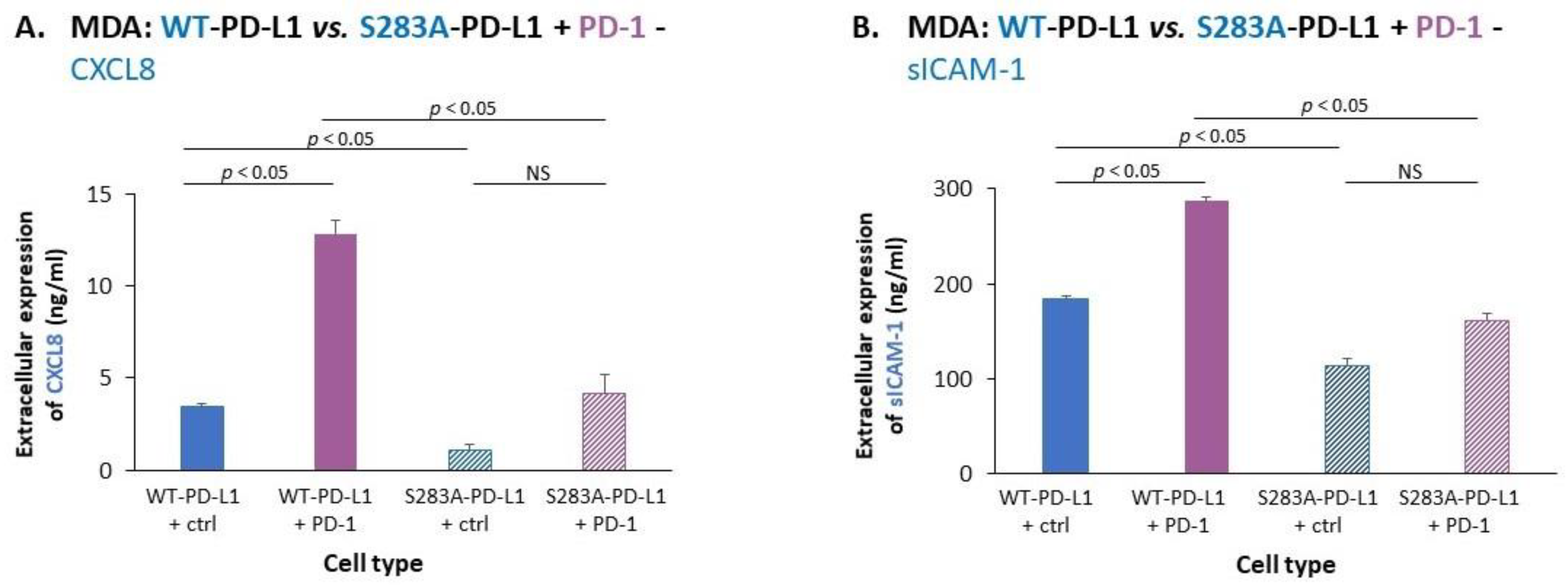
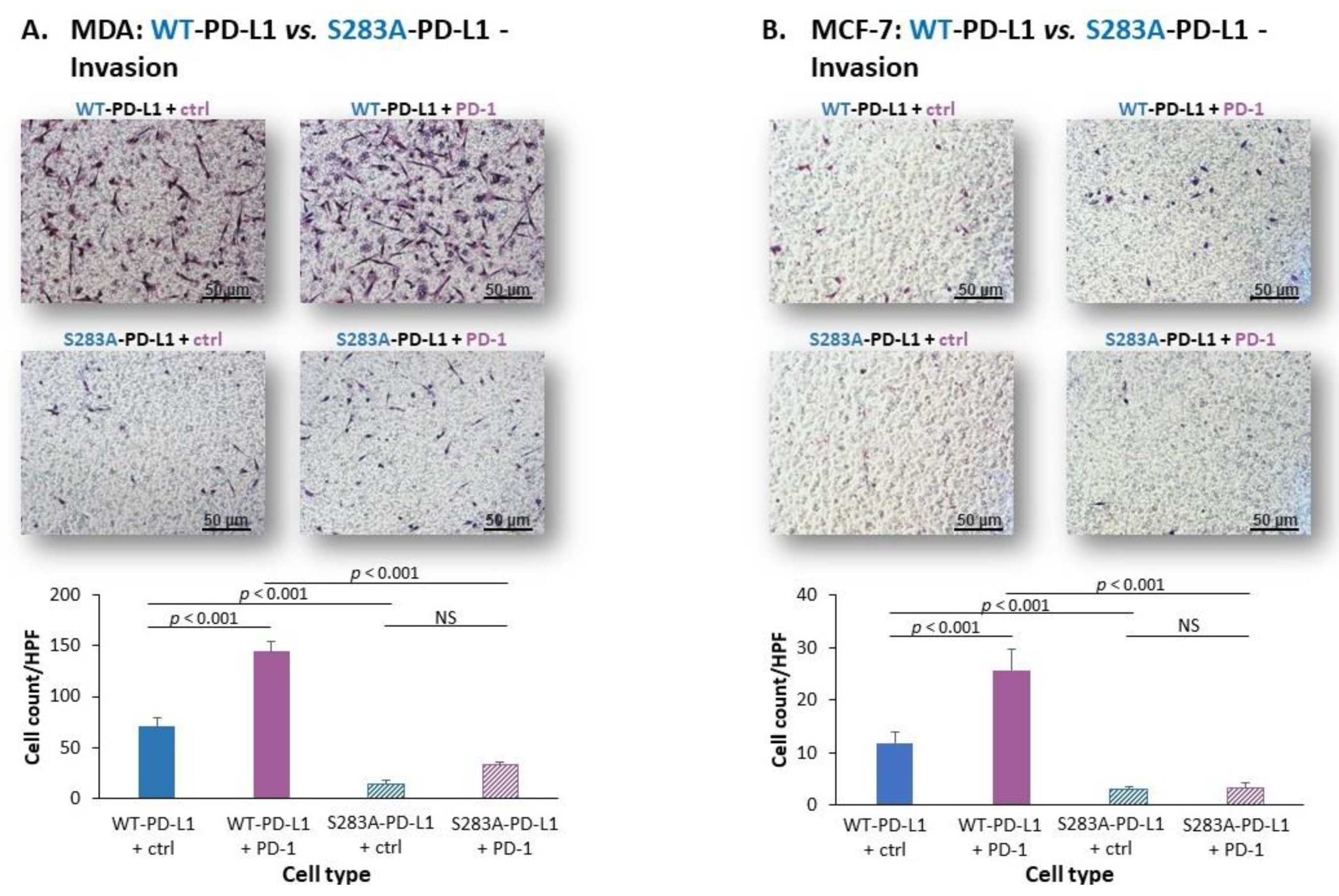
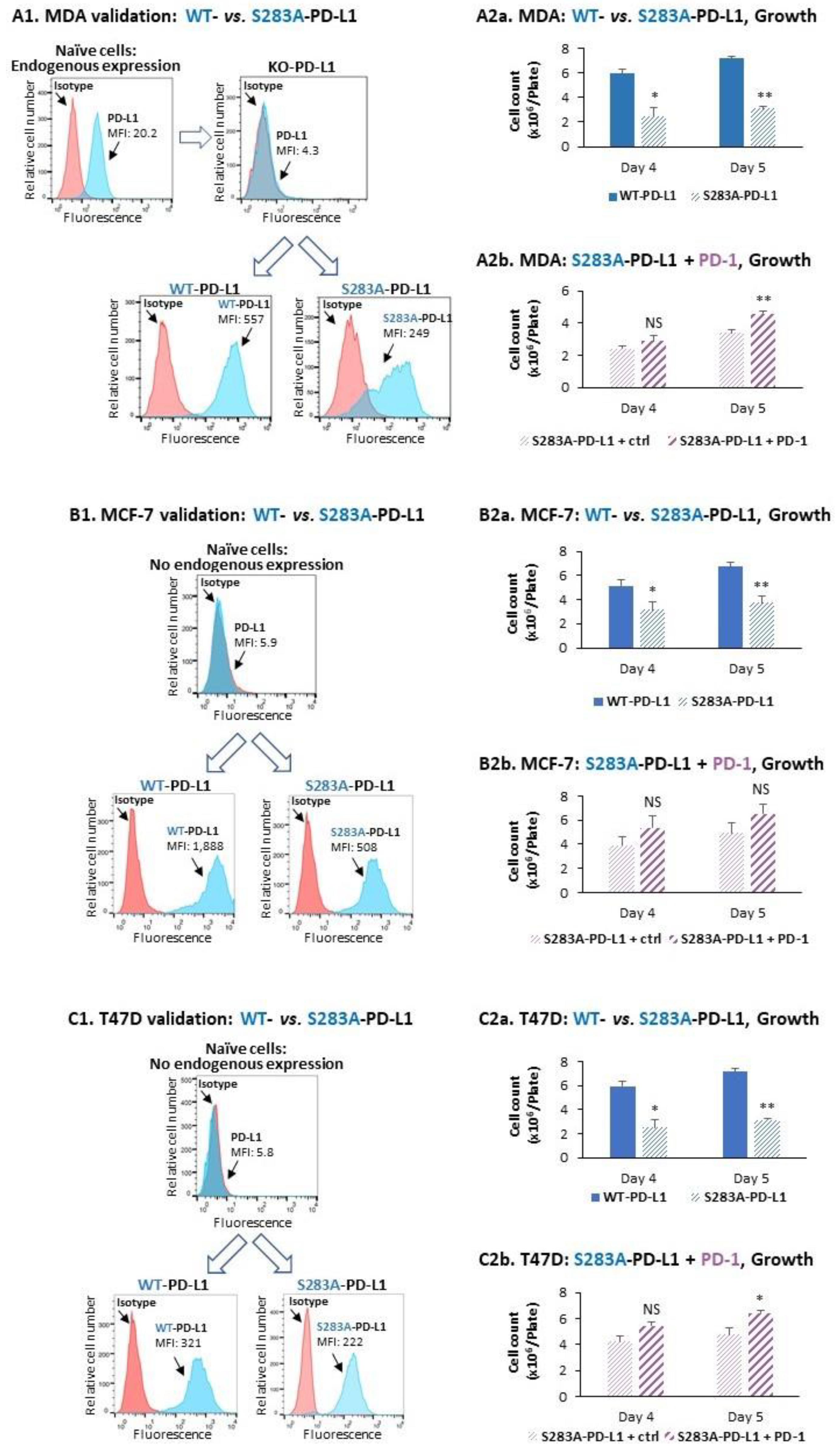
2.3. The Cell-Autonomous and PD-1-Induced Activities of PD-L1 in Breast Tumor Cells Depend on Integrity of the S283 Residue
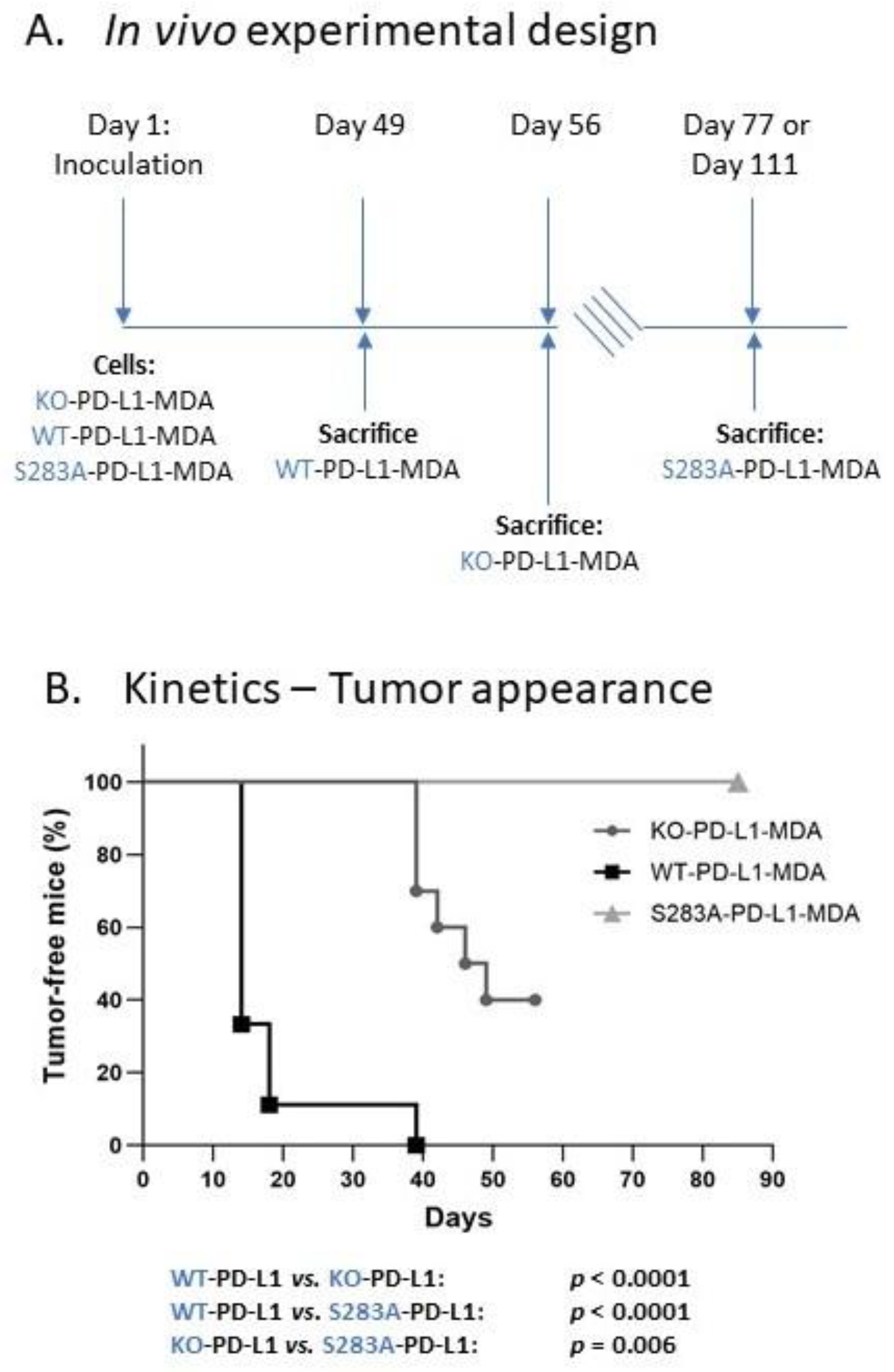
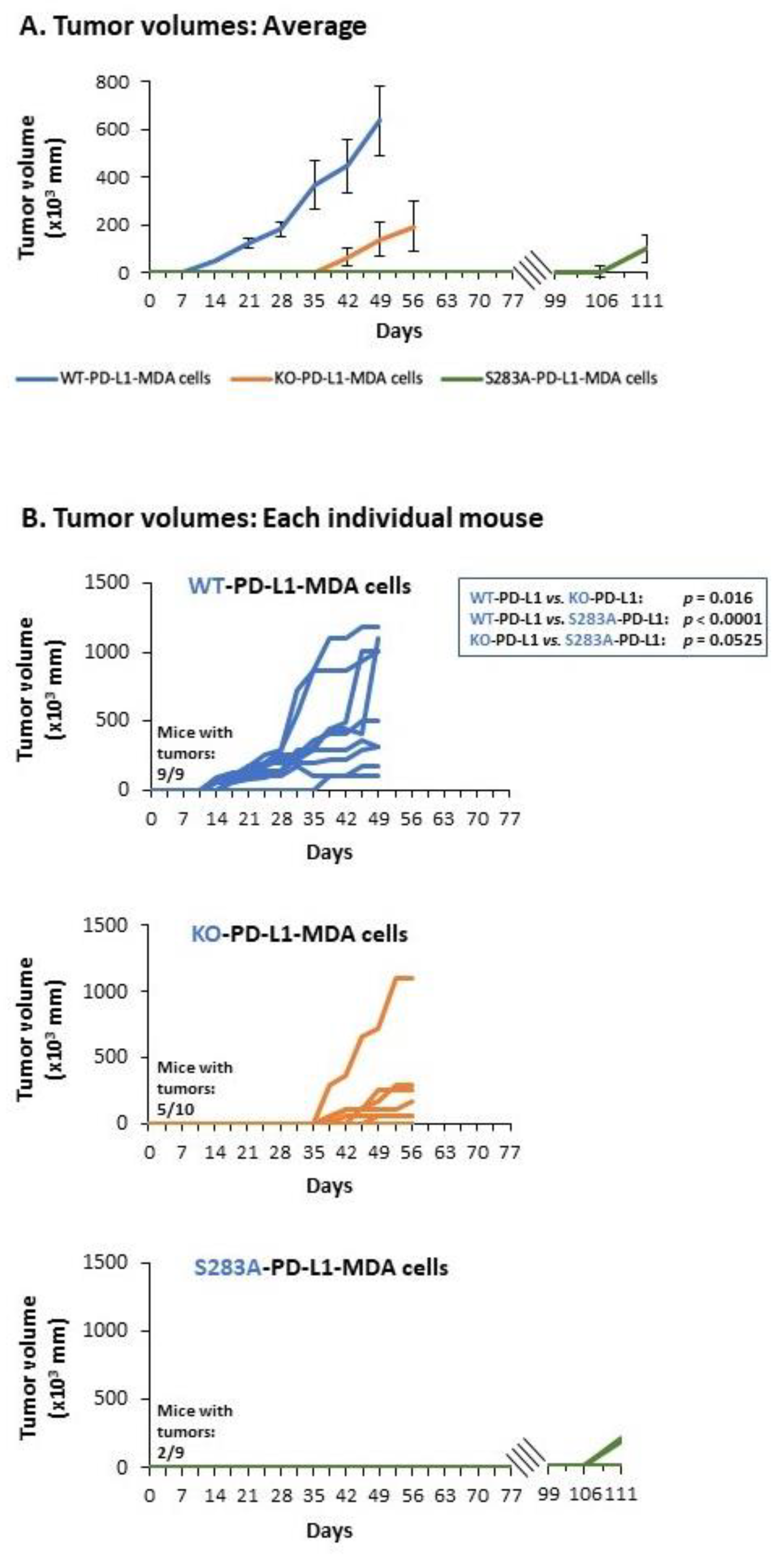
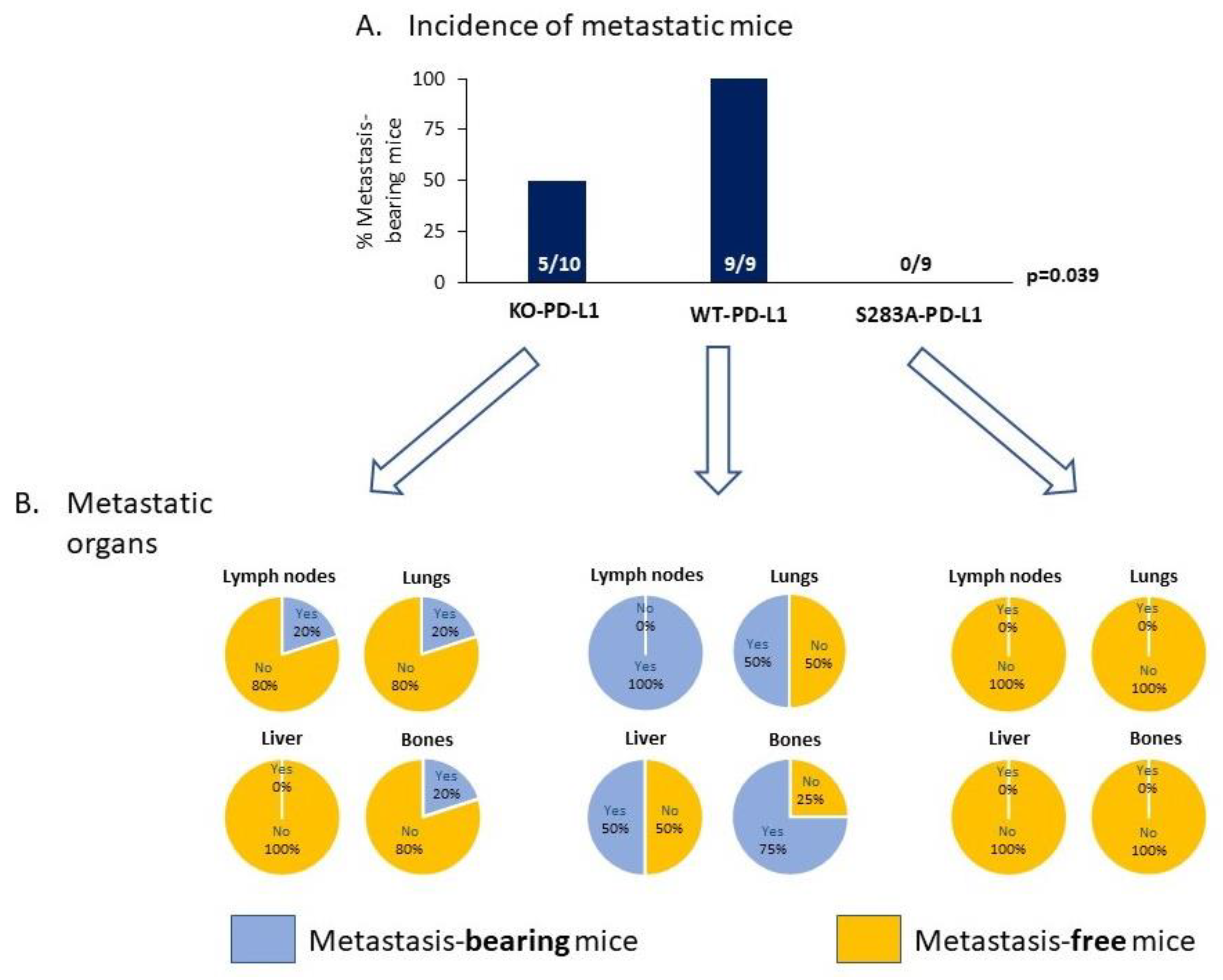
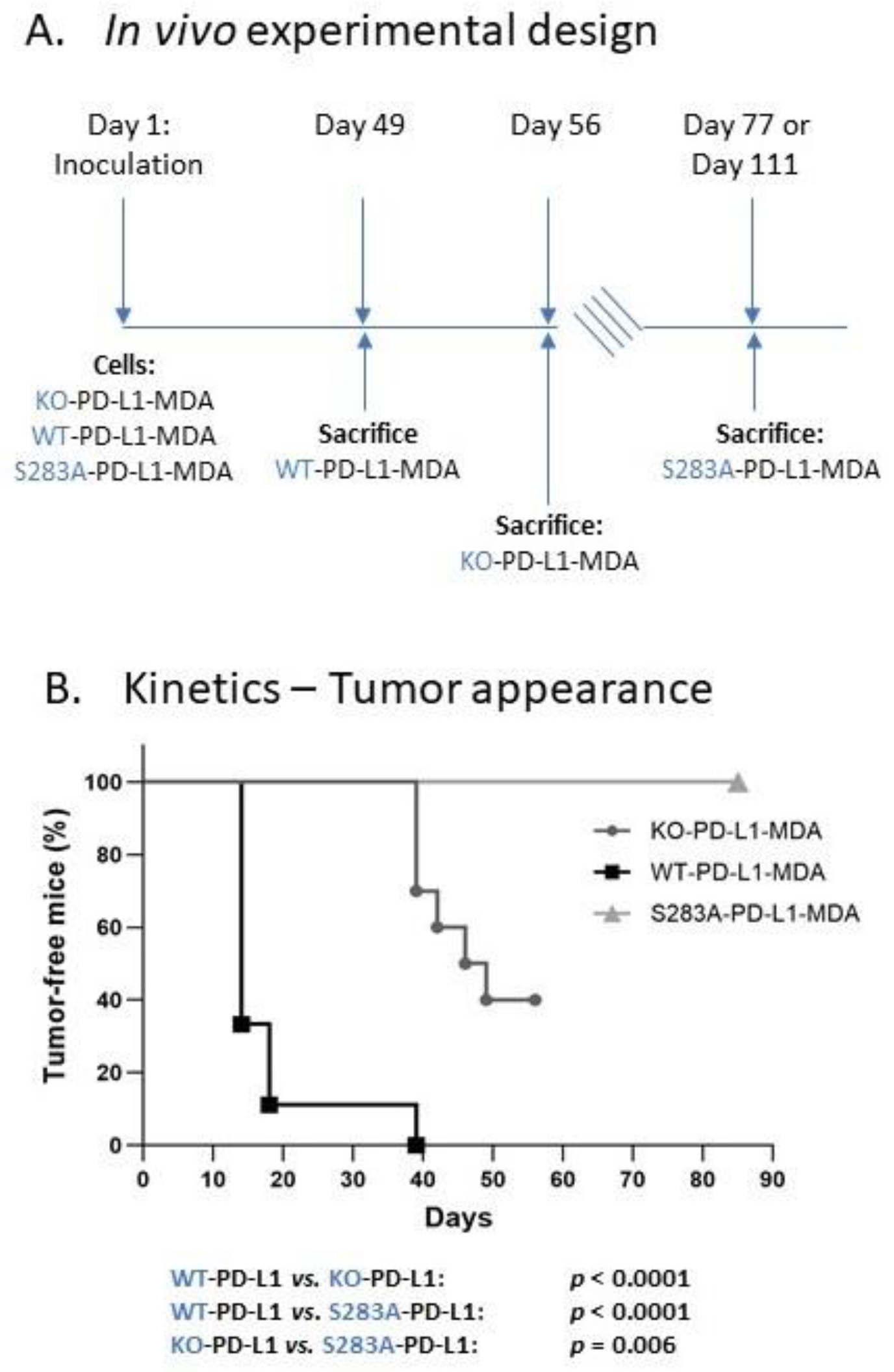
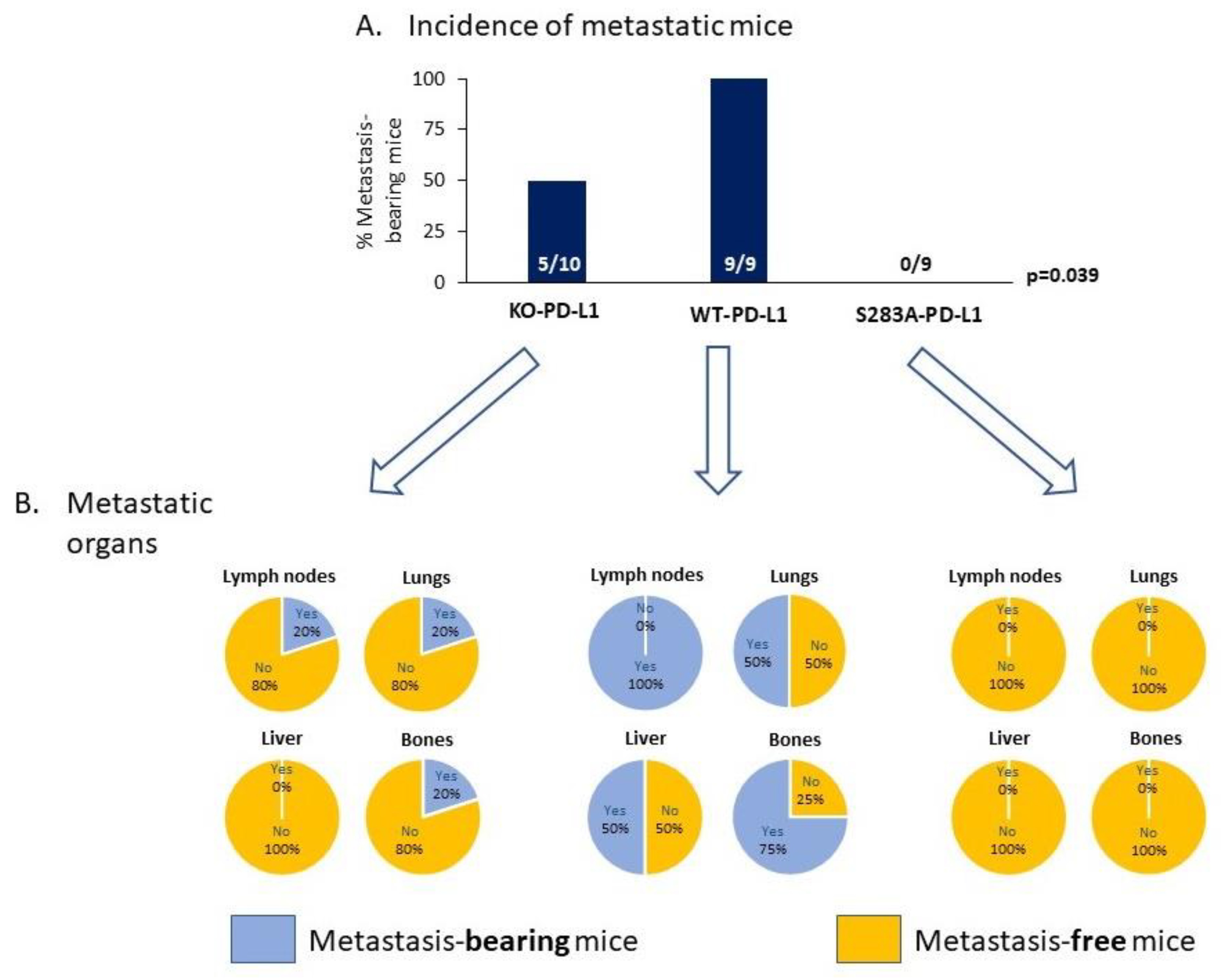
2.4. The Cell-Autonomous Activities of PD-L1 Promote Tumor Progression In Vivo, in a S283-Dependent Manner
3. Discussion
- PD-L1 exerted tumor-promoting functions in breast cancer, which are not connected to its ability to induce immune suppression but rather to potentiation of intrinsic tumor cell activities that support tumor progression: cancer cell proliferation, release of soluble pro-metastatic factors and invasion through matrigel and organ-relevant ECM. Moreover, these cell-intrinsic pro-metastatic activities of PD-L1 were more potent when its levels were increased, indicating that not only the incidence of PD-L1-expressing cells but also its expression levels by the cells dictates the efficacy of PD-L1 in promoting metastasis-supporting intrinsic activities in the cancer cells. Furthermore, PD-1 enhanced all of these activities and the cell-autonomous activities of PD-L1 potently increased tumor growth and metastasis in the in vivo setting, independently of its immune suppressive activities.
- We demonstrated, for the first time, that the S283 residue of PD-L1, which is expressed at the intracellular domain of the protein, is absolutely essential for PD-L1-induced intracellular signaling that leads to increased tumor cell proliferation, release of pro-metastatic soluble factors and invasion. The cardinal roles of S283 were reinforced by in vivo studies, demonstrating the critical roles of the S283 residue in regulating the intrinsic activities of PD-L1 that lead to increased tumor growth and metastasis.
- We identified novel intracellular signaling cascades that mediate PD-L1 intrinsic activities in TNBC cells. We demonstrated that chemokine axes can establish positive autocrine feedback loops that connect intracellular signaling events with the increased ability of TNBC cells to invade. Here, our research identified the chemokine receptors CXCR1/2, CCR2, CCR5 and their ligands to stand in the center of the regulatory circuits that control PD-L1 pro-tumoral activities in TNBC cells.
4. Materials and Methods
4.1. Cell Growth and Exposure to PD-1
4.2. Generation of Cells Over-Expressing WT-PD-L1 or S283A-PD-L1
4.3. Flow Cytometry Analyses of PD-L1 Expression
4.4. Determination of Cancer Cell Growth
4.5. ELISA Assays
4.6. Transwell Invasion Assays
4.7. Inhibitors: Titration and Selected Concentrations
4.8. TCGA Dataset Analyses
4.9. Modeling of PD-L1-Peptide Binding to AMPK
4.10. In Vivo Studies
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molnar, I.A.; Molnar, B.A.; Vizkeleti, L.; Fekete, K.; Tamas, J.; Deak, P.; Szundi, C.; Szekely, B.; Moldvay, J.; Vari-Kakas, S.; et al. Breast carcinoma subtypes show different patterns of metastatic behavior. Virchows Arch. 2017, 470, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Abdelhakiem, M.K.; Johnstone, C.; Bergom, C.; Currey, A.; Robbins, J.R. The influence of breast cancer subtype on survival after palliative radiation for osseous metastases. Cancer Med. 2020, 9, 8979–8988. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Eroles, P.; Bosch, A.; Perez-Fidalgo, J.A.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Fanotto, V.; Bonotto, M.; Bolzonello, S.; Andreetta, C.; Moroso, S.; Pascoletti, G.; Fasole, G.; Puglisi, F. Pattern of metastatic spread and prognosis of breast cancer biologic subtypes. J. Clin. Oncol. 2014, 32, e12532. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Makhoul, I.; Atiq, M.; Alwbari, A.; Kieber-Emmons, T. Breast Cancer Immunotherapy: An Update. Breast Cancer 2018, 12, 1178223418774802. [Google Scholar] [CrossRef]
- Szekely, B.; Silber, A.L.; Pusztai, L. New Therapeutic Strategies for Triple-Negative Breast Cancer. Oncology 2017, 31, 130–137. [Google Scholar]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Farshbafnadi, M.; Pastaki Khoshbin, A.; Rezaei, N. Immune checkpoint inhibitors for triple-negative breast cancer: From immunological mechanisms to clinical evidence. Int. Immunopharmacol. 2021, 98, 107876. [Google Scholar] [CrossRef] [PubMed]
- Dushyanthen, S.; Beavis, P.A.; Savas, P.; Teo, Z.L.; Zhou, C.; Mansour, M.; Darcy, P.K.; Loi, S. Relevance of tumor-infiltrating lymphocytes in breast cancer. BMC Med. 2015, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Savas, P.; Salgado, R.; Loi, S. Seeing the forest and the tree: TILs and PD-L1 as immune biomarkers. Breast Cancer Res. Treat. 2021, 189, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, N.; Gao, Y.; Jia, L.; Lyu, Z.; Cui, J. Clinical Progress of PD-1/L1 Inhibitors in Breast Cancer Immunotherapy. Front. Oncol. 2021, 11, 724424. [Google Scholar] [CrossRef] [PubMed]
- Mediratta, K.; El-Sahli, S.; D’Costa, V.; Wang, L. Current Progresses and Challenges of Immunotherapy in Triple-Negative Breast Cancer. Cancers 2020, 12, 3529. [Google Scholar] [CrossRef]
- Force, J.; Leal, J.H.S.; McArthur, H.L. Checkpoint Blockade Strategies in the Treatment of Breast Cancer: Where We Are and Where We Are Heading. Curr Treat. Options Oncol. 2019, 20, 35. [Google Scholar] [CrossRef]
- Heimes, A.S.; Schmidt, M. Atezolizumab for the treatment of triple-negative breast cancer. Expert Opin. Investig. Drugs 2019, 28, 1–5. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef]
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Munoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499–511. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Savas, P.; Loi, S. Expanding the Role for Immunotherapy in Triple-Negative Breast Cancer. Cancer Cell 2020, 37, 623–624. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-atezolizumab-pd-l1-positive-unresectable-locally-advanced-or-metastatic-triple-negative (accessed on 12 December 2021).
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef]
- Karasar, P.; Esendagli, G. T helper responses are maintained by basal-like breast cancer cells and confer to immune modulation via upregulation of PD-1 ligands. Breast Cancer Res. Treat. 2014, 145, 605–614. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Matikas, A.; Zerdes, I.; Lovrot, J.; Richard, F.; Sotiriou, C.; Bergh, J.; Valachis, A.; Foukakis, T. Prognostic Implications of PD-L1 Expression in Breast Cancer: Systematic Review and Meta-analysis of Immunohistochemistry and Pooled Analysis of Transcriptomic Data. Clin. Cancer Res. 2019, 25, 5717–5726. [Google Scholar] [CrossRef]
- Li, Z.; Dong, P.; Ren, M.; Song, Y.; Qian, X.; Yang, Y.; Li, S.; Zhang, X.; Liu, F. PD-L1 Expression Is Associated with Tumor FOXP3(+) Regulatory T-Cell Infiltration of Breast Cancer and Poor Prognosis of Patient. J. Cancer 2016, 7, 784–793. [Google Scholar] [CrossRef]
- Almozyan, S.; Colak, D.; Mansour, F.; Alaiya, A.; Al-Harazi, O.; Qattan, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int. J. Cancer 2017, 141, 1402–1412. [Google Scholar] [CrossRef]
- Saleh, R.; Taha, R.Z.; Sasidharan Nair, V.; Alajez, N.M.; Elkord, E. PD-L1 Blockade by Atezolizumab Downregulates Signaling Pathways Associated with Tumor Growth, Metastasis, and Hypoxia in Human Triple Negative Breast Cancer. Cancers 2019, 11, 1050. [Google Scholar] [CrossRef]
- Chen, C.; Li, S.; Xue, J.; Qi, M.; Liu, X.; Huang, Y.; Hu, J.; Dong, H.; Ling, K. PD-L1 tumor-intrinsic signaling and its therapeutic implication in triple-negative breast cancer. JCI Insight 2021, 6, e131458. [Google Scholar] [CrossRef] [PubMed]
- Lotfinejad, P.; Kazemi, T.; Safaei, S.; Amini, M.; Roshani Asl, E.; Baghbani, E.; Sandoghchian Shotorbani, S.; Jadidi Niaragh, F.; Derakhshani, A.; Abdoli Shadbad, M.; et al. PD-L1 silencing inhibits triple-negative breast cancer development and upregulates T-cell-induced pro-inflammatory cytokines. Biomed. Pharm. 2021, 138, 111436. [Google Scholar] [CrossRef] [PubMed]
- Montemagno, C.; Hagege, A.; Borchiellini, D.; Thamphya, B.; Rastoin, O.; Ambrosetti, D.; Iovanna, J.; Rioux-Leclercq, N.; Porta, C.; Negrier, S.; et al. Soluble forms of PD-L1 and PD-1 as prognostic and predictive markers of sunitinib efficacy in patients with metastatic clear cell renal cell carcinoma. Oncoimmunology 2020, 9, 1846901. [Google Scholar] [CrossRef] [PubMed]
- Vajavaara, H.; Bondgaard Mortensen, J.; Leivonen, S.K.; Monrad Hansen, I.; Ludvigsen, M.; Holte, H.; Jørgensen, J.; Bjerre, M.; d’Amore, F.; Leppä, S. Soluble PD-1 but Not PD-L1 Levels Predict Poor Outcome in Patients with High-Risk Diffuse Large B-Cell Lymphoma. Cancers 2021, 13, 398. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Zhao, Z.; Arooj, S.; Fu, Y.; Liao, G. Soluble PD-1: Predictive, Prognostic, and Therapeutic Value for Cancer Immunotherapy. Front. Immunol. 2020, 11, 587460. [Google Scholar] [CrossRef] [PubMed]
- Black, M.; Barsoum, I.B.; Truesdell, P.; Cotechini, T.; Macdonald-Goodfellow, S.K.; Petroff, M.; Siemens, D.R.; Koti, M.; Craig, A.W.B.; Graham, C.H. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget 2016, 7, 10557–10567. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Ao, X.; Yang, Y.; Chen, Z.; Xu, X. Soluble immune checkpoints in cancer: Production, function and biological significance. J. Immunother. Cancer 2018, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Morein, D.; Rubinstein-Achiasaf, L.; Brayer, H.; Dorot, O.; Pichinuk, E.; Ben-Yaakov, H.; Meshel, T.; Pasmanik-Chor, M.; Ben-Baruch, A. Continuous Inflammatory Stimulation Leads via Metabolic Plasticity to a Prometastatic Phenotype in Triple-Negative Breast Cancer Cells. Cells 2021, 10, 1356. [Google Scholar] [CrossRef]
- Morein, D.; Erlichman, N.; Ben-Baruch, A. Beyond Cell Motility: The Expanding Roles of Chemokines and Their Receptors in Malignancy. Front. Immunol. 2020, 11, 952. [Google Scholar] [CrossRef]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, C.; Sanmamed, M.F.; Rodriguez-Ruiz, M.E.; Teijeira, A.; Onate, C.; Gonzalez, A.; Ponz, M.; Schalper, K.A.; Perez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Todorovic-Rakovic, N.; Milovanovic, J. Interleukin-8 in breast cancer progression. J. Interferon Cytokine Res. 2013, 33, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein-Achiasaf, L.; Morein, D.; Ben-Yaakov, H.; Liubomirski, Y.; Meshel, T.; Elbaz, E.; Dorot, O.; Pichinuk, E.; Gershovits, M.; Weil, M.; et al. Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers 2021, 13, 1472. [Google Scholar] [CrossRef] [PubMed]
- Baram, T.; Rubinstein-Achiasaf, L.; Ben-Yaakov, H.; Ben-Baruch, A. Inflammation-Driven Breast Tumor Cell Plasticity: Stemness/EMT, Therapy Resistance and Dormancy. Front. Oncol. 2020, 10, 614468. [Google Scholar] [CrossRef]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Korner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef]
- Karin, N. Chemokines in the Landscape of Cancer Immunotherapy: How They and Their Receptors Can Be Used to Turn Cold Tumors into Hot Ones? Cancers 2021, 13, 6317. [Google Scholar] [CrossRef]
- Bewick, M.; Conlon, M.; Lee, H.; Parissenti, A.M.; Zhang, L.; Gluck, S.; LaFrenie, R.M. Evaluation of sICAM-1, sVCAM-1, and sE-Selectin levels in patients with metastatic breast cancer receiving high-dose chemotherapy. Stem Cells Dev. 2004, 13, 281–294. [Google Scholar] [CrossRef]
- Thielemann, A.; Baszczuk, A.; Kopczynski, Z.; Nowak, A.; Grodecka-Gazdecka, S. The clinical usefulness of assessing the concentration of cell adhesion molecules sVCAM-1 and sICAM-1 in the serum of women with primary breast cancer. Contemp. Oncol. 2014, 18, 252–259. [Google Scholar] [CrossRef]
- Touvier, M.; Fezeu, L.; Ahluwalia, N.; Julia, C.; Charnaux, N.; Sutton, A.; Mejean, C.; Latino-Martel, P.; Hercberg, S.; Galan, P.; et al. Association between prediagnostic biomarkers of inflammation and endothelial function and cancer risk: A nested case-control study. Am. J. Epidemiol. 2013, 177, 3–13. [Google Scholar] [CrossRef]
- Chiodoni, C.; Sangaletti, S.; Colombo, M.P. Matricellular proteins tune myeloid-derived suppressor cell recruitment and function in breast cancer. J. Leukoc Biol. 2017, 102, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Lew, A.M.; Chopin, M. The Pleiotropic Effects of the GM-CSF Rheostat on Myeloid Cell Differentiation and Function: More Than a Numbers Game. Front. Immunol. 2019, 10, 2679. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.A.; Mulligan, K.T.; Maxwell-Jones, H.; Adams, M.; Walker, R.A.; Jones, J.L. Breast cell invasive potential relates to the myoepithelial phenotype. Int. J. Cancer 2003, 106, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Furtado, C.M.; Marcondes, M.C.; Sola-Penna, M.; de Souza, M.L.; Zancan, P. Clotrimazole preferentially inhibits human breast cancer cell proliferation, viability and glycolysis. PLoS ONE 2012, 7, e30462. [Google Scholar] [CrossRef]
- Sihto, H.; Lundin, J.; Lundin, M.; Lehtimaki, T.; Ristimaki, A.; Holli, K.; Sailas, L.; Kataja, V.; Turpeenniemi-Hujanen, T.; Isola, J.; et al. Breast cancer biological subtypes and protein expression predict for the preferential distant metastasis sites: A nationwide cohort study. Breast Cancer Res. 2011, 13, R87. [Google Scholar] [CrossRef]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef]
- Arang, N.; Gutkind, J.S. G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett. 2020, 594, 4201–4232. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Chen, H.; Smaill, J.B.; Liu, T.; Ding, K.; Lu, X. Small-Molecule Inhibitors Directly Targeting KRAS as Anticancer Therapeutics. J. Med. Chem. 2020, 63, 14404–14424. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Gross, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L.; Wolf, M.J.; Roblek, M.; Lorentzen, A.; Heikenwalder, M. Inflammatory chemokines and metastasis-tracing the accessory. Oncogene 2013, 33, 3217–3224. [Google Scholar] [CrossRef] [PubMed]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef]
- Lacalle, R.A.; Blanco, R.; Carmona-Rodriguez, L.; Martin-Leal, A.; Mira, E.; Manes, S. Chemokine Receptor Signaling and the Hallmarks of Cancer. Int. Rev. Cell Mol. Biol. 2017, 331, 181–244. [Google Scholar] [CrossRef]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef]
- Weitzenfeld, P.; Ben-Baruch, A. The chemokine system, and its CCR5 and CXCR4 receptors, as potential targets for personalized therapy in cancer. Cancer Lett. 2014, 352, 36–53. [Google Scholar] [CrossRef]
- Yoshie, O. CCR4 as a Therapeutic Target for Cancer Immunotherapy. Cancers 2021, 13, 5542. [Google Scholar] [CrossRef]
- Ksiazkiewicz, M.; Gottfried, E.; Kreutz, M.; Mack, M.; Hofstaedter, F.; Kunz-Schughart, L.A. Importance of CCL2-CCR2A/2B signaling for monocyte migration into spheroids of breast cancer-derived fibroblasts. Immunobiology 2010, 215, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M. Chemokine receptor CCR5: Insights into structure, function, and regulation. Cell. Signal. 2004, 16, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.M.; Zou, Y.; Sharma, T.; Gatla, H.R.; Vancurova, I. Proteasome inhibition induces IKK-dependent interleukin-8 expression in triple negative breast cancer cells: Opportunity for combination therapy. PLoS ONE 2018, 13, e0201858. [Google Scholar] [CrossRef]
- Gu, W.; Dong, N.; Wang, P.; Shi, C.; Yang, J.; Wang, J. Tamoxifen resistance and metastasis of human breast cancer cells were mediated by the membrane-associated estrogen receptor ER-alpha36 signaling in vitro. Cell Biol. Toxicol. 2017, 33, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.B.; Jokar, I.; Zou, A.; Lambert, D.; Dendukuri, P.; Cheng, N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J. Biol. Chem. 2012, 287, 36593–36608. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Velazquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef]
- Manes, S.; Mira, E.; Colomer, R.; Montero, S.; Real, L.M.; Gomez-Mouton, C.; Jimenez-Baranda, S.; Garzon, A.; Lacalle, R.A.; Harshman, K.; et al. CCR5 expression influences the progression of human breast cancer in a p53-dependent manner. J. Exp. Med. 2003, 198, 1381–1389. [Google Scholar] [CrossRef]
- Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Ibanez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef]
- Dai, X.; Bu, X.; Gao, Y.; Guo, J.; Hu, J.; Jiang, C.; Zhang, Z.; Xu, K.; Duan, J.; He, S.; et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol. Cell 2021, 81, 2317–2331.e6. [Google Scholar] [CrossRef]
- Wen, M.; Cao, Y.; Wu, B.; Xiao, T.; Cao, R.; Wang, Q.; Liu, X.; Xue, H.; Yu, Y.; Lin, J.; et al. PD-L1 degradation is regulated by electrostatic membrane association of its cytoplasmic domain. Nat. Commun. 2021, 12, 5106. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, H.J.; Yoon, S.; Ryu, H.M.; Lee, E.; Jo, Y.; Seo, S.; Kim, D.; Lee, C.H.; Kim, W.; et al. Blockade of CCL2 expression overcomes intrinsic PD-1/PD-L1 inhibitor-resistance in transglutaminase 2-induced PD-L1 positive triple negative breast cancer. Am. J. Cancer Res. 2020, 10, 2878–2894. [Google Scholar]
- Cimas, F.J.; Manzano, A.; Baliu-Pique, M.; Garcia-Gil, E.; Perez-Segura, P.; Nagy, A.; Pandiella, A.; Gyorffy, B.; Ocana, A. Genomic Mapping Identifies Mutations in RYR2 and AHNAK as Associated with Favorable Outcome in Basal-Like Breast Tumors Expressing PD1/PD-L1. Cancers 2020, 12, 2243. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Keydar, I.; Chen, L.; Karby, S.; Weiss, F.R.; Delarea, J.; Radu, M.; Chaitcik, S.; Brenner, H.J. Establishment and characterization of a cell line of human breast carcinoma origin. Eur. J. Cancer 1979, 15, 659–670. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef]
- Brown, N.R.; Noble, M.E.; Endicott, J.A.; Johnson, L.N. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 438–443. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erlichman, N.; Baram, T.; Meshel, T.; Morein, D.; Da’adoosh, B.; Ben-Baruch, A. Tumor Cell-Autonomous Pro-Metastatic Activities of PD-L1 in Human Breast Cancer Are Mediated by PD-L1-S283 and Chemokine Axes. Cancers 2022, 14, 1042. https://doi.org/10.3390/cancers14041042
Erlichman N, Baram T, Meshel T, Morein D, Da’adoosh B, Ben-Baruch A. Tumor Cell-Autonomous Pro-Metastatic Activities of PD-L1 in Human Breast Cancer Are Mediated by PD-L1-S283 and Chemokine Axes. Cancers. 2022; 14(4):1042. https://doi.org/10.3390/cancers14041042
Chicago/Turabian StyleErlichman, Nofar, Tamir Baram, Tsipi Meshel, Dina Morein, Benny Da’adoosh, and Adit Ben-Baruch. 2022. "Tumor Cell-Autonomous Pro-Metastatic Activities of PD-L1 in Human Breast Cancer Are Mediated by PD-L1-S283 and Chemokine Axes" Cancers 14, no. 4: 1042. https://doi.org/10.3390/cancers14041042
APA StyleErlichman, N., Baram, T., Meshel, T., Morein, D., Da’adoosh, B., & Ben-Baruch, A. (2022). Tumor Cell-Autonomous Pro-Metastatic Activities of PD-L1 in Human Breast Cancer Are Mediated by PD-L1-S283 and Chemokine Axes. Cancers, 14(4), 1042. https://doi.org/10.3390/cancers14041042