
A Comprehensive Review of the Current and Future Role of the Microbiome in Pancreatic Ductal Adenocarcinoma

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
Definitions
2. Discussion
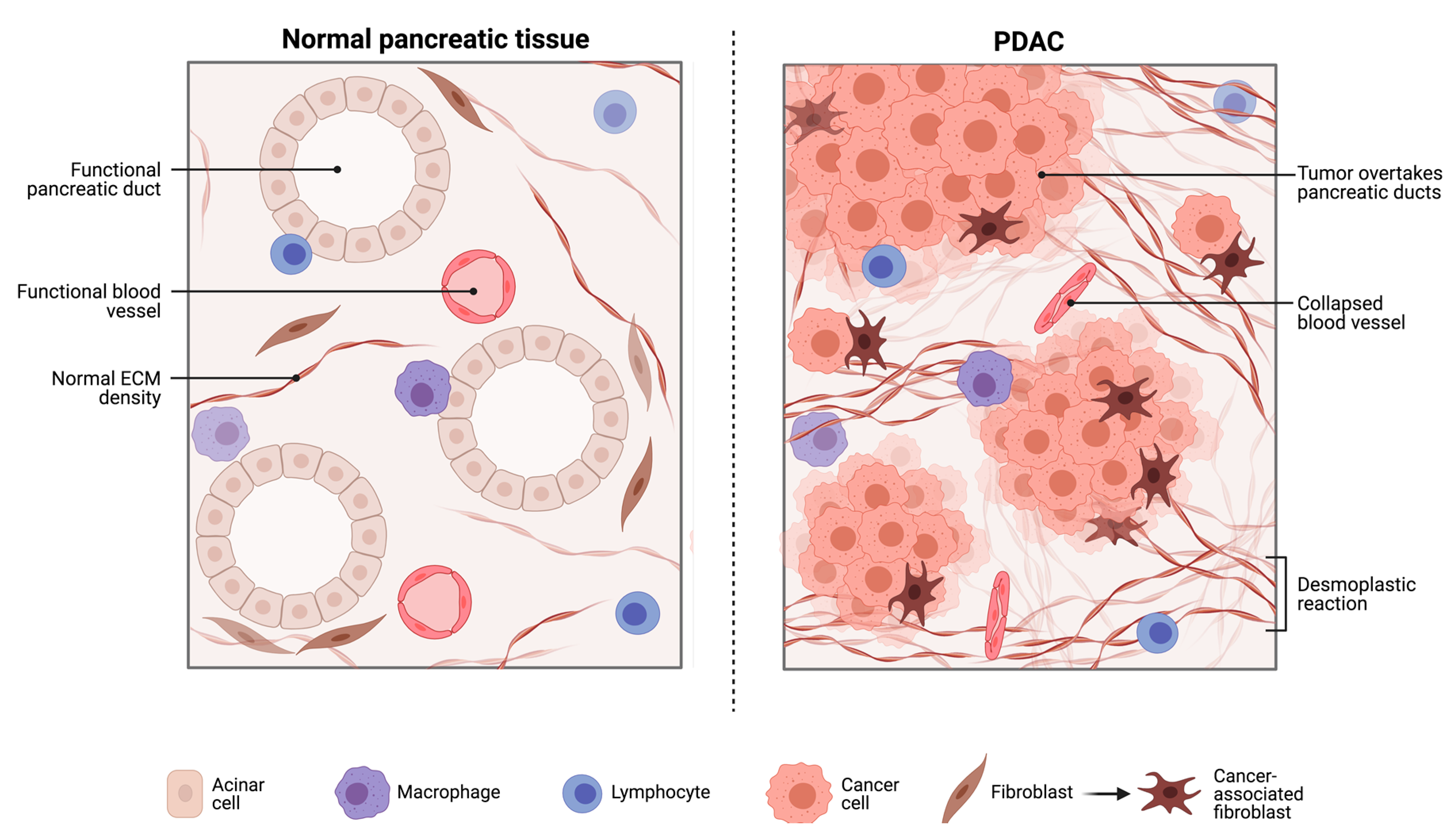
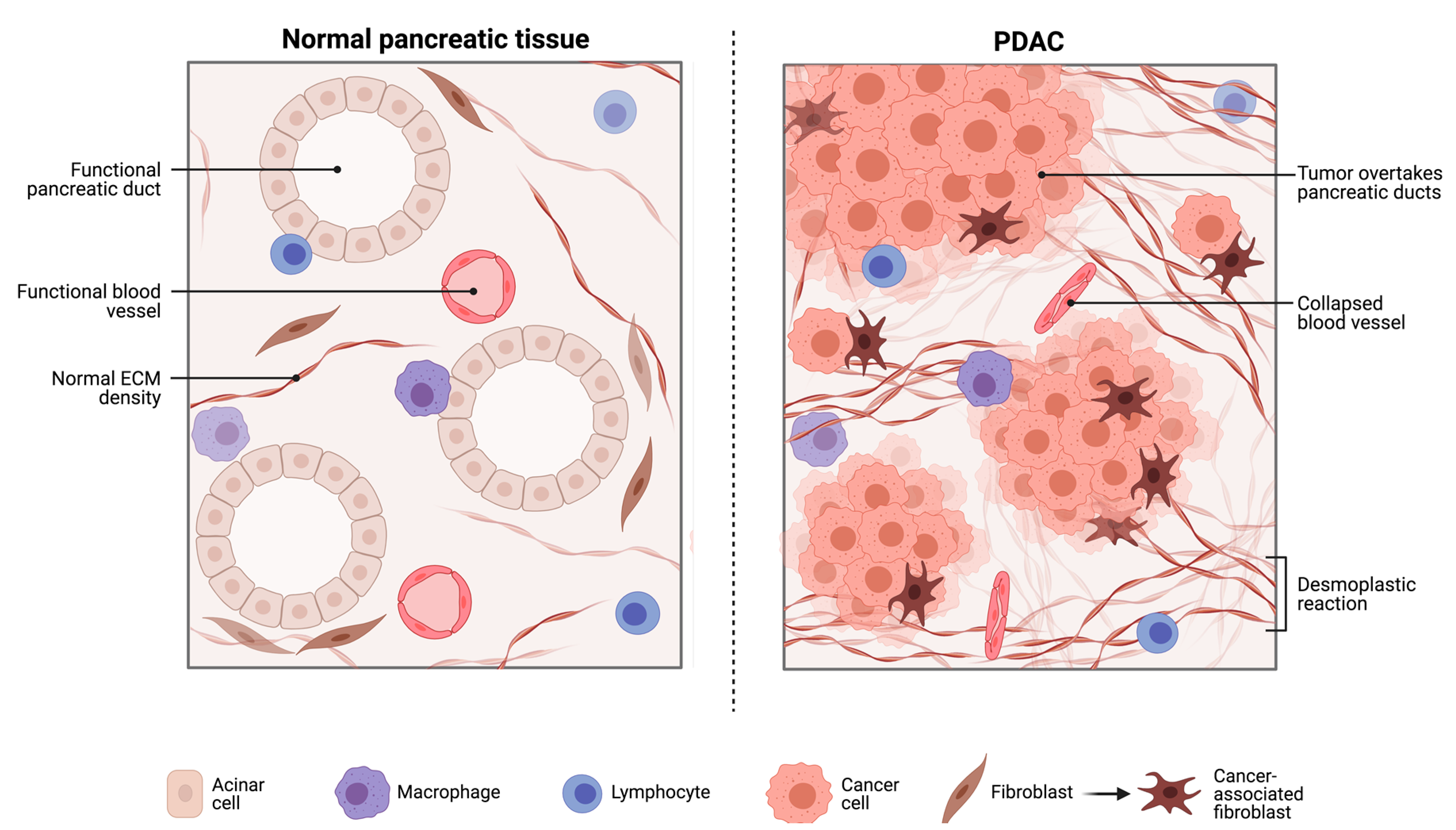
2.1. The Pancreatic Intra-Tumoural Microbiome and Its’ Relationship with Metabolism, Immune Response and Survival Outcomes
2.1.1. The Bacterial Microbiome in PDAC
2.1.2. The Fungal Community in PDAC
2.1.3. The Effect of the PDAC Microbiome on Metabolism
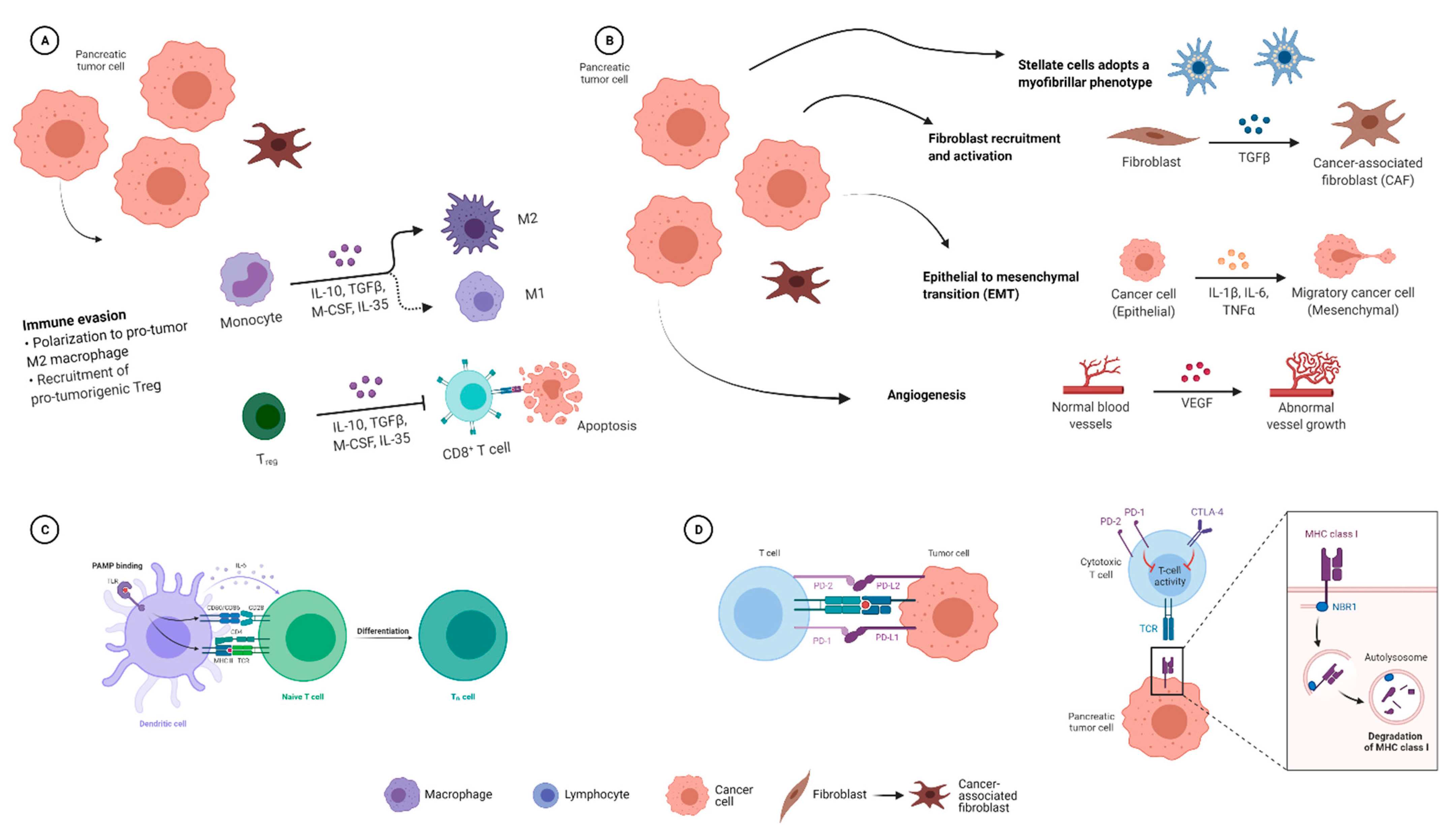
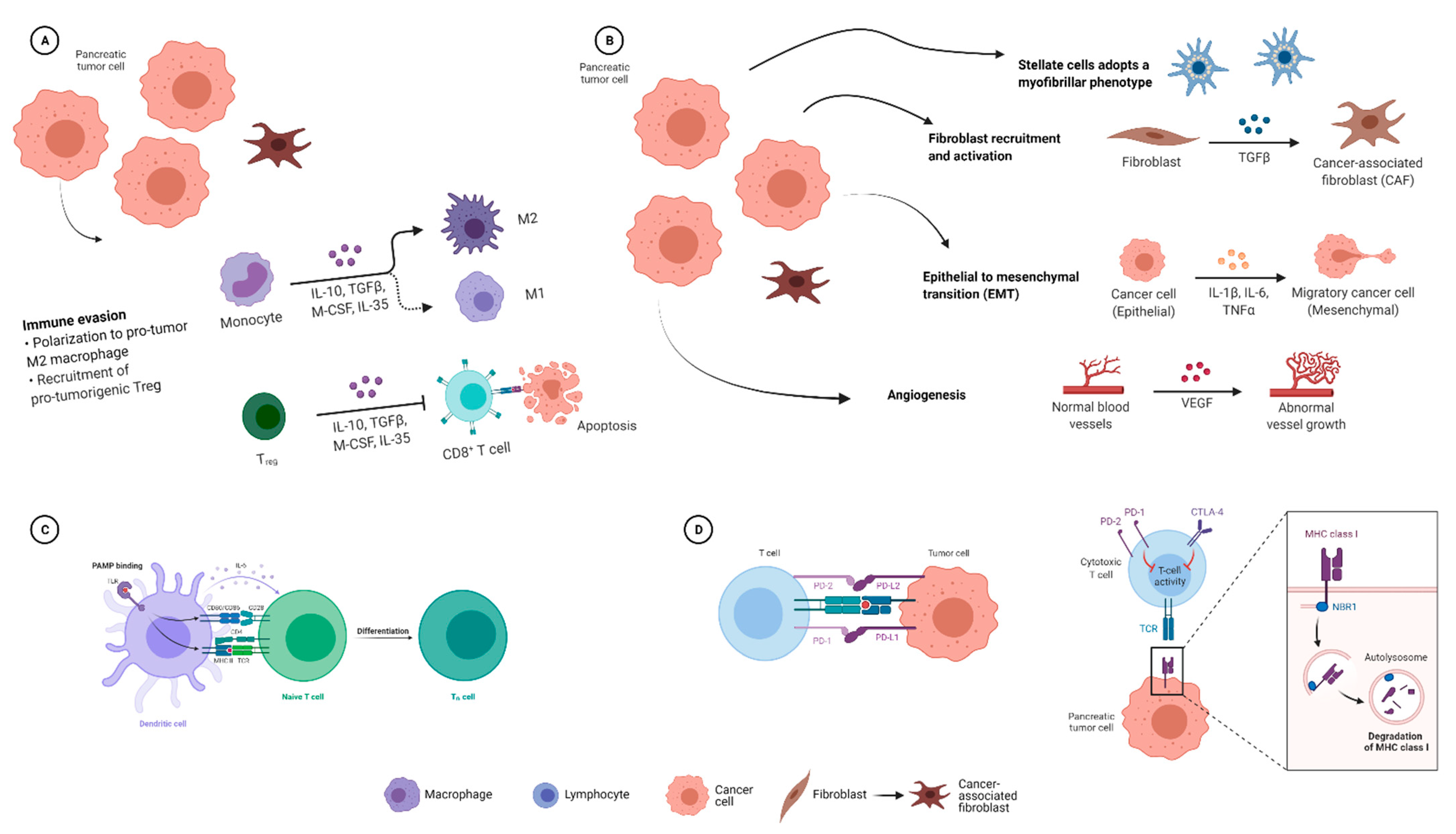
2.1.4. Effects of the Microbiome on the Immune Response in PDAC
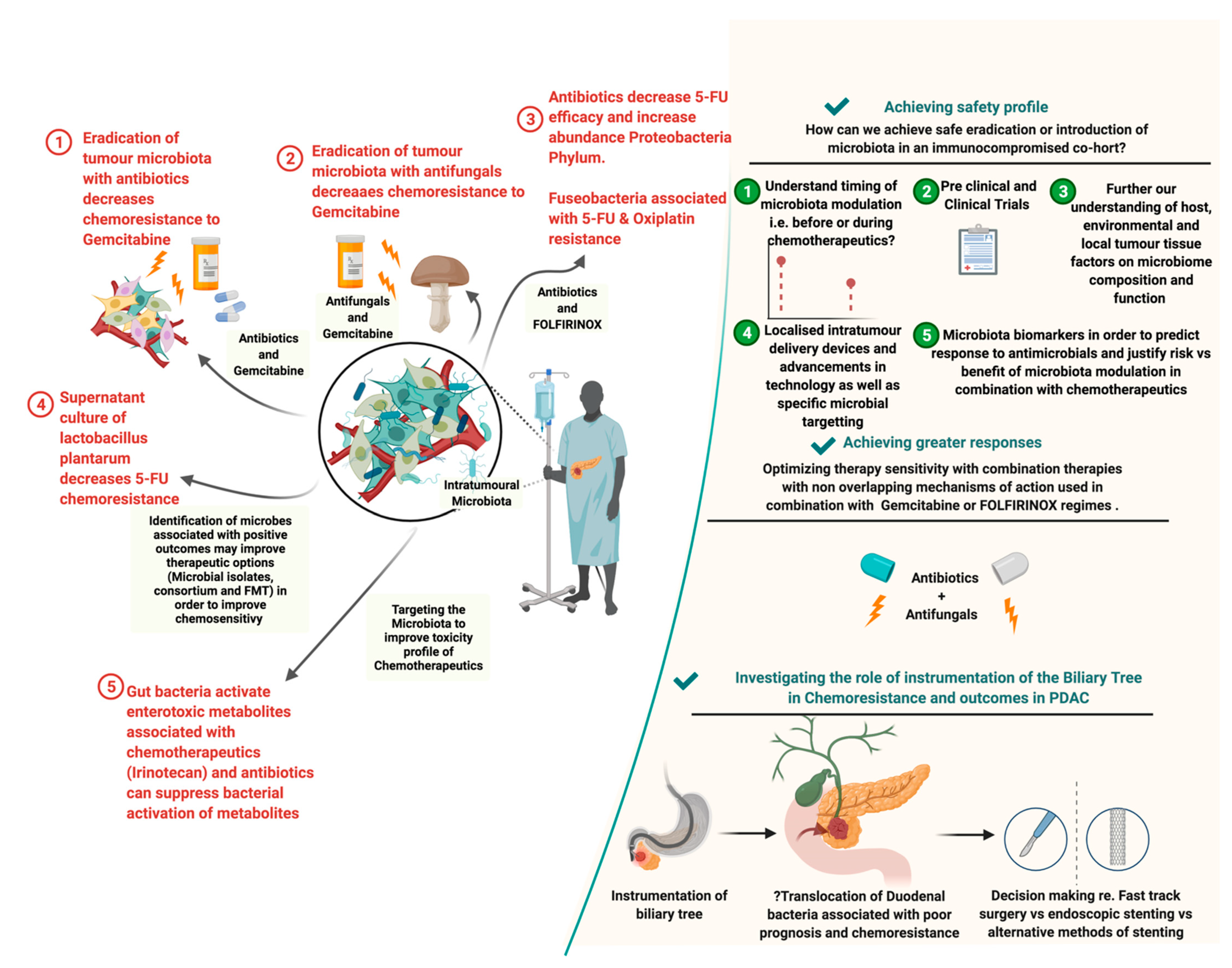
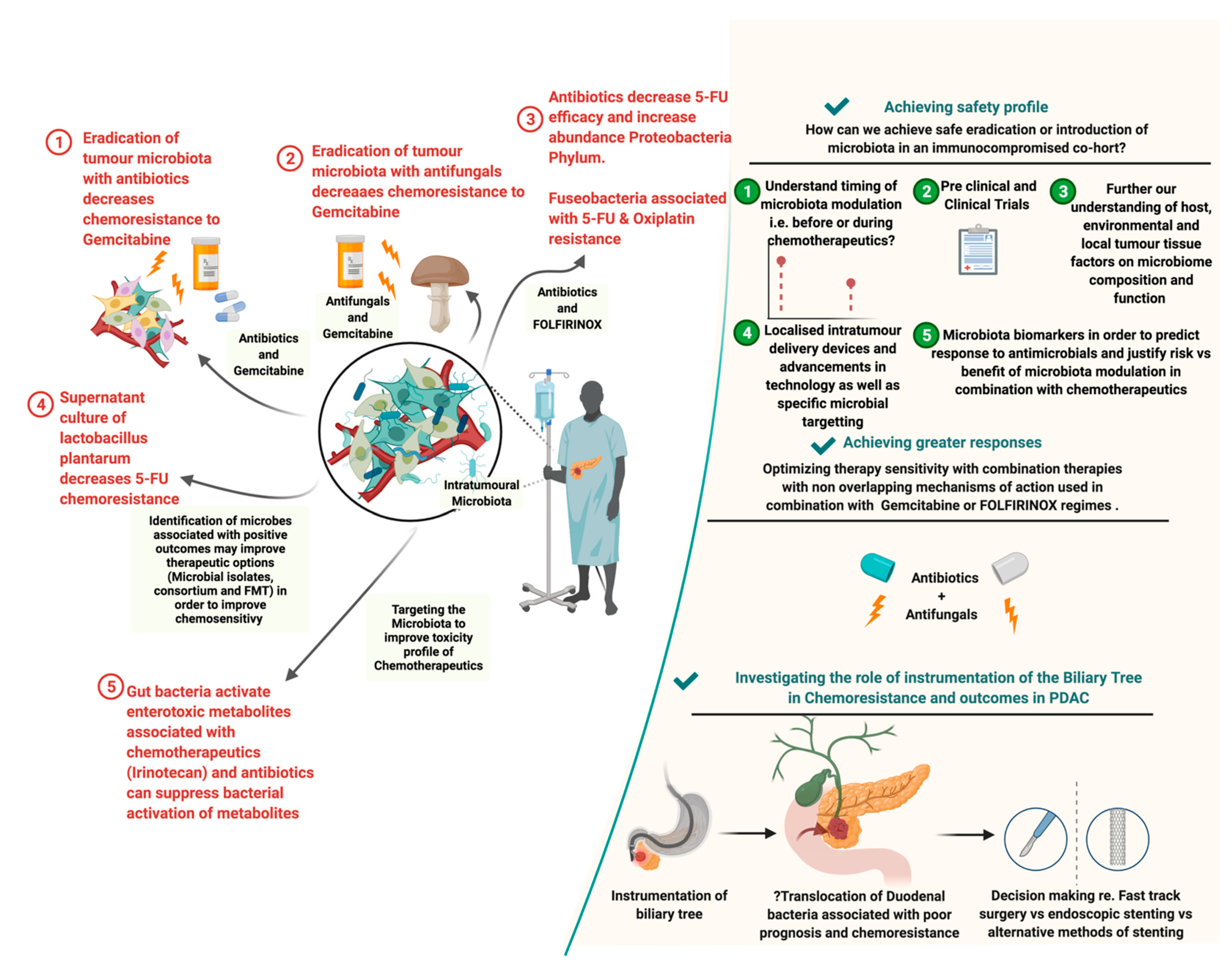
2.2. Influence of the PDAC Microbiome on Chemotherapy
2.3. Faecal Microbiota Transplant in PDAC
2.3.1. Evidence for Faecal Microbial Transplant in PDAC
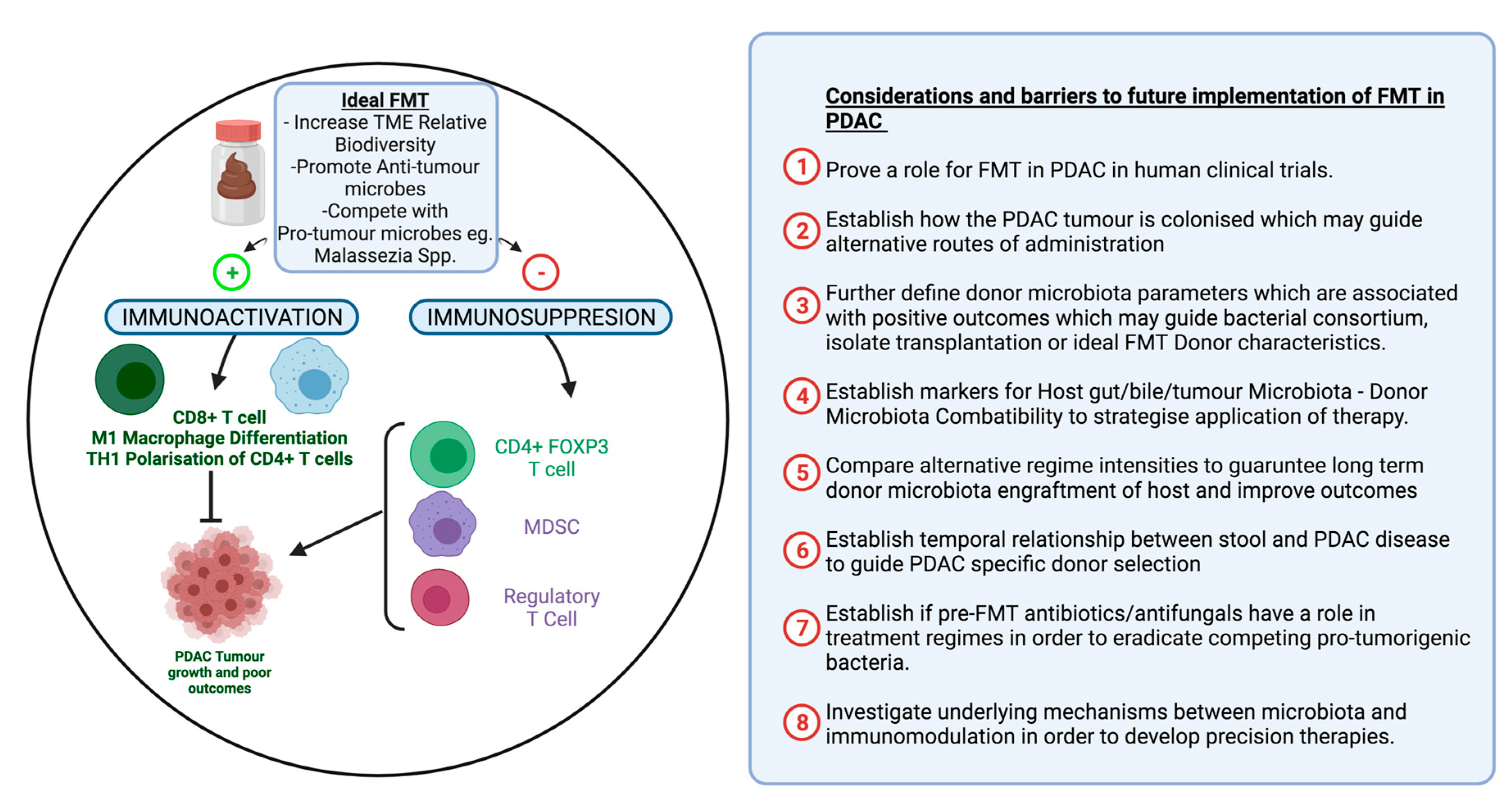
2.3.2. Future Role of FMT in PDAC
Optimizing FMT in Order to Improve Outcomes
Routes of FMT Administration
FMT Donor Selection
Role of Antibiotics in FMT
Future of FMT in PDAC Conclusion
2.4. Role of Antimicrobials in PDAC
2.5. Modulation in the Perioperative Period
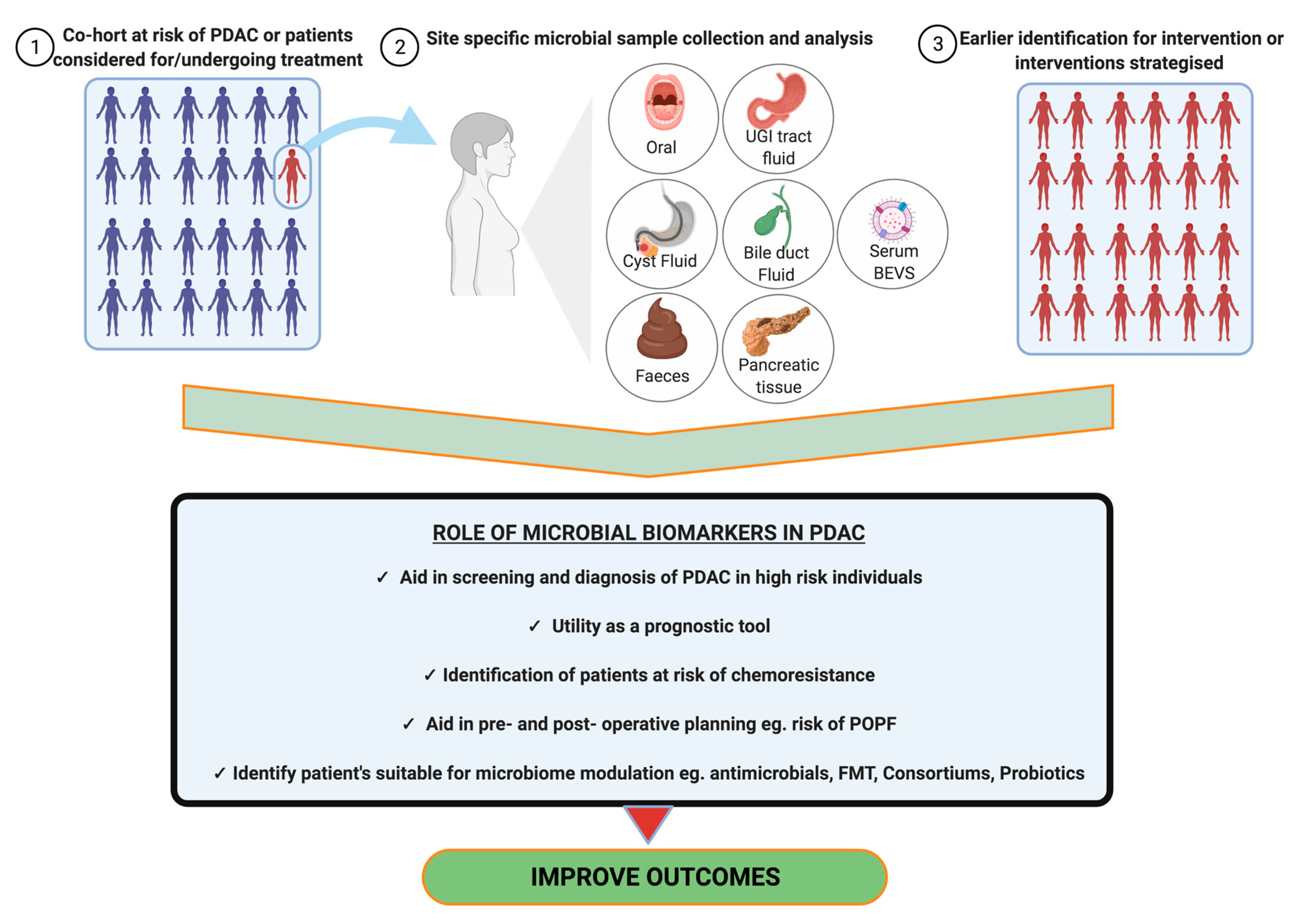
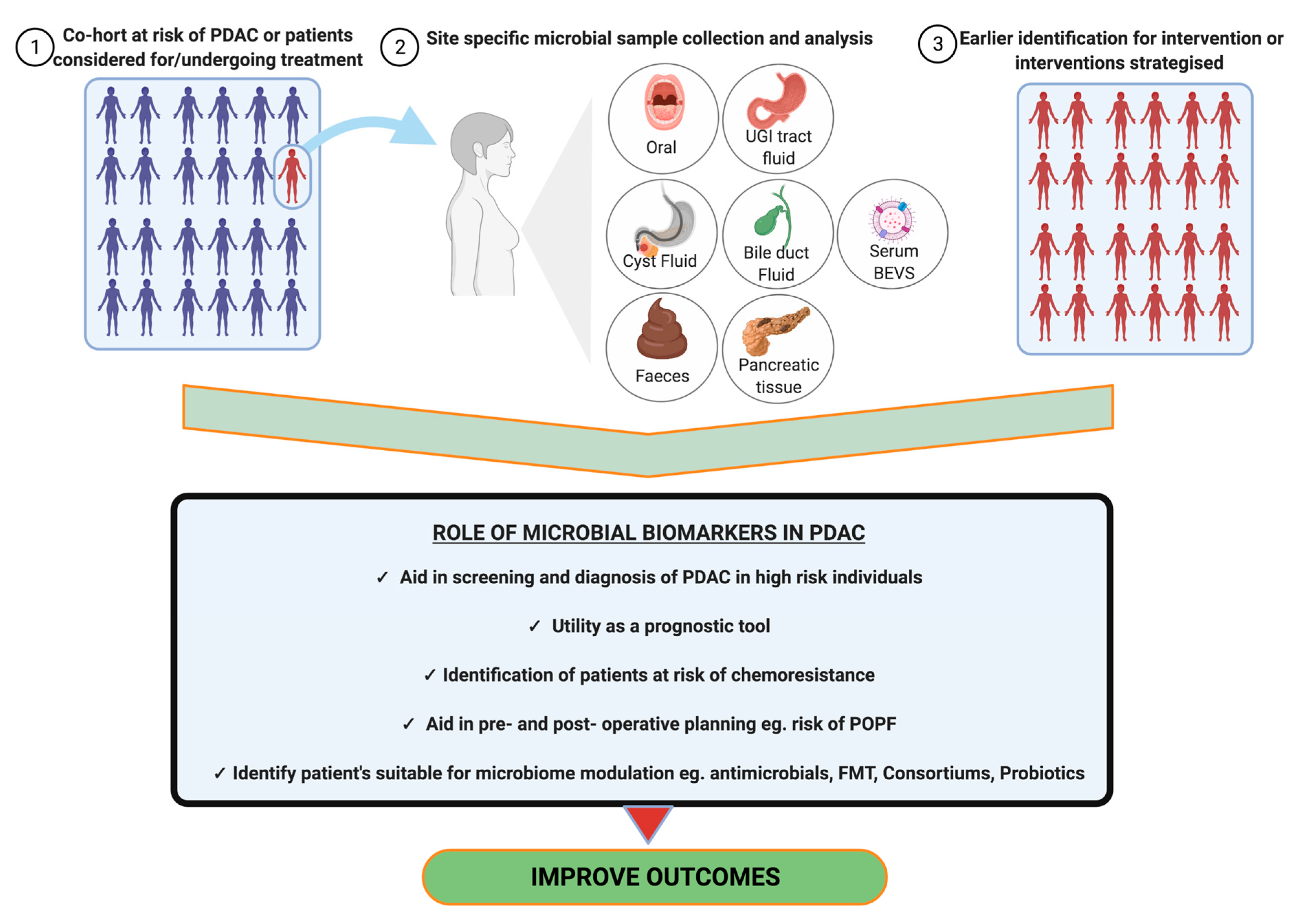
2.6. Biomarkers and Diagnostics
2.6.1. Oral Microbiota
2.6.2. Duodenal Microbiota
2.6.3. Bile Microbiota
2.6.4. Stool Microbiota
2.6.5. Cyst Fluid Biomarkers in IPMN
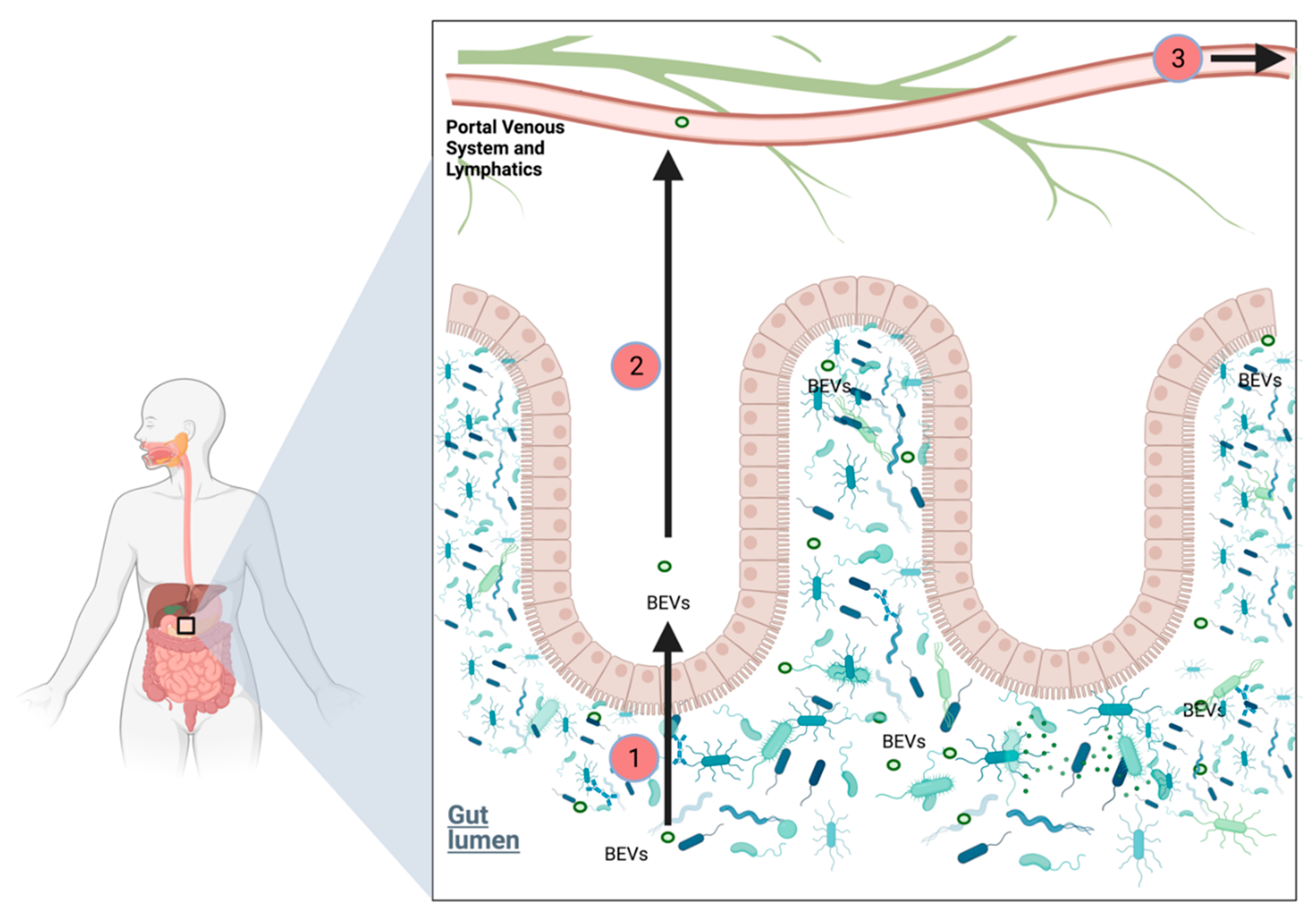
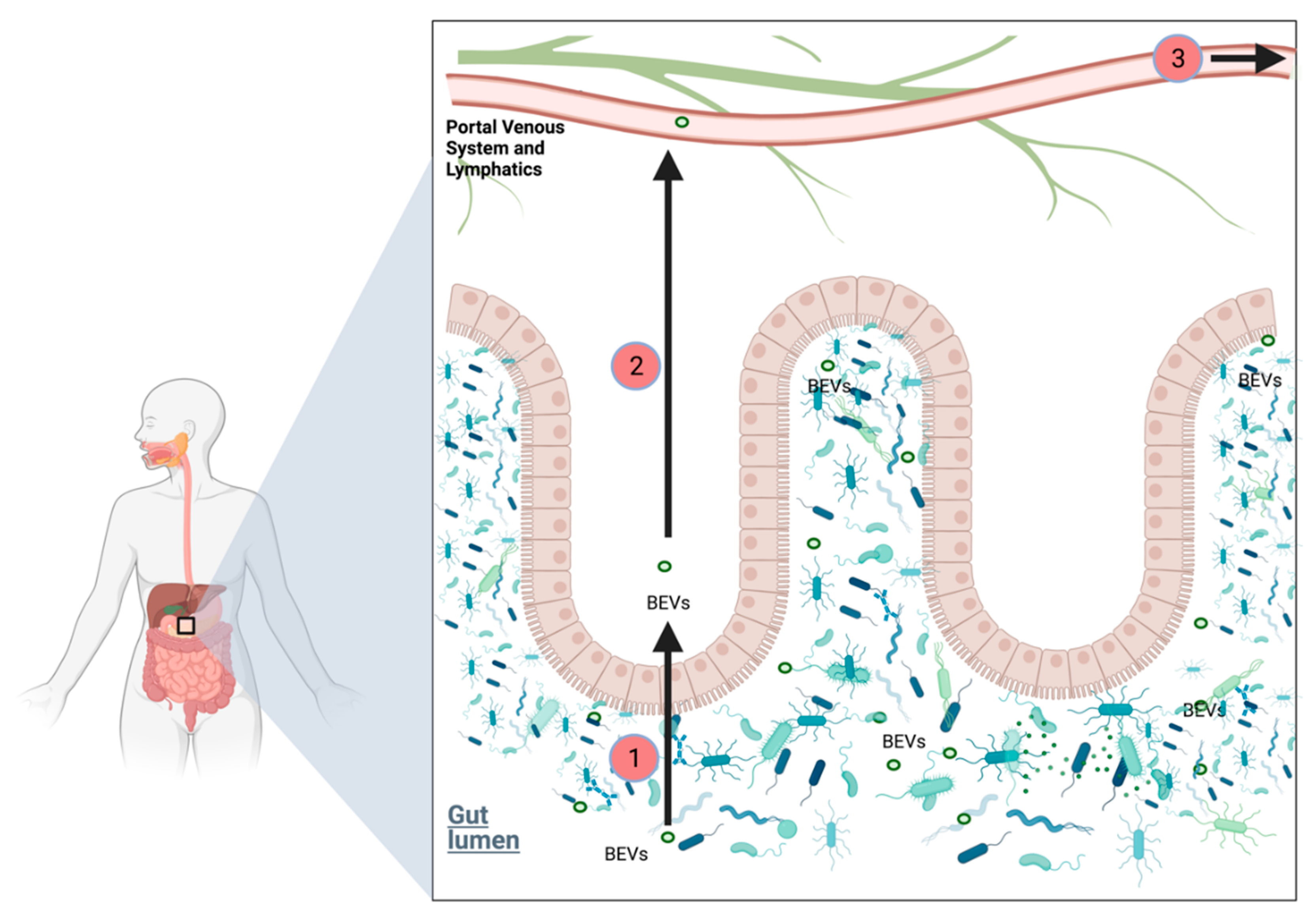
2.6.6. Role of Bacterial Extracellular Vesicles in PDAC
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Pancreatic Cancer Survival Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer/survival#heading-Two (accessed on 23 June 2021).
- Dal Molin, M.; Zhang, M.; de Wilde, R.F.; Ottenhof, N.A.; Rezaee, N.; Wolfgang, C.L.; Blackford, A.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; et al. Very Long-term Survival Following Resection for Pancreatic Cancer Is Not Explained by Commonly Mutated Genes: Results of Whole-Exome Sequencing Analysis. Clin. Cancer Res. 2015, 21, 1944–1950. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bleich, R.M.; Arthur, J.C. Microbiome and the Hallmarks of Cancer. In Inflammation, Infection, and Microbiome in Cancers: Evidence, Mechanisms, and Implications; Sun, J., Society, T.A.P., Eds.; Physiology in Health and Disease; Springer Nature AG: Cham, Switzerland, 2021; Volume 11, pp. 1–27. [Google Scholar]
- Fulbright, L.E.; Ellermann, M.; Arthur, J.C. The microbiome and the hallmarks of cancer. PLoS Pathog. 2017, 13, e1006480. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [Green Version]
- Whisner, C.M.; Athena Aktipis, C. The Role of the Microbiome in Cancer Initiation and Progression: How Microbes and Cancer Cells Utilize Excess Energy and Promote One Another’s Growth. Curr. Nutr. Rep. 2019, 8, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Thomas, R.M.; Jobin, C. Microbiota in pancreatic health and disease: The next frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 53–64. [Google Scholar] [CrossRef] [PubMed]
- McAllister, F.; Khan, M.A.W.; Helmink, B.; Wargo, J.A. The Tumor Microbiome in Pancreatic Cancer: Bacteria and Beyond. Cancer Cell 2019, 36, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Zhu, M.; Kashyap, P.C.; Chia, N.; Tran, N.H.; McWilliams, R.R.; Bekaii-Saab, T.S.; Ma, W.W. The role of microbiome in pancreatic cancer. Cancer Metastasis Rev. 2021, 40, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 2019, 178, 795–806.e12. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Whipps, J.M.; Lewis, K.; Cooke, R.C. Fungi in Biological Control Systems; Burge, M.N., Ed.; Manchester University Press: Manchester, UK, 1988; p. 176. [Google Scholar]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Chronopoulos, A.; Kalluri, R. Emerging role of bacterial extracellular vesicles in cancer. Oncogene 2020, 39, 6951–6960. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Del Castillo, E.; Meier, R.; Chung, M.; Koestler, D.C.; Chen, T.; Paster, B.J.; Charpentier, K.P.; Kelsey, K.T.; Izard, J.; Michaud, D.S. The Microbiomes of Pancreatic and Duodenum Tissue Overlap and Are Highly Subject Specific but Differ between Pancreatic Cancer and Noncancer Subjects. Cancer Epidemiol. Biomark. Prev. 2019, 28, 370–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, M.B.; Aveson, V.; Firek, B.; Yeh, A.; Brooks, B.; Brower-Sinning, R.; Steve, J.; Banfield, J.F.; Zureikat, A.; Hogg, M.; et al. Disturbances of the Perioperative Microbiome Across Multiple Body Sites in Patients Undergoing Pancreaticoduodenectomy. Pancreas 2017, 46, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Q.X.; Huang, C.L.; Luo, S.Z.; Zhang, X.M.; Zeng, Y.; Lu, Y.Y. Characterization of the duodenal bacterial microbiota in patients with pancreatic head cancer vs. healthy controls. Pancreatology 2018, 18, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Jiang, J.; Xie, H.; Li, A.; Lu, H.; Xu, S.; Zhou, L.; Zhang, H.; Cui, G.; Chen, X.; et al. Gut microbial profile analysis by MiSeq sequencing of pancreatic carcinoma patients in China. Oncotarget 2017, 8, 95176–95191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleeson, F.C.; Jeraldo, P.; Levy, M.J.; Murphy, S.J.; Mendes-Soares, H.; Karagouga, G.; Mccune, A.F.; Garcia Garcia Deparedes, A.; Kipp, B.R.; Song, S.D.; et al. Composition, diversity and potential utility of intervention-naïve pancreatic cancer intratumoral microbiome signature profiling via endoscopic ultrasound. Gut 2022, 71, 441–443. [Google Scholar] [CrossRef]
- Chung, M.; Zhao, N.; Meier, R.; Koestler, D.C.; Wu, G.; de Castillo, E.; Paster, B.J.; Charpentier, K.; Izard, J.; Kelsey, K.T.; et al. Comparisons of oral, intestinal, and pancreatic bacterial microbiomes in patients with pancreatic cancer and other gastrointestinal diseases. J. Oral Microbiol. 2021, 13, 1887680. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Chakladar, J.; Kuo, S.Z.; Castaneda, G.; Li, W.T.; Gnanasekar, A.; Yu, M.A.; Chang, E.Y.; Wang, X.Q.; Ongkeko, W.M. The Pancreatic Microbiome is Associated with Carcinogenesis and Worse Prognosis in Males and Smokers. Cancers 2020, 12, 2672. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, Y.; Guo, S.; Mei, Z.; Liao, H.; Dong, H.; Wu, K.; Ye, H.; Zhang, Y.; Zhu, Y.; et al. Tumor microbiome contributes to an aggressive phenotype in the basal-like subtype of pancreatic cancer. Commun. Biol. 2021, 4, 1019. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, L.; Gupta, S.; Daily, J.; Kelly, L. Human microbiome signatures of differential colorectal cancer drug metabolism. NPJ Biofilm. Microbiomes 2017, 3, 27. [Google Scholar] [CrossRef]
- Langheinrich, M.; Wirtz, S.; Kneis, B.; Gittler, M.M.; Tyc, O.; Schierwagen, R.; Brunner, M.; Krautz, C.; Weber, G.F.; Pilarsky, C.; et al. Microbiome Patterns in Matched Bile, Duodenal, Pancreatic Tumor Tissue, Drainage, and Stool Samples: Association with Preoperative Stenting and Postoperative Pancreatic Fistula Development. J. Clin. Med. 2020, 9, 2785. [Google Scholar] [CrossRef]
- Picardo, S.L.; Coburn, B.; Hansen, A.R. The microbiome and cancer for clinicians. Crit. Rev. Oncol. Hematol. 2019, 141, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Gharaibeh, R.Z.; Gauthier, J.; Beveridge, M.; Pope, J.L.; Guijarro, M.V.; Yu, Q.; He, Z.; Ohland, C.; Newsome, R.; et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 2018, 39, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.D. Rarefaction, Alpha Diversity, and Statistics. Front. Microbiol. 2019, 10, 2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraoka, N.; Ino, Y.; Yamazaki-Itoh, R.; Kanai, Y.; Kosuge, T.; Shimada, K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br. J. Cancer 2015, 112, 1782–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Ariston Gabriel, A.N.; Jiao, Q.; Yvette, U.; Yang, X.; Al-Ameri, S.A.; Du, L.; Wang, Y.-s.; Wang, C. Differences between KC and KPC pancreatic ductal adenocarcinoma mice models, in terms of their modeling biology and their clinical relevance. Pancreatology 2020, 20, 79–88. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F., III; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Kinkead, H.L.; Hopkins, A.; Lutz, E.; Wu, A.A.; Yarchoan, M.; Cruz, K.; Woolman, S.; Vithayathil, T.; Glickman, L.H.; Ndubaku, C.O.; et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018, 3, e122857. [Google Scholar] [CrossRef] [Green Version]
- Li, K.-Y.; Yuan, J.-L.; Trafton, D.; Wang, J.-X.; Niu, N.; Yuan, C.-H.; Liu, X.-B.; Zheng, L. Pancreatic ductal adenocarcinoma immune microenvironment and immunotherapy prospects. Chronic Dis. Transl. Med. 2020, 6, 6–17. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Kohi, S.; Macgregor-Das, A.; Dbouk, M.; Yoshida, T.; Chuidian, M.; Abe, T.; Borges, M.; Lennon, A.M.; Shin, E.J.; Canto, M.I.; et al. Alterations in the Duodenal Fluid Microbiome of Patients With Pancreatic Cancer. Clin. Gastroenterol. Hepatol. 2020, 20, e196–e227. [Google Scholar] [CrossRef]
- van Asbeck, E.C.; Hoepelman, A.I.; Scharringa, J.; Herpers, B.L.; Verhoef, J. Mannose binding lectin plays a crucial role in innate immunity against yeast by enhanced complement activation and enhanced uptake of polymorphonuclear cells. BMC Microbiol. 2008, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.S.; Vasquez, H.G.; Rupaimoole, R.; Pradeep, S.; Wu, S.; Zand, B.; Han, H.D.; Rodriguez-Aguayo, C.; Bottsford-Miller, J.; Huang, J.; et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014, 6, 1085–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, Q.H.; Chang, M.W.; Chai, L.Y. The Fungal Mycobiome and Its Interaction with Gut Bacteria in the Host. Int. J. Mol. Sci. 2017, 18, 330. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Cani, P.D. Gut cell metabolism shapes the microbiome. Science 2017, 357, 548–549. [Google Scholar] [CrossRef]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Dzutsev, A.; Badger, J.H.; Perez-Chanona, E.; Roy, S.; Salcedo, R.; Smith, C.K.; Trinchieri, G. Microbes and Cancer. Annu. Rev. Immunol. 2017, 35, 199–228. [Google Scholar] [CrossRef]
- McQuade, J.L.; Daniel, C.R.; Helmink, B.A.; Wargo, J.A. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019, 20, e77–e91. [Google Scholar] [CrossRef]
- Xu, J.; Peng, J.-J.; Yang, W.; Fu, K.; Zhang, Y. Vaginal microbiomes and ovarian cancer: A review. Am. J. Cancer Res. 2020, 10, 743–756. [Google Scholar] [PubMed]
- Zitvogel, L.; Daillère, R.; Roberti, M.P.; Routy, B.; Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 2017, 15, 465–478. [Google Scholar] [CrossRef]
- Swaney, M.H.; Kalan, L.R.; Richardson, A.R. Living in Your Skin: Microbes, Molecules, and Mechanisms. Infect. Immun. 2021, 89, e00695-20. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.R.; Gabaldón, T. The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems. Microorganisms 2020, 8, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, C.M.; Moran, G.P. The microbiome and oral cancer: More questions than answers. Oral Oncol. 2019, 89, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef]
- Ansaldo, E.; Farley, T.K.; Belkaid, Y. Control of Immunity by the Microbiota. Annu. Rev. Immunol. 2021, 39, 449–479. [Google Scholar] [CrossRef]
- Ansaldo, E.; Belkaid, Y. How microbiota improve immunotherapy. Science 2021, 373, 966–967. [Google Scholar] [CrossRef]
- Sears, C.L.; Pardoll, D.M. Perspective: Alpha-bugs, their microbial partners, and the link to colon cancer. J. Infect. Dis. 2011, 203, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver–passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol. Asp. Med. 2019, 69, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Torphy, R.J.; Schulick, R.D.; Zhu, Y. Understanding the immune landscape and tumor microenvironment of pancreatic cancer to improve immunotherapy. Mol. Carcinog. 2020, 59, 775–782. [Google Scholar] [CrossRef]
- Tan, E.; El-Rayes, B. Pancreatic Cancer and Immunotherapy: Resistance Mechanisms and Proposed Solutions. J. Gastrointest. Cancer 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 2017, 18, 851–860. [Google Scholar] [CrossRef]
- Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer. Oncology 2002, 16, 217–226, 229. [Google Scholar]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, H.; Bording-Jorgensen, M.; Dijk, S.; Wine, E. The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It. Cancers 2018, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Redelman-Sidi, G.; Binyamin, A.; Gaeta, I.; Palm, W.; Thompson, C.B.; Romesser, P.B.; Lowe, S.W.; Bagul, M.; Doench, J.G.; Root, D.E.; et al. The Canonical Wnt Pathway Drives Macropinocytosis in Cancer. Cancer Res. 2018, 78, 4658–4670. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, C.W.; Kang, Y.B.; Pichika, M.R. Should a Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: Its expression and effects in the ten most common cancers. OncoTargets 2013, 6, 1573–1587. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Daniluk, J.; Liu, Y.; Chu, J.; Li, Z.; Ji, B.; Logsdon, C.D. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 2014, 33, 532–535. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.A.; Shaw, H.M.; Bataille, V.; Nathan, P.; Spector, T.D. Role of the gut microbiome for cancer patients receiving immunotherapy: Dietary and treatment implications. Eur. J. Cancer 2020, 138, 149–155. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Das, S.; Handler, J.S.; Hajdu, C.H.; Coffre, M.; Koralov, S.B.; Bar-Sagi, D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016, 6, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307.e9. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFNγ and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinwand, J.; Miller, G. Regulation and modulation of antitumor immunity in pancreatic cancer. Nat. Immunol. 2020, 21, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Vande Voorde, J.; Sabuncuoğlu, S.; Noppen, S.; Hofer, A.; Ranjbarian, F.; Fieuws, S.; Balzarini, J.; Liekens, S. Nucleoside-catabolizing enzymes in mycoplasma-infected tumor cell cultures compromise the cytostatic activity of the anticancer drug gemcitabine. J. Biol. Chem. 2014, 289, 13054–13065. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- García-González, A.P.; Ritter, A.D.; Shrestha, S.; Andersen, E.C.; Yilmaz, L.S.; Walhout, A.J.M. Bacterial Metabolism Affects the C. elegans Response to Cancer Chemotherapeutics. Cell 2017, 169, 431–441.e8. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Zhang, S.; Li, H.; Yang, F.; Mushtaq, N.; Ullah, S.; Shi, Y.; An, C.; Xu, J. The influence of gut microbiota dysbiosis to the efficacy of 5-Fluorouracil treatment on colorectal cancer. Biomed. Pharm. 2018, 108, 184–193. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Y.; Weng, W.; Guo, B.; Cai, G.; Ma, Y.; Cai, S. Fusobacterium nucleatum promotes chemoresistance to 5-fluorouracil by upregulation of BIRC3 expression in colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 14. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Ha, E.M. Combination Therapy of Lactobacillus plantarum Supernatant and 5-Fluouracil Increases Chemosensitivity in Colorectal Cancer Cells. J. Microbiol. Biotechnol. 2016, 26, 1490–1503. [Google Scholar] [CrossRef]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Elting, L.S.; Cooksley, C.; Chambers, M.; Cantor, S.B.; Manzullo, E.; Rubenstein, E.B. The burdens of cancer therapy. Clinical and economic outcomes of chemotherapy-induced mucositis. Cancer 2003, 98, 1531–1539. [Google Scholar] [CrossRef]
- Kodawara, T.; Higashi, T.; Negoro, Y.; Kamitani, Y.; Igarashi, T.; Watanabe, K.; Tsukamoto, H.; Yano, R.; Masada, M.; Iwasaki, H.; et al. The Inhibitory Effect of Ciprofloxacin on the β-Glucuronidase-mediated Deconjugation of the Irinotecan Metabolite SN-38-G. Basic Clin. Pharm. Toxicol. 2016, 118, 333–337. [Google Scholar] [CrossRef]
- Choy, A.T.F.; Carnevale, I.; Coppola, S.; Meijer, L.L.; Kazemier, G.; Zaura, E.; Deng, D.; Giovannetti, E. The microbiome of pancreatic cancer: From molecular diagnostics to new therapeutic approaches to overcome chemoresistance caused by metabolic inactivation of gemcitabine. Expert Rev. Mol. Diagn. 2018, 18, 1005–1009. [Google Scholar] [CrossRef]
- Org, E.; Mehrabian, M.; Parks, B.W.; Shipkova, P.; Liu, X.; Drake, T.A.; Lusis, A.J. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; de Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.; Fitzgerald, G.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4586–4591. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Wilson, B.C.; Vatanen, T.; Cutfield, W.S.; O’Sullivan, J.M. The Super-Donor Phenomenon in Fecal Microbiota Transplantation. Front. Cell Infect. Microbiol. 2019, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.S.; Zhu, A.; Benes, V.; Costea, P.I.; Hercog, R.; Hildebrand, F.; Huerta-Cepas, J.; Nieuwdorp, M.; Salojärvi, J.; Voigt, A.Y.; et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 2016, 352, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramsothy, S.; Paramsothy, R.; Rubin, D.T.; Kamm, M.A.; Kaakoush, N.O.; Mitchell, H.M.; Castaño-Rodríguez, N. Faecal Microbiota Transplantation for Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. J. Crohns Colitis 2017, 11, 1180–1199. [Google Scholar] [CrossRef] [Green Version]
- Kootte, R.S.; Levin, E.; Salojärvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619.e16. [Google Scholar] [CrossRef] [Green Version]
- Staley, C.; Vaughn, B.P.; Graiziger, C.T.; Singroy, S.; Hamilton, M.J.; Yao, D.; Chen, C.; Khoruts, A.; Sadowsky, M.J. Community dynamics drive punctuated engraftment of the fecal microbiome following transplantation using freeze-dried, encapsulated fecal microbiota. Gut Microbes 2017, 8, 276–288. [Google Scholar] [CrossRef] [Green Version]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Baunwall, S.M.D.; Lee, M.M.; Eriksen, M.K.; Mullish, B.H.; Marchesi, J.R.; Dahlerup, J.F.; Hvas, C.L. Faecal microbiota transplantation for recurrent Clostridioides difficile infection: An updated systematic review and meta-analysis. EClinicalMedicine 2020, 29–30, 100642. [Google Scholar] [CrossRef]
- Gaiser, R.A.; Halimi, A.; Alkharaan, H.; Lu, L.; Davanian, H.; Healy, K.; Hugerth, L.W.; Ateeb, Z.; Valente, R.; Fernández Moro, C.; et al. Enrichment of oral microbiota in early cystic precursors to invasive pancreatic cancer. Gut 2019, 68, 2186–2194. [Google Scholar] [CrossRef] [Green Version]
- DeFilipp, Z.; Bloom, P.P.; Torres Soto, M.; Mansour, M.K.; Sater, M.R.A.; Huntley, M.H.; Turbett, S.; Chung, R.T.; Chen, Y.B.; Hohmann, E.L. Drug-Resistant, E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N. Engl. J. Med. 2019, 381, 2043–2050. [Google Scholar] [CrossRef]
- Guo, M.; Tao, W.; Flavell, R.A.; Zhu, S. Potential intestinal infection and faecal–oral transmission of SARS-CoV-2. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Keshteli, A.H.; Millan, B.; Madsen, K.L. Pretreatment with antibiotics may enhance the efficacy of fecal microbiota transplantation in ulcerative colitis: A meta-analysis. Mucosal. Immunol. 2017, 10, 565–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugenholtz, F.; de Vos, W.M. Mouse models for human intestinal microbiota research: A critical evaluation. Cell Mol. Life Sci. 2018, 75, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, V.; Kurtom, S.; Tarique, M.; Lavania, S.; Malchiodi, Z.; Hellmund, L.; Zhang, L.; Sharma, U.; Giri, B.; Garg, B.; et al. Gut Microbiota Promotes Tumor Growth in Mice by Modulating Immune Response. Gastroenterology 2018, 155, 33–37.e6. [Google Scholar] [CrossRef]
- Mohindroo, C.; Rogers, J.E.; Hasanov, M.; Mizrahi, J.; Overman, M.J.; Varadhachary, G.R.; Wolff, R.A.; Javle, M.M.; Fogelman, D.R.; Pant, S.; et al. A retrospective analysis of antibiotics usage and effect on overall survival and progressive free survival in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2019, 37, e15781. [Google Scholar] [CrossRef]
- Boursi, B.; Mamtani, R.; Haynes, K.; Yang, Y.X. Recurrent antibiotic exposure may promote cancer formation—Another step in understanding the role of the human microbiota? Eur. J. Cancer 2015, 51, 2655–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasanov, M.; Mohindroo, C.; Rogers, J.; Prakash, L.; Overman, M.J.; Varadhachary, G.R.; Wolff, R.A.; Javle, M.M.; Fogelman, D.R.; Pant, S.; et al. The effect of antibiotic use on survival of patients with resected pancreatic ductal adenocarcinoma. J. Clin. Oncol. 2019, 37, e15773. [Google Scholar] [CrossRef]
- Cao, Y.; Wu, K.; Mehta, R.; Drew, D.A.; Song, M.; Lochhead, P.; Nguyen, L.H.; Izard, J.; Fuchs, C.S.; Garrett, W.S.; et al. Long-term use of antibiotics and risk of colorectal adenoma. Gut 2018, 67, 672–678. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.Z.; Gao, P.; Song, Y.X.; Xu, Y.; Sun, J.X.; Chen, X.W.; Zhao, J.H.; Wang, Z.N. Antibiotic use and the efficacy of immune checkpoint inhibitors in cancer patients: A pooled analysis of 2740 cancer patients. Oncoimmunology 2019, 8, e1665973. [Google Scholar] [CrossRef] [Green Version]
- van Praagh, J.B.; de Goffau, M.C.; Bakker, I.S.; van Goor, H.; Harmsen, H.J.M.; Olinga, P.; Havenga, K. Mucus Microbiome of Anastomotic Tissue During Surgery Has Predictive Value for Colorectal Anastomotic Leakage. Ann. Surg. 2019, 269, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Shogan, B.D.; Belogortseva, N.; Luong, P.M.; Zaborin, A.; Lax, S.; Bethel, C.; Ward, M.; Muldoon, J.P.; Singer, M.; An, G.; et al. Collagen degradation and MMP9 activation by Enterococcus faecalis contribute to intestinal anastomotic leak. Sci. Transl. Med. 2015, 7, 286ra268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, F.C.F.; Brenner, T.; Uhle, F.; Loesch, S.; Hackert, T.; Ulrich, A.; Hofer, S.; Dalpke, A.H.; Weigand, M.A.; Boutin, S. Gut microbiome patterns correlate with higher postoperative complication rates after pancreatic surgery. BMC Microbiol. 2019, 19, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belmouhand, M.; Krohn, P.S.; Svendsen, L.B.; Henriksen, A.; Hansen, C.P.; Achiam, M.P. The occurrence of Enterococcus faecium and faecalis Is significantly associated With anastomotic leakage After pancreaticoduodenectomy. Scand. J. Surg. 2018, 107, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Rashid, T.; Ebringer, A. Autoimmunity in Rheumatic Diseases Is Induced by Microbial Infections via Crossreactivity or Molecular Mimicry. Autoimmune Dis. 2012, 2012, 539282. [Google Scholar] [CrossRef] [Green Version]
- The Microbiome of Pancreatic Cancer: “PANDEMIC” Study—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04274972 (accessed on 1 August 2021).
- Jia, G.; Zhi, A.; Lai, P.F.H.; Wang, G.; Xia, Y.; Xiong, Z.; Zhang, H.; Che, N.; Ai, L. The oral microbiota—A mechanistic role for systemic diseases. Br. Dent. J. 2018, 224, 447–455. [Google Scholar] [CrossRef]
- García-Castillo, V.; Sanhueza, E.; McNerney, E.; Onate, S.A.; García, A. Microbiota dysbiosis: A new piece in the understanding of the carcinogenesis puzzle. J. Med. Microbiol. 2016, 65, 1347–1362. [Google Scholar] [CrossRef] [Green Version]
- Maisonneuve, P.; Amar, S.; Lowenfels, A.B. Periodontal disease, edentulism, and pancreatic cancer: A meta-analysis. Ann. Oncol. 2017, 28, 985–995. [Google Scholar] [CrossRef]
- Hiraki, A.; Matsuo, K.; Suzuki, T.; Kawase, T.; Tajima, K. Teeth loss and risk of cancer at 14 common sites in Japanese. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1222–1227. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.S.; Joshipura, K.; Giovannucci, E.; Michaud, D.S. A review of the relationship between tooth loss, periodontal disease, and cancer. Cancer Causes Control 2008, 19, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Stolzenberg-Solomon, R.Z.; Dodd, K.W.; Blaser, M.J.; Virtamo, J.; Taylor, P.R.; Albanes, D. Tooth loss, pancreatic cancer, and Helicobacter pylori. Am. J. Clin. Nutr. 2003, 78, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.J.; Zhang, L.; Zhou, H.; Chia, D.; Elashoff, D.; Akin, D.; Paster, B.J.; Joshipura, K.; Wong, D.T. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 2012, 61, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Izard, J.; Wilhelm-Benartzi, C.S.; You, D.H.; Grote, V.A.; Tjønneland, A.; Dahm, C.C.; Overvad, K.; Jenab, M.; Fedirko, V.; et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 2013, 62, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öğrendik, M. Periodontal Pathogens in the Etiology of Pancreatic Cancer. Gastrointest. Tumors 2017, 3, 125–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, P.J.; Fletcher, E.M.; Gibbons, S.M.; Bouvet, M.; Doran, K.S.; Kelley, S.T. Characterization of the salivary microbiome in patients with pancreatic cancer. PeerJ 2015, 3, e1373. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Ren, Z.; Li, A.; Li, J.; Xu, S.; Zhang, H.; Jiang, J.; Yang, J.; Luo, Q.; Zhou, K.; et al. Tongue coating microbiome data distinguish patients with pancreatic head cancer from healthy controls. J. Oral Microbiol. 2019, 11, 1563409. [Google Scholar] [CrossRef]
- Fritz, S.; Hackert, T.; Hartwig, W.; Rossmanith, F.; Strobel, O.; Schneider, L.; Will-Schweiger, K.; Kommerell, M.; Büchler, M.W.; Werner, J. Bacterial translocation and infected pancreatic necrosis in acute necrotizing pancreatitis derives from small bowel rather than from colon. Am. J. Surg. 2010, 200, 111–117. [Google Scholar] [CrossRef]
- Risch, H.A. Etiology of pancreatic cancer, with a hypothesis concerning the role of N-nitroso compounds and excess gastric acidity. J. Natl. Cancer Inst. 2003, 95, 948–960. [Google Scholar] [CrossRef]
- Blaser, M.J. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology 1992, 102, 720–727. [Google Scholar] [CrossRef]
- Akshintala, V.S.; Talukdar, R.; Singh, V.K.; Goggins, M. The Gut Microbiome in Pancreatic Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Sasaki, T.; Itoh, R.; Kato, D.; Hatano, N.; Soejima, T.; Ishii, K.; Takenawa, T.; Hiromatsu, K.; Yamashita, Y. Pancreatic fistulae secondary to trypsinogen activation by Pseudomonas aeruginosa infection after pancreatoduodenectomy. J. Hepato-Biliary-Pancreat. Sci. 2015, 22, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Fukaya, R.; Takamatsu, S.; Itoyama, S.; Fukuoka, T.; Yamada, M.; Hata, T.; Nagaoka, S.; Kawamoto, K.; Eguchi, H.; et al. Possible involvement of Enterococcus infection in the pathogenesis of chronic pancreatitis and cancer. Biochem. Biophys. Res. Commun. 2018, 506, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Serra, N.; Di Carlo, P.; Gulotta, G.; d’Arpa, F.; Giammanco, A.; Colomba, C.; Melfa, G.; Fasciana, T.; Sergi, C. Bactibilia in women affected with diseases of the biliary tract and pancreas. A STROBE guidelines-adherent cross-sectional study in Southern Italy. J. Med. Microbiol. 2018, 67, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Liwinski, T.; Zenouzi, R.; John, C.; Ehlken, H.; Rühlemann, M.C.; Bang, C.; Groth, S.; Lieb, W.; Kantowski, M.; Andersen, N.; et al. Alterations of the bile microbiome in primary sclerosing cholangitis. Gut 2020, 69, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrader, H.R.; Miller, A.M.; Tomanek-Chalkley, A.; McCarthy, A.; Coleman, K.L.; Ear, P.H.; Mangalam, A.K.; Salem, A.K.; Chan, C.H.F. Effect of bacterial contamination in bile on pancreatic cancer cell survival. Surgery 2021, 169, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Half, E.; Keren, N.; Reshef, L.; Dorfman, T.; Lachter, I.; Kluger, Y.; Reshef, N.; Knobler, H.; Maor, Y.; Stein, A.; et al. Fecal microbiome signatures of pancreatic cancer patients. Sci. Rep. 2019, 9, 16801. [Google Scholar] [CrossRef]
- Aronsson, L.; Andersson, R.; Ansari, D. Intraductal papillary mucinous neoplasm of the pancreas—Epidemiology, risk factors, diagnosis, and management. Scand. J. Gastroenterol. 2017, 52, 803–815. [Google Scholar] [CrossRef]
- Crippa, S.; Capurso, G.; Cammà, C.; Fave, G.D.; Castillo, C.F.; Falconi, M. Risk of pancreatic malignancy and mortality in branch-duct IPMNs undergoing surveillance: A systematic review and meta-analysis. Dig. Liver Dis. 2016, 48, 473–479. [Google Scholar] [CrossRef]
- European Study Group on Cystic Tumours of the Pancreas. European evidence-based guidelines on pancreatic cystic neoplasms. Gut 2018, 67, 789–804. [Google Scholar] [CrossRef]
- Valsangkar, N.P.; Morales-Oyarvide, V.; Thayer, S.P.; Ferrone, C.R.; Wargo, J.A.; Warshaw, A.L.; Fernández-del Castillo, C. 851 resected cystic tumors of the pancreas: A 33-year experience at the Massachusetts General Hospital. Surgery 2012, 152, S4–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Chiaro, M.; Segersvärd, R.; Pozzi Mucelli, R.; Rangelova, E.; Kartalis, N.; Ansorge, C.; Arnelo, U.; Blomberg, J.; Löhr, M.; Verbeke, C. Comparison of preoperative conference-based diagnosis with histology of cystic tumors of the pancreas. Ann. Surg. Oncol. 2014, 21, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Del Chiaro, M.; Segersvärd, R.; Lohr, M.; Verbeke, C. Early detection and prevention of pancreatic cancer: Is it really possible today? World J. Gastroenterol. 2014, 20, 12118–12131. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fernández-Del Castillo, C.; Kamisawa, T.; Jang, J.Y.; Levy, P.; Ohtsuka, T.; Salvia, R.; Shimizu, Y.; Tada, M.; Wolfgang, C.L. Revisions of international consensus Fukuoka guidelines for the management of IPMN of the pancreas. Pancreatology 2017, 17, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fuhler, G.M.; Bn, N.; Jose, T.; Bruno, M.J.; Peppelenbosch, M.P.; Konstantinov, S.R. Pancreatic cyst fluid harbors a unique microbiome. Microbiome 2017, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.H.; Satagopan, J.; Xu, Y.; Ling, L.; Leong, S.; Orlow, I.; Saldia, A.; Li, P.; Nunes, P.; Madonia, V.; et al. The oral microbiota in patients with pancreatic cancer, patients with IPMNs, and controls: A pilot study. Cancer Causes Control 2017, 28, 959–969. [Google Scholar] [CrossRef]
- Maker, A.V.; Katabi, N.; Qin, L.-X.; Klimstra, D.S.; Schattner, M.; Brennan, M.F.; Jarnagin, W.R.; Allen, P.J. Cyst fluid interleukin-1beta (IL1beta) levels predict the risk of carcinoma in intraductal papillary mucinous neoplasms of the pancreas. Clin. Cancer Res. 2011, 17, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 2015, 13, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, M.; Nomura, N.; Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef]
- Tulkens, J.; De Wever, O.; Hendrix, A. Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nat. Protoc. 2020, 15, 40–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.W.; Boulton, I.C.; Reddin, K.; Wong, H.; Halliwell, D.; Mandelboim, O.; Gorringe, A.R.; Gray-Owen, S.D. Neisserial outer membrane vesicles bind the coinhibitory receptor carcinoembryonic antigen-related cellular adhesion molecule 1 and suppress CD4+ T lymphocyte function. Infect. Immun. 2007, 75, 4449–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peek, R.M.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Tsatsaronis, J.A.; Franch-Arroyo, S.; Resch, U.; Charpentier, E. Extracellular Vesicle RNA: A Universal Mediator of Microbial Communication? Trends Microbiol. 2018, 26, 401–410. [Google Scholar] [CrossRef]
- Shah, B.; Sullivan, C.J.; Lonergan, N.E.; Stanley, S.; Soult, M.C.; Britt, L.D. Circulating bacterial membrane vesicles cause sepsis in rats. Shock 2012, 37, 621–628. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Microbiol. 2015, 13, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Gu, L.; Meng, R.; Tang, Y.; Zhao, K.; Liang, F.; Zhang, R.; Xue, Q.; Chen, F.; Xiao, X.; Wang, H.; et al. Toll-Like Receptor 4 Signaling Licenses the Cytosolic Transport of Lipopolysaccharide From Bacterial Outer Membrane Vesicles. Shock 2019, 51, 256–265. [Google Scholar] [CrossRef]
- Cecil, J.D.; O’Brien-Simpson, N.M.; Lenzo, J.C.; Holden, J.A.; Chen, Y.-Y.; Singleton, W.; Gause, K.T.; Yan, Y.; Caruso, F.; Reynolds, E.C. Differential Responses of Pattern Recognition Receptors to Outer Membrane Vesicles of Three Periodontal Pathogens. PLoS ONE 2016, 11, e0151967. [Google Scholar] [CrossRef]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B.; Lippens, L.; De Scheerder, M.-A.; Miinalainen, I.; Rappu, P.; De Geest, B.G.; et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020, 69, 191–193. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.R.; Han, K.; Han, Y.; Kang, N.; Shin, T.-S.; Park, H.J.; Kim, H.; Kwon, W.; Lee, S.; Kim, Y.-K.; et al. Microbiome Markers of Pancreatic Cancer Based on Bacteria-Derived Extracellular Vesicles Acquired from Blood Samples: A Retrospective Propensity Score Matching Analysis. Biology 2021, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Kunovsky, L.; Tesarikova, P.; Kala, Z.; Kroupa, R.; Kysela, P.; Dolina, J.; Trna, J. The Use of Biomarkers in Early Diagnostics of Pancreatic Cancer. Can. J. Gastroenterol. Hepatol. 2018, 2018, 5389820. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merali, N.; Chouari, T.; Kayani, K.; Rayner, C.J.; Jiménez, J.I.; Krell, J.; Giovannetti, E.; Bagwan, I.; Relph, K.; Rockall, T.A.; et al. A Comprehensive Review of the Current and Future Role of the Microbiome in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 1020. https://doi.org/10.3390/cancers14041020
Merali N, Chouari T, Kayani K, Rayner CJ, Jiménez JI, Krell J, Giovannetti E, Bagwan I, Relph K, Rockall TA, et al. A Comprehensive Review of the Current and Future Role of the Microbiome in Pancreatic Ductal Adenocarcinoma. Cancers. 2022; 14(4):1020. https://doi.org/10.3390/cancers14041020
Chicago/Turabian StyleMerali, Nabeel, Tarak Chouari, Kayani Kayani, Charles J. Rayner, José I. Jiménez, Jonathan Krell, Elisa Giovannetti, Izhar Bagwan, Kate Relph, Timothy A. Rockall, and et al. 2022. "A Comprehensive Review of the Current and Future Role of the Microbiome in Pancreatic Ductal Adenocarcinoma" Cancers 14, no. 4: 1020. https://doi.org/10.3390/cancers14041020
APA StyleMerali, N., Chouari, T., Kayani, K., Rayner, C. J., Jiménez, J. I., Krell, J., Giovannetti, E., Bagwan, I., Relph, K., Rockall, T. A., Dhillon, T., Pandha, H., Annels, N. E., & Frampton, A. E. (2022). A Comprehensive Review of the Current and Future Role of the Microbiome in Pancreatic Ductal Adenocarcinoma. Cancers, 14(4), 1020. https://doi.org/10.3390/cancers14041020