

The METTL3 RNA Methyltransferase Regulates Transcriptional Networks in Prostate Cancer

 , , , ,
, , , ,  , , , , , and add
Show full author list
, , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analysis of METTL3 in Clinical Specimens
2.2. Ethics Statement, Tissue Specimens, and Immunohistochemistry
2.3. Cell Lines and Culture Conditions
2.4. siRNA Mediated Functional Depletion and Pharmaco-Inhibition of METTL3
2.5. Gene Expression Analysis and Western Blotting
2.6. RNA-seq and Splicing Analysis of siRNA Depletion or Pharmaco-Inhibition of METTL3
2.7. Evaluation of Phenotypic Effects
2.8. Statistical Analysis
3. Results
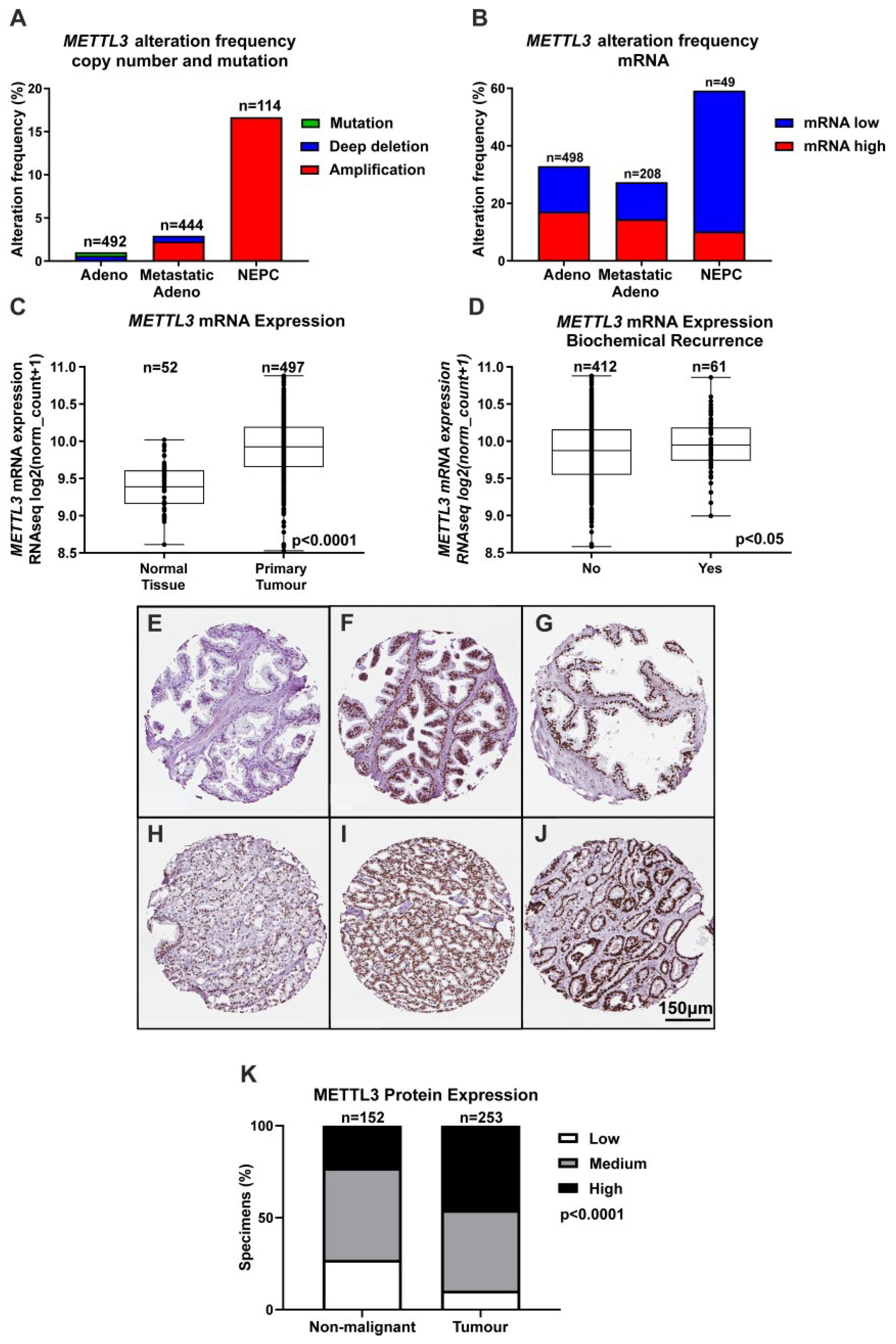
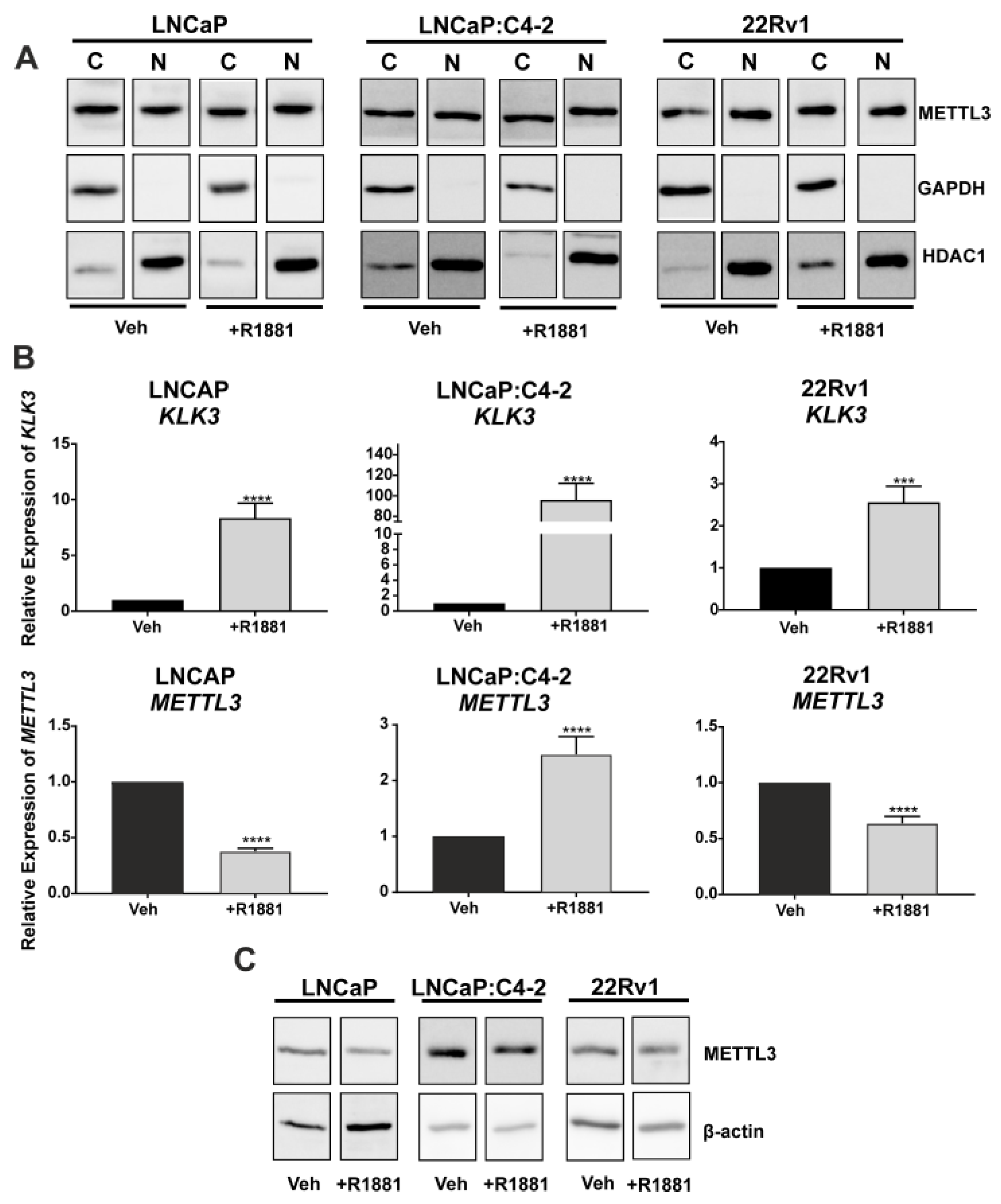
3.1. Expression of METTL3 in PCa Patients and Cell Lines
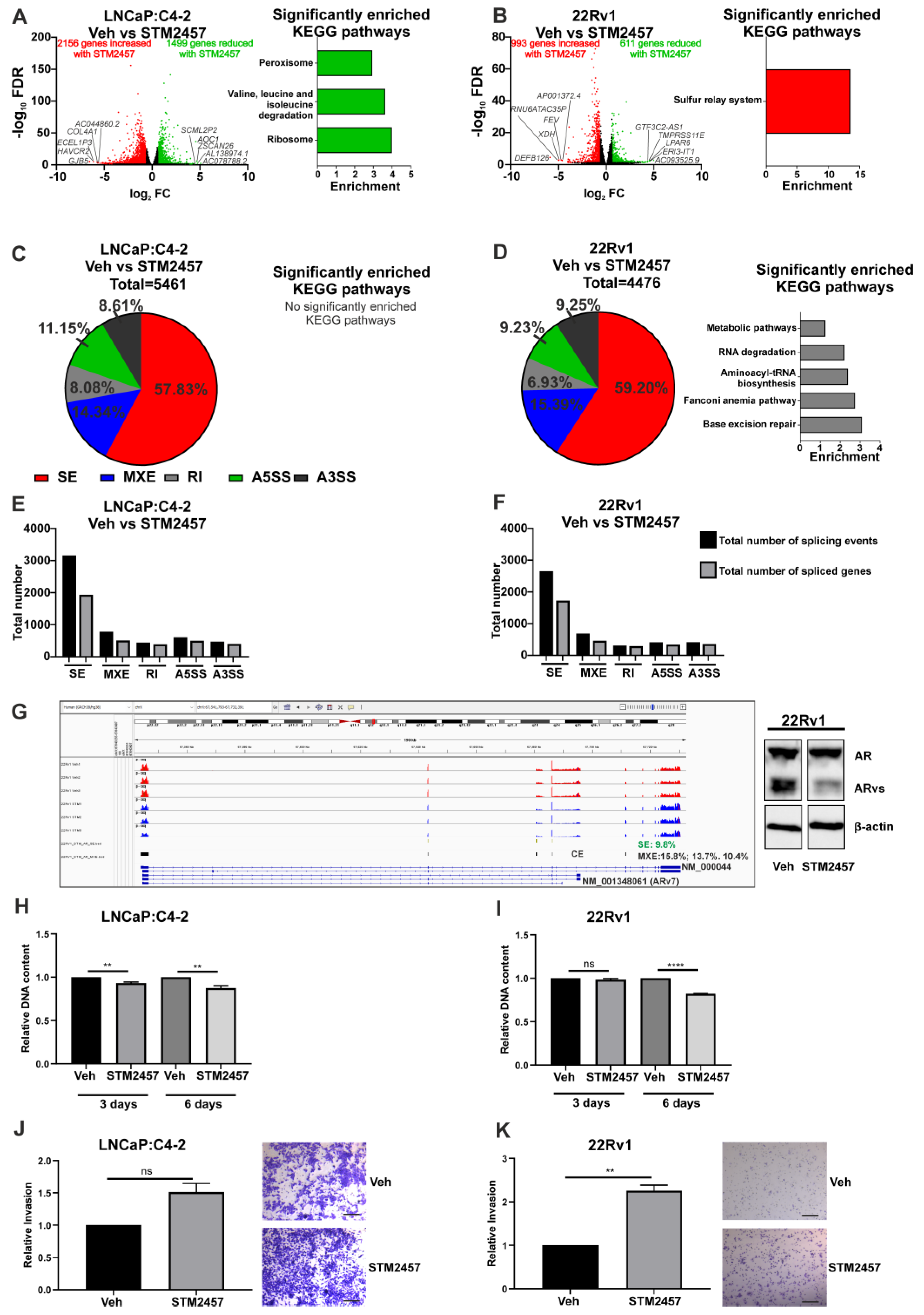
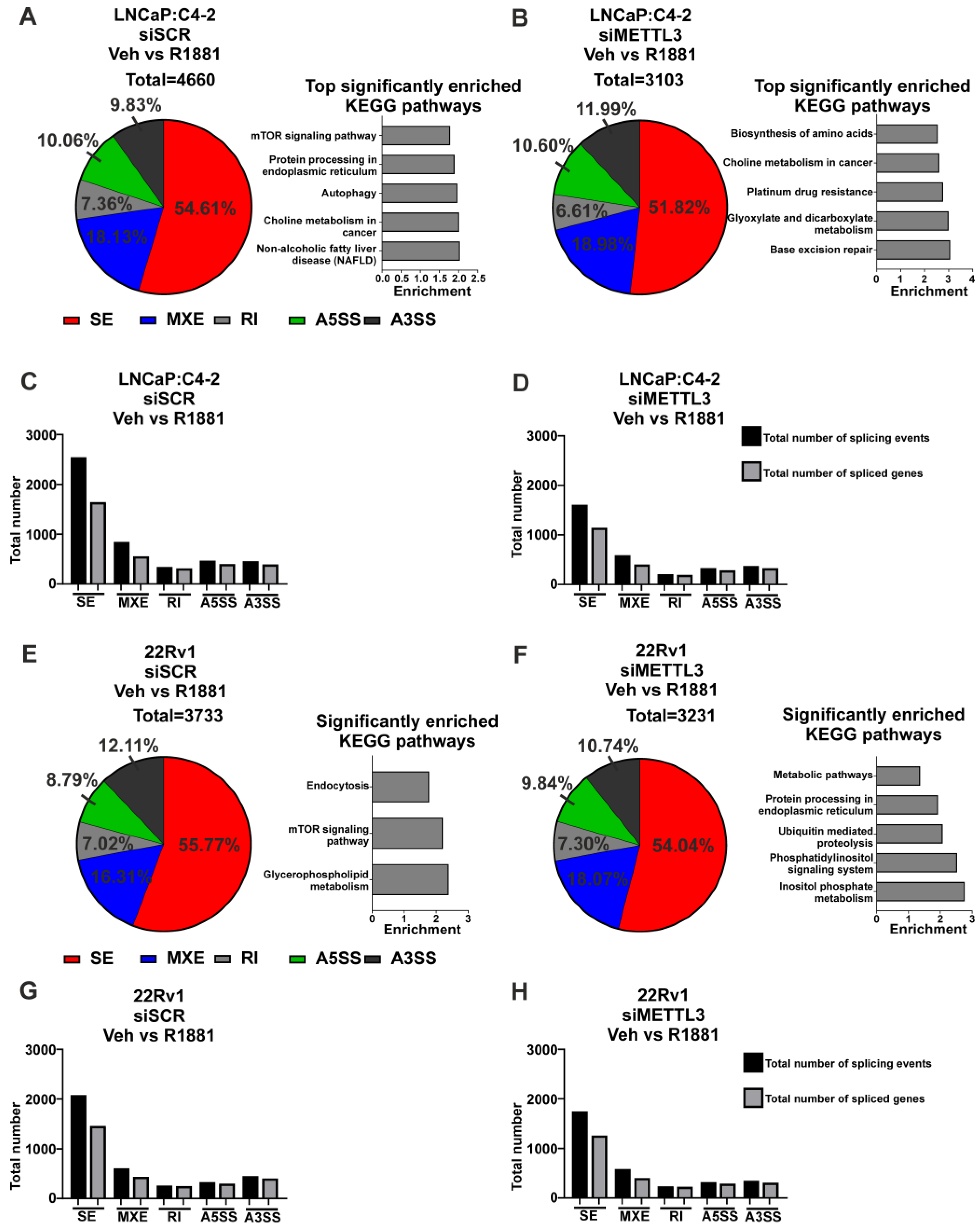
3.2. Functional Inhibition of METTL3 Alters PCa Cell Line Transcriptome and Splicing
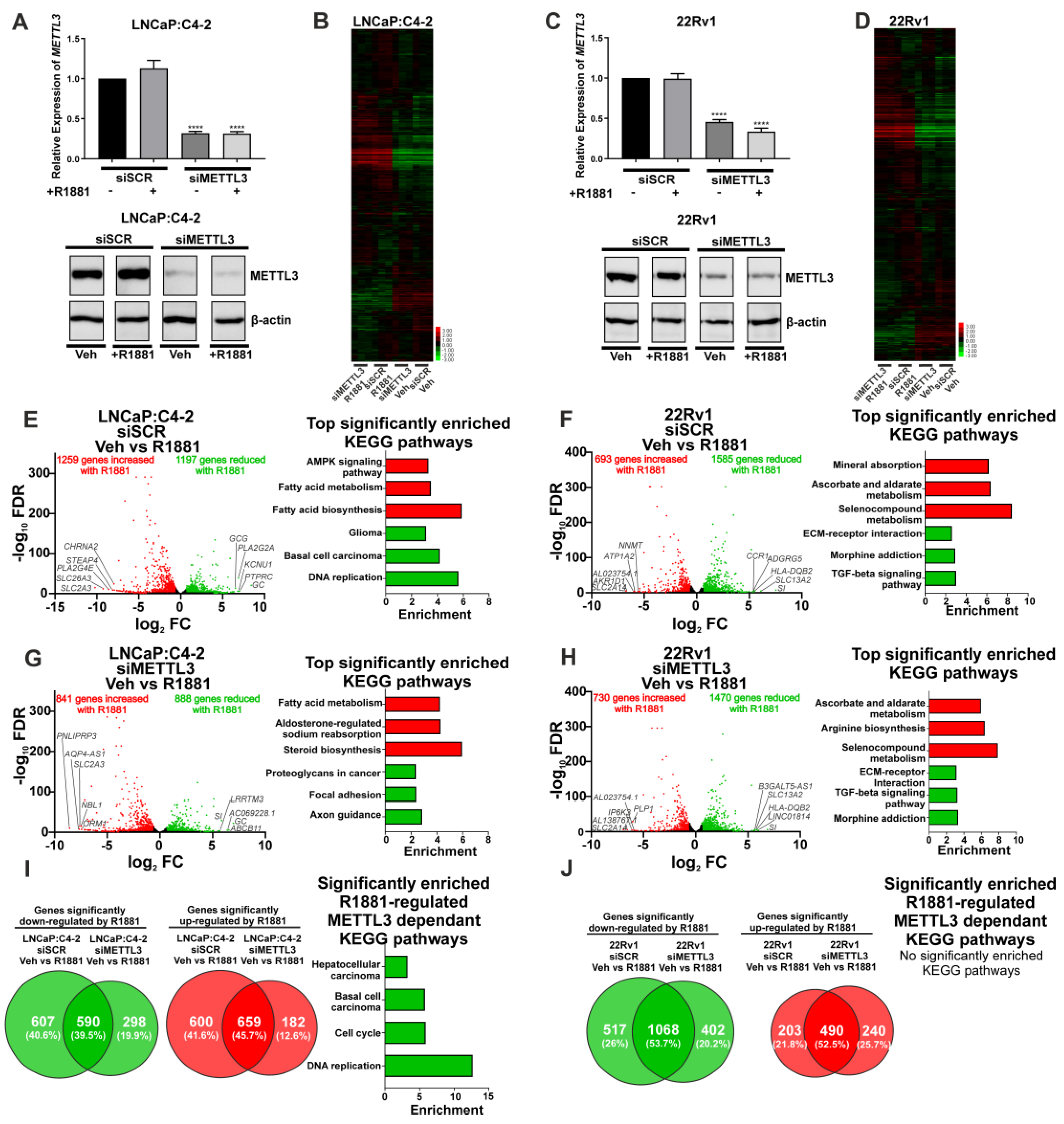
3.3. Depletion of METTL3 Alters the Androgen-Regulated PCa Transcriptome and Splicing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Rottman, F.M. An in vitro system for accurate methylation of internal adenosine residues in messenger RNA. Science 1988, 242, 1159–1162. [Google Scholar] [CrossRef]
- Bokar, J.A.; Rath-Shambaugh, M.E.; Ludwiczak, R.; Narayan, P.; Rottman, F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 1994, 269, 17697–17704. [Google Scholar] [CrossRef]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A census of human soluble protein complexes. Cell 2012, 150, 1068–1081. [Google Scholar] [CrossRef]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell. Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Lv, R.; Ma, H.; Shen, H.; He, C.; Wang, J.; Jiao, F.; Liu, H.; Yang, P.; Tan, L.; et al. Zc3h13 Regulates Nuclear RNA m(6)A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol. Cell. 2018, 69, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m(6)A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell. Discov. 2018, 4, 10. [Google Scholar] [CrossRef]
- Ruzicka, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m(6) A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017, 215, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Bodi, Z.; Mackinnon, K.; Zhong, S.; Archer, N.; Mongan, N.P.; Simpson, G.G.; Fray, R.G. Two zinc finger proteins with functions in m(6)A writing interact with HAKAI. Nat. Commun. 2022, 13, 1127. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Kawamura, T.; Iwanari, H.; Ohashi, R.; Naito, M.; Kodama, T.; Hamakubo, T. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J. Biol. Chem. 2013, 288, 33292–33302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Theler, D.; Kaminska, K.H.; Hiller, M.; de la Grange, P.; Pudimat, R.; Rafalska, I.; Heinrich, B.; Bujnicki, J.M.; Allain, F.H.; et al. The YTH domain is a novel RNA binding domain. J. Biol. Chem. 2010, 285, 14701–14710. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Louloupi, A.; Ntini, E.; Conrad, T.; Ørom, U.A.V. Transient N-6-methyladenosine transcriptome sequencing reveals a regulatory role of m6a in splicing efficiency. Cell Rep. 2018, 23, 3429–3437. [Google Scholar] [CrossRef] [PubMed]
- Haussmann, I.U.; Bodi, Z.; Sanchez-Moran, E.; Mongan, N.P.; Archer, N.; Fray, R.G.; Soller, M. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 2016, 540, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Klukovich, R.; Peng, H.; Wang, Z.; Yu, T.; Zhang, Y.; Zheng, H.; Klungland, A.; Yan, W. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA 2018, 115, E325–E333. [Google Scholar] [CrossRef] [PubMed]
- Coots, R.A.; Liu, X.M.; Mao, Y.; Dong, L.; Zhou, J.; Wan, J.; Zhang, X.; Qian, S.B. m(6)A Facilitates eIF4F-Independent mRNA Translation. Mol. Cell 2017, 68, 504–514.e7. [Google Scholar] [CrossRef]
- Li, J.; Meng, S.; Xu, M.; Wang, S.; He, L.; Xu, X.; Wang, X.; Xie, L. Downregulation of N(6)-methyladenosine binding YTHDF2 protein mediated by miR-493-3p suppresses prostate cancer by elevating N(6)-methyladenosine levels. Oncotarget 2018, 9, 3752–3764. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Du, Y.; Wang, L.; Liu, X. The M6A methyltransferase METTL3 promotes the development and progression of prostate carcinoma via mediating MYC methylation. J. Cancer 2020, 11, 3588–3595. [Google Scholar] [CrossRef]
- Cai, J.; Yang, F.; Zhan, H.; Situ, J.; Li, W.; Mao, Y.; Luo, Y. RNA m(6)A Methyltransferase METTL3 Promotes The Growth Of Prostate Cancer By Regulating Hedgehog Pathway. Onco. Targets Ther. 2019, 12, 9143–9152. [Google Scholar] [CrossRef]
- Ma, X.X.; Cao, Z.G.; Zhao, S.L. m6A methyltransferase METTL3 promotes the progression of prostate cancer via m6A-modified LEF1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3565–3571. [Google Scholar] [CrossRef]
- Li, J.; Xie, H.; Ying, Y.; Chen, H.; Yan, H.; He, L.; Xu, M.; Xu, X.; Liang, Z.; Liu, B.; et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6-methyladenosine-dependent way in prostate cancer. Mol. Cancer 2020, 19, 152. [Google Scholar] [CrossRef] [PubMed]
- Cotter, K.A.; Gallon, J.; Uebersax, N.; Rubin, P.; Meyer, K.D.; Piscuoglio, S.; Jaffrey, S.R.; Rubin, M.A. Mapping of m(6)A and Its Regulatory Targets in Prostate Cancer Reveals a METTL3-Low Induction of Therapy Resistance. Mol. Cancer Res. 2021, 19, 1398–1411. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Ahmad, S.; Nilsson, E.M.; Helczynski, L.; Kenna, S.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol. Oncol. 2013, 7, 555–566. [Google Scholar] [CrossRef] [PubMed]
- McCarty, K.S.; Jr Miller, L.S.; Cox, E.B.; Konrath, J.; McCarty, K.S., Sr. Estrogen receptor analyses. Correlation of biochemical and immunohistochemical methods using monoclonal antireceptor antibodies. Arch. Pathol. Lab. Med. 1985, 109, 716–721. [Google Scholar]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.-X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013, 23, 35–47. [Google Scholar] [CrossRef]
- de Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Ji, G.; Huang, C.; He, S.; Gong, Y.; Song, G.; Li, X.; Zhou, L. Comprehensive analysis of m6A regulators prognostic value in prostate cancer. Aging 2020, 12, 14863–14884. [Google Scholar] [CrossRef]
- Li, E.; Wei, B.; Wang, X.; Kang, R. METTL3 enhances cell adhesion through stabilizing integrin β1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am. J. Cancer Res. 2020, 10, 1012–1025. [Google Scholar]
- Chen, Y.; Pan, C.; Wang, X.; Xu, D.; Ma, Y.; Hu, J.; Chen, P.; Xiang, Z.; Rao, Q.; Han, X. Silencing of METTL3 effectively hinders invasion and metastasis of prostate cancer cells. Theranostics 2021, 11, 7640–7657. [Google Scholar] [CrossRef] [PubMed]
- Barros-Silva, D.; Lobo, J.; Guimarães-Teixeira, C.; Carneiro, I.; Oliveira, J.; Martens-Uzunova, E.S.; Henrique, R.; Jerónimo, C. VIRMA-Dependent N6-Methyladenosine Modifications Regulate the Expression of Long Non-Coding RNAs CCAT1 and CCAT2 in Prostate Cancer. Cancers 2020, 12, 771. [Google Scholar] [CrossRef]
- Xu, J.; Liu, Y.; Liu, J.; Xu, T.; Cheng, G.; Shou, Y.; Tong, J.; Liu, L.; Zhou, L.; Xiao, W.; et al. The Identification of Critical m(6)A RNA Methylation Regulators as Malignant Prognosis Factors in Prostate Adenocarcinoma. Front. Genet. 2020, 11, 602485. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef]
- Luxton, H.J.; Simpson, B.S.; Mills, I.G.; Brindle, N.R.; Ahmed, Z.; Stavrinides, V.; Heavey, S.; Stamm, S.; Whitaker, H.C. The Oncogene Metadherin Interacts with the Known Splicing Proteins YTHDC1, Sam68 and T-STAR and Plays a Novel Role in Alternative mRNA Splicing. Cancers 2019, 11, 1233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Auboeuf, D.; Honig, A.; Berget, S.M.; O’Malley, B.W. Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science 2002, 298, 416–419. [Google Scholar] [CrossRef]
- Auboeuf, D.; Dowhan, D.H.; Dutertre, M.; Martin, N.; Berget, S.M.; O’Malley, B.W. A subset of nuclear receptor coregulators act as coupling proteins during synthesis and maturation of RNA transcripts. Mol. Cell. Biol. 2005, 25, 5307–5316. [Google Scholar] [CrossRef]
- Rana, M.; Dong, J.; Robertson, M.J.; Basil, P.; Coarfa, C.; Weigel, N.L. Androgen receptor and its splice variant, AR-V7, differentially induce mRNA splicing in prostate cancer cells. Sci. Rep. 2021, 11, 1393. [Google Scholar] [CrossRef]
- Darnell, R.B.; Ke, S.; Darnell, J.E., Jr. Pre-mRNA processing includes N(6) methylation of adenosine residues that are retained in mRNA exons and the fallacy of “RNA epigenetics”. RNA 2018, 24, 262–267. [Google Scholar] [CrossRef]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vagbo, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E., Jr.; Darnell, R.B. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; Nachtergaele, S.; Roundtree, I.A.; He, C. Our views of dynamic N(6)-methyladenosine RNA methylation. RNA 2018, 24, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Jaffrey, S.R. Hidden codes in mRNA: Control of gene expression by m(6)A. Mol. Cell 2022, 82, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Lence, T.; Akhtar, J.; Bayer, M.; Schmid, K.; Spindler, L.; Ho, C.H.; Kreim, N.; Andrade-Navarro, M.A.; Poeck, B.; Helm, M.; et al. m(6)A modulates neuronal functions and sex determination in Drosophila. Nature 2016, 540, 242–247. [Google Scholar] [CrossRef]
- Ke, S.; Alemu, E.A.; Mertens, C.; Gantman, E.C.; Fak, J.J.; Mele, A.; Haripal, B.; Zucker-Scharff, I.; Moore, M.J.; Park, C.Y.; et al. A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev. 2015, 29, 2037–2053. [Google Scholar] [CrossRef]
- Wei, G.; Almeida, M.; Pintacuda, G.; Coker, H.; Bowness, J.S.; Ule, J.; Brockdorff, N. Acute depletion of METTL3 implicates N (6)-methyladenosine in alternative intron/exon inclusion in the nascent transcriptome. Genome Res. 2021, 31, 1395–1408. [Google Scholar] [CrossRef]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Avgan, N.; Wang, J.I.; Fernandez-Chamorro, J.; Weatheritt, R.J. Multilayered control of exon acquisition permits the emergence of novel forms of regulatory control. Genome Biol. 2019, 20, 141. [Google Scholar] [CrossRef]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646–1656. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Antonarakis, E.S.; Gjyrezi, A.; Galletti, G.; Kim, S.; Worroll, D.; Stewart, J.; Zaher, A.; Szatrowski, T.P.; Ballman, K.V.; et al. Expression of AR-V7 and ARv(567es) in Circulating Tumor Cells Correlates with Outcomes to Taxane Therapy in Men with Metastatic Prostate Cancer Treated in TAXYNERGY. Clin. Cancer Res. 2019, 25, 1880–1888. [Google Scholar] [CrossRef]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Attard, G.; Balk, S.P.; Bevan, C.; Burnstein, K.; Cato, L.; Cherkasov, A.; De Bono, J.S.; Dong, Y.; Gao, A.C.; et al. Role of Androgen Receptor Variants in Prostate Cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. Eur. Urol. 2018, 73, 715–723. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haigh, D.B.; Woodcock, C.L.; Lothion-Roy, J.; Harris, A.E.; Metzler, V.M.; Persson, J.L.; Robinson, B.D.; Khani, F.; Alsaleem, M.; Ntekim, A.; et al. The METTL3 RNA Methyltransferase Regulates Transcriptional Networks in Prostate Cancer. Cancers 2022, 14, 5148. https://doi.org/10.3390/cancers14205148
Haigh DB, Woodcock CL, Lothion-Roy J, Harris AE, Metzler VM, Persson JL, Robinson BD, Khani F, Alsaleem M, Ntekim A, et al. The METTL3 RNA Methyltransferase Regulates Transcriptional Networks in Prostate Cancer. Cancers. 2022; 14(20):5148. https://doi.org/10.3390/cancers14205148
Chicago/Turabian StyleHaigh, Daisy B., Corinne L. Woodcock, Jennifer Lothion-Roy, Anna E. Harris, Veronika M. Metzler, Jenny L. Persson, Brian D. Robinson, Francesca Khani, Mansour Alsaleem, Atara Ntekim, and et al. 2022. "The METTL3 RNA Methyltransferase Regulates Transcriptional Networks in Prostate Cancer" Cancers 14, no. 20: 5148. https://doi.org/10.3390/cancers14205148
APA StyleHaigh, D. B., Woodcock, C. L., Lothion-Roy, J., Harris, A. E., Metzler, V. M., Persson, J. L., Robinson, B. D., Khani, F., Alsaleem, M., Ntekim, A., Madhusudan, S., Davis, M. B., Laursen, K. B., Gudas, L. J., Rutland, C. S., Toss, M. S., Archer, N., Bodi, Z., Rakha, E. A., ... Mongan, N. P. (2022). The METTL3 RNA Methyltransferase Regulates Transcriptional Networks in Prostate Cancer. Cancers, 14(20), 5148. https://doi.org/10.3390/cancers14205148