
Advances in Promoting the Efficacy of Chimeric Antigen Receptor T Cells in the Treatment of Hepatocellular Carcinoma


Abstract
Simple Summary
Abstract
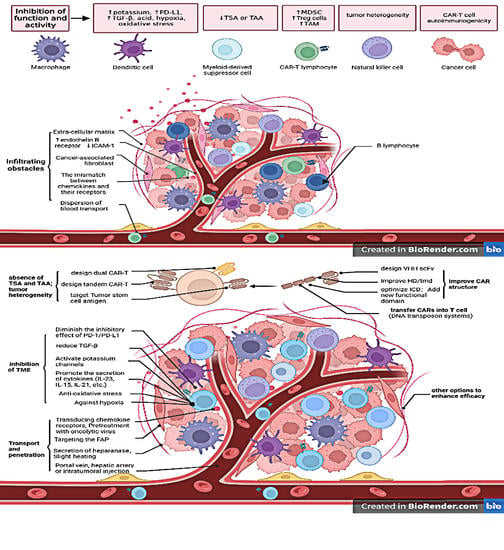
1. Introduction
2. A Brief Introduction to CAR-T Therapy
3. Transport and Penetration
3.1. Local Administration of CAR Cells to the Tumor
3.2. Improve the Penetration of CAR Cells
4. Function and Activity of CAR-T Cells in the TME
4.1. Against Hypoxia
4.2. Antioxidative Stress
4.3. Diminish the Inhibitory Effect of PD-1/PD-L1
4.4. Promote the Secretion of Cytokines
4.5. Other Solutions to Bypass TME Inhibition
5. CAR Structure
5.1. Improve HD/TMD
5.2. Intracellular Domain Optimization
6. Development of Combinatorial Targeting CAR-T Cells
7. Targeting Multiple Antigens or Tumor Stem Cell Antigens
8. Transduction of CARs into T Cells
9. Other Options to Enhance Efficacy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Latest Global Cancer Data: Cancer Burden Rises to 19.3 Million New Cases and 10.0 Million Cancer Deaths in 2020; IARC: Lyon, France, 2020. [Google Scholar]
- Chakraborty, E.; Sarkar, D. Emerging Therapies for Hepatocellular Carcinoma (HCC). Cancers 2022, 14, 2798. [Google Scholar] [CrossRef]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef]
- Sun, B.; Yang, D.; Dai, H.; Liu, X.; Jia, R.; Cui, X.; Li, W.; Cai, C.; Xu, J.; Zhao, X. Eradication of Hepatocellular Carcinoma by NKG2D-Based CAR-T Cells. Cancer Immunol. Res. 2019, 7, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef]
- Wu, X.; Luo, H.; Shi, B.; Di, S.; Sun, R.; Su, J.; Liu, Y.; Li, H.; Jiang, H.; Li, Z. Combined Antitumor Effects of Sorafenib and GPC3-CAR T Cells in Mouse Models of Hepatocellular Carcinoma. Mol. Ther. 2019, 27, 1483–1494. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Klein Wolterink, R.G.; Wang, J.; Bos, G.M.; Germeraad, W.T. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced traf-ficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]
- Meng, M.; Wu, Y.C. Combination of AAV-CCL19 and GPC3 CAR-T Cells in the Treatment of Hepatocellular Carcinoma. J. Immunol. Res. 2021, 2021, 1782728. [Google Scholar] [CrossRef]
- Chen, Y.; Chang-Yong, E.; Gong, Z.W.; Liu, S.; Wang, Z.X.; Yang, Y.S.; Zhang, X.W. Chimeric antigen receptor-engineered T-cell therapy for liver cancer. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 301–309. [Google Scholar] [CrossRef]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef]
- Bughda, R.; Dimou, P.; D’Souza, R.R.; Klampatsa, A. Fibroblast Activation Protein (FAP)-Targeted CAR-T Cells: Launching an Attack on Tumor Stroma. Immunotargets Ther. 2021, 10, 313–323. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coex-pressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Greenshpan, Y.; Sharabi, O.; Ottolenghi, A.; Cahana, A.; Kundu, K.; Yegodayev, K.M.; Elkabets, M.; Gazit, R.; Porgador, A. Synthetic promoters to induce immune-effectors into the tumor microenvironment. Commun. Biol. 2021, 4, 143. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liao, Q.; Zhao, C.; Zhu, C.; Feng, M.; Liu, Z.; Jiang, L.; Zhang, L.; Ding, X.; Yuan, M.; et al. Conditioned CAR-T cells by hypoxia-inducible transcription amplification (HiTA) system significantly enhances systemic safety and retains antitumor ef-ficacy. J. Immunother. Cancer 2021, 9, e002755. [Google Scholar] [CrossRef] [PubMed]
- Renken, S.; Nakajima, T.; Magalhaes, I.; Mattsson, J.; Lundqvist, A.; Arnér, E.S.; Kiessling, R.; Wickström, S.L. Targeting of Nrf2 improves antitumoral responses by human NK cells, TIL and CAR T cells during oxidative stress. J. Immunother. Cancer 2022, 10, e004458. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Yang, X.C.; Barca, T.; Lucas, A.; Turkoz, M.; Wong, J.T.; Nishimoto, K.P.; Brodey, M.M.; Tabrizizad, M.; Gundurao, S.R.; et al. Off-the-shelf Vδ1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, e003441. [Google Scholar] [CrossRef] [PubMed]
- Dal Bo, M.; De Mattia, E.; Baboci, L.; Mezzalira, S.; Cecchin, E.; Assaraf, Y.G.; Toffoli, G. New insights into the pharmacological, im-munological, and CAR-T-cell approaches in the treatment of hepatocellular carcinoma. Drug Resist. Updates 2020, 51, 100702. [Google Scholar] [CrossRef] [PubMed]
- Gacerez, A.T.; Sentman, C.L. T-bet promotes potent antitumor activity of CD4+ CAR T cells. Cancer Gene Ther. 2018, 25, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef]
- Boomer, J.S.; Green, J.M. An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002436. [Google Scholar] [CrossRef]
- Wan, Z.; Shao, X.; Ji, X.; Dong, L.; Wei, J.; Xiong, Z.; Liu, W.; Qi, H. Transmembrane domain-mediated Lck association underlies by-stander and costimulatory ICOS signaling. Cell Mol. Immunol. 2020, 17, 143–152. [Google Scholar] [CrossRef]
- Tipanee, J.; VandenDriessche, T.; Chuah, M.K. Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum. Gene Ther. 2017, 28, 1087–1104. [Google Scholar] [CrossRef]
- Hudecek, M.; Ivics, Z. Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr. Opin. Genet. Dev. 2018, 52, 100–108. [Google Scholar] [CrossRef]
- Chen, C.; Li, K.; Jiang, H.; Song, F.; Gao, H.; Pan, X.; Shi, B.; Bi, Y.; Wang, H.; Wang, H.; et al. Development of T cells carrying two com-plementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol. Immunother. 2017, 66, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Elahi, R.; Khosh, E.; Esmaeilzadeh, A. Programmable and multi-targeted CARs: A new breakthrough in cancer CAR-T cell therapy. Clin. Transl. Oncol. 2021, 23, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: A single-arm, open-label, phase II trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef]
- Pan, D.S.; Feng, S.Z.; Cao, P.; Li, J.J. Endothelin B receptor promotes the proliferation and immune escape of malignant gliomas. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1230–1235. [Google Scholar] [CrossRef]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef]
- Vignali, D.; Kallikourdis, M. Improving homing in T cell therapy. Cytokine Growth Factor Rev. 2017, 36, 107–116. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Park, A.K.; Fong, Y.; Kim, S.I.; Yang, J.; Murad, J.P.; Lu, J.; Jeang, B.; Chang, W.C.; Chen, N.G.; Thomas, S.H.; et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020, 12, eaaz1863. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, Q.; Dukhovlinova, E.; Chen, G.; Ahn, S.; Wang, C.; Ogunnaike, E.A.; Ligler, F.S.; Dotti, G.; Gu, Z. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv. Mater. 2019, 31, e1900192. [Google Scholar] [CrossRef]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef]
- Zhao, Y.; Qiu, W.; Liu, J.; Yuan, X.; Mao, W.; Yin, J.; Peng, B.; Liu, W.; Han, S.; He, X. Blockade of Kv1.3 potassium channel inhibits CD8+ T cell-mediated neuroinflammation via PD-1/Blimp-1 signaling. FASEB J. 2020, 34, 15492–15503. [Google Scholar] [CrossRef]
- Ong, S.T.; Ng, A.S.; Ng, X.R.; Zhuang, Z.; Wong, B.H.S.; Prasannan, P.; Kok, Y.J.; Bi, X.; Shim, H.; Wulff, H.; et al. Ex-tracellular K+ Dampens T Cell Functions: Implications for Immune Suppression in the Tumor Microenvironment. Bioelectricity 2019, 1, 169–179. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.F. Hypoxia and the Tumor Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 15330338211036304. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Pan, Z.; Di, S.; Shi, B.; Jiang, H.; Shi, Z.; Liu, Y.; Wang, Y.; Luo, H.; Yu, M.; Wu, X.; et al. Increased antitumor activities of glypican-3-specific chimeric antigen receptor-modified T cells by coexpression of a soluble PD1-CH3 fusion protein. Cancer Immunol. Immunother. 2018, 67, 1621–1634. [Google Scholar] [CrossRef]
- Prosser, M.E.; Brown, C.E.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef]
- Dong, C. Cytokine Regulation and Function in T Cells. Annu. Rev. Immunol. 2021, 39, 51–76. [Google Scholar] [CrossRef]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.F.; Bulsara, S.; et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Osman, A.; Yan, B.; Li, Y.; Pavelko, K.D.; Quandt, J.; Saadalla, A.; Singh, M.P.; Kazemian, M.; Gounari, F.; Khazaie, K. TCF-1 controls Treg cell functions that regulate inflammation, CD8+ T cell cytotoxicity and severity of colon cancer. Nat. Immunol. 2021, 22, 1152–1162. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 Inverted Cytokine Receptor Improving CAR-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front. Immunol. 2019, 10, 1691. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- McKenna, M.K.; Englisch, A.; Brenner, B.; Smith, T.; Hoyos, V.; Suzuki, M.; Brenner, M.K. Mesenchymal stromal cell delivery of oncolytic immunotherapy improves CAR-T cell antitumor activity. Mol. Ther. 2021, 29, 1808–1820. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, L.; Luo, Y.; Cai, Q.; Jiang, X.; Liao, C.; Wei, H. Optimization of GPC3-specific chimeric antigen receptor structure and its effect on killing hepatocellular carcinoma cells. Bioengineered 2021, 12, 3674–3683. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N. The Landscape of CAR-T Cell Clinical Trials against Solid Tumors-A Comprehensive Overview. Cancers 2020, 12, 2567. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, C.; Lu, P.; Lou, Y.; Liu, H.; Wang, T.; Yang, S.; Bao, Z.; Han, L.; Liang, X.; et al. Switch receptor T3/28 improves long-term persistence and antitumor efficacy of CAR-T cells. J. Immunother. Cancer 2021, 9, e003176. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Julamanee, J.; Terakura, S.; Umemura, K.; Adachi, Y.; Miyao, K.; Okuno, S.; Takagi, E.; Sakai, T.; Koyama, D.; Goto, T.; et al. Composite CD79A/CD40 co-stimulatory endodomain enhances CD19CAR-T cell proliferation and survival. Mol. Ther. 2021, 29, 2677–2690. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Z.; Ren, Z.; Li, Y. Reactions Related to CAR-T Cell Therapy. Front. Immunol. 2021, 12, 663201. [Google Scholar] [CrossRef]
- Yu, L.; Yang, X.; Huang, N.; Lang, Q.L.; He, Q.L.; Jian-Hua, W.; Liang-Peng, G. A novel targeted GPC3/CD3 bispecific antibody for the treatment hepatocellular carcinoma. Cancer Biol. Ther. 2020, 21, 597–603. [Google Scholar] [CrossRef]
- Du, K.; Li, Y.; Liu, J.; Chen, W.; Wei, Z.; Luo, Y.; Liu, H.; Qi, Y.; Wang, F.; Sui, J. A bispecific antibody targeting GPC3 and CD47 induced enhanced antitumor efficacy against dual antigen-expressing HCC. Mol. Ther. 2021, 29, 1572–1584. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Sun, L.; Guo, H.; Jiang, R.; Lu, L.; Liu, T.; He, X. Engineered cytotoxic T lymphocytes with AFP-specific TCR gene for adoptive im-munotherapy in hepatocellular carcinoma. Tumour Biol. 2016, 37, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Mizukoshi, E.; Kobayashi, E.; Tamai, T.; Hamana, H.; Ozawa, T.; Kishi, H.; Kitahara, M.; Yamashita, T.; Arai, K.; et al. Association Between High-Avidity T-Cell Receptors, Induced by α-Fetoprotein-Derived Peptides, and Anti-Tumor Effects in Patients with Hepatocellular Carcinoma. Gastroenterology 2017, 152, 1395–1406.e10. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Hanafi, L.A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Karschnia, P.; Jordan, J.T.; Forst, D.A.; Arrillaga-Romany, I.C.; Batchelor, T.T.; Baehring, J.M.; Clement, N.F.; Gonzalez Castro, L.N.; Herlopian, A.; Maus, M.V.; et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood 2019, 133, 2212–2221. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Xiong, W.; Yin, B.; Huang, Y.; Chu, J.; Xing, C.; Qian, C.; Du, Y.; Duan, T.; et al. Development of a TCR-like antibody and chimeric antigen receptor against NY-ESO-1/HLA-A2 for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004035. [Google Scholar] [CrossRef]
- Rajabzadeh, A.; Rahbarizadeh, F.; Ahmadvand, D.; Kabir Salmani, M.; Hamidieh, A.A. A VHH-Based Anti-MUC1 Chimeric An-tigen Receptor for Specific Retargeting of Human Primary T Cells to MUC1-Positive Cancer Cells. Cell J. 2021, 22, 502–513. [Google Scholar] [CrossRef]
- Zhai, X.; You, F.; Xiang, S.; Jiang, L.; Chen, D.; Li, Y.; Fan, S.; Han, Z.; Zhang, T.; An, G.; et al. MUC1-Tn-targeting chimeric antigen receptor-modified Vγ9Vδ2 T cells with enhanced antigen-specific anti-tumor activity. Am. J. Cancer Res. 2021, 11, 79–91. [Google Scholar]
- Shirasu, N.; Yamada, H.; Shibaguchi, H.; Kuroki, M.; Kuroki, M. Molecular characterization of a fully human chimeric T-cell antigen receptor for tumor-associated antigen EpCAM. J. Biomed. Biotechnol. 2012, 2012, 853879. [Google Scholar] [CrossRef]
- Pe’er, D.; Ogawa, S.; Elhanani, O.; Keren, L.; Oliver, T.G.; Wedge, D. Tumor heterogeneity. Cancer Cell. 2021, 39, 1015–1017. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Scholler, J.; Kubicka, E.; Bliemeister, E.T.; Schalk, D.L.; June, C.H.; Lum, L.G. Bispecific Antibody Armed Metabolically Enhanced Headless CAR T Cells. Front. Immunol. 2021, 12, 690437. [Google Scholar] [CrossRef]
- Lamers, C.H.; Willemsen, R.; van Elzakker, P.; van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef]
- Yusa, K. piggyBac Transposon. Microbiol. Spectr. 2015, 3, MDNA3-0028-2014. [Google Scholar] [CrossRef]
- Becklin, K.L.; Smeester, B.A.; Moriarity, B.S. Cancer Gene Discovery Utilizing Sleeping Beauty Transposon Mutagenesis. Methods Mol. Biol. 2019, 1907, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nano-body-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef]
- Tang, L.; Zheng, Y.; Melo, M.B.; Mabardi, L.; Castaño, A.P.; Xie, Y.Q.; Li, N.; Kudchodkar, S.B.; Wong, H.C.; Jeng, E.K.; et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 2018, 36, 707–716. [Google Scholar] [CrossRef]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef]
- Kallies, A.; Good-Jacobson, K.L. Transcription Factor T-bet Orchestrates Lineage Development and Function in the Immune System. Trends Immunol. 2017, 38, 287–297. [Google Scholar] [CrossRef]
- Sengupta, S.; Katz, S.C.; Sengupta, S.; Sampath, P. Glycogen synthase kinase 3 inhibition lowers PD-1 expression, promotes long-term survival and memory generation in antigen-specific CAR-T cells. Cancer Lett. 2018, 433, 131–139. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Limitations of CAR-T for HCC | Reasons | Available Strategies |
|---|---|---|
| Infiltration obstacles for CAR-T cells | Physical barrier of tumor stroma (extracellular matrix, disturbed vasculature), obstacles to T-cell penetration of blood vessels, lack of chemokine receptors or mismatch with chemokines | Design CAR-T cells expressing chemokines or chemokine receptors [9,10], pretreat with oncolytic virus expressing chemokines [11], local administration (within the tumor, etc.) [12,13], disruption of degraded extracellular matrix [14,15] |
| Inhibition of CAR T cell function and activity | Tumor microenvironment of HCC (Inhibitory receptor-ligand interaction, acid, hypoxia, oxidative stress, high potassium level, immunosuppression of regulatory T cells and myeloid-derived suppressor cells, negative regulation of cytokines), structural design flaws in CAR itself | Block PD-L1/PD-1 (expresses PD-1 antibody in CAR-T [5,16], modifies the sequence encoding PD-1 [17], develop switch-receptor of PD-1) [18], activate K channels, secrete catalase in CAR-T [19], develop antihypoxia systems [20,21], enhance expression of Nrf2 [22], secrete pro-inflammatory cytokines [23,24], consume regulatory T cells [25] |
| Viability and persistence of CAR-T cells | Immunogenicity of CAR-T cells themselves, nutritional competition for tumor cells, harsh tumor microenvironment | Increase T-bet expression [26], suppress the function of transforming growth factor-β [27], optimize the intracellular domain [28,29], use the DNA transposon system [30,31] |
| Antigen escape | Impairment of antigen-presenting ability, lack or downregulation of tumor-specific or associated antigens, lack of costimulatory molecules | Combinatorial targeted CAR-T cells [32,33], two or more CAR-T cells in combination |
| Tumor heterogeneity | Striking physiological and morphological heterogeneity of HCC | Co-express several CARs on a single T-cell (dual CAR-T), express a chimeric receptor with two or more antigen recognition domains (tandem CAR-T) [34], target tumor stem cell antigens [35,36] |
| Available Targets for Liver Cancer | Brief Introduction | Clinical Trials | Current Research Results in Liver Cancer | Reference |
|---|---|---|---|---|
| AFP (alpha-fetoprotein) | A glycoprotein, is usually overexpressed in HCC, germ cell neoplasms, pancreatic cancer, gastrointestinal cancer, or lung cancer | NCT03349255 (phase1) NCT03971747 (phase1) NCT04368182 (phase1) | Sun confirmed that engineered cytotoxic T cells with AFP158-166 specific TCR were effective against liver cancer [72]; 15 patients with liver cancer were enrolled in a phase 1 trial: AFP-derived peptide (AFP357 and AFP403) vaccine can induce the increase of AFP-specific T cells. No severe adverse events related to the drug were observed. 1 patient developed fever, which resolved spontaneously after 1 day, and 1 patient developed gastrointestinal symptoms. Injection-site reactions at the injection site occurred in almost all patients, but they were not severe. After treatment, 1 patient had a complete response, 8 had stable disease [73]. | [72,73] |
| GPC3 (glypican 3) | A 70 kDa heparan sulfate proteoglycan that is undetectable in the liver of healthy adults, but overexpression has been detected in patients with HCC, also expressed in adult tissues ovary, breast, mesothelium, lung and kidney | NCT02395250 (phase1) NCT03130712 (phase1, 2) NCT02715362 (phase 1, 2) NCT03084380 (phase1, 2) | GPC3 has been demonstrated in several trials (liver cancer cell lines, mouse tumor models and liver cancer patients) to be relatively safe and effective in killing tumor cells and reducing tumor size. However, it may cause side effects such as cytokine release syndrome [74], neurotoxicity [75], etc. | [74,75] |
| NY-ESO-1 (New York esophageal squamous cell carcinoma-1) | Expression is mainly restricted to testicular germ cells and placental trophoblast cells, no or low expression in normal adult somatic cells but ectopic expression in many tumor types | NCT03175705 (phase1) NCT03941626 (phase1, 2) | HLA-A2-restricted NY-ESO-1-specific T cell receptor engineered T cells can kill tumor cells, smaller tumor weight in mice after CAR-T cell treatment compared to control group (p = 0.0018) [76] | [76] |
| MUC1 (mucin-1) | A transmembrane glycoprotein, can expose epitopes that are normally hidden, is widely distributed and exceptionally abundant on the surface of cancer cells (breast cancer, stomach cancer, colon cancer, and liver cancer) | NCT02587689 (phase1, 2) NCT02839954 (phase1, 2) | Anti-MUC1 CAR based on variable domain of heavy chain of heavy chain antibody was effective against MUC1-positive cell lines [77,78] | [77,78] |
| EpCAM (epithelial cell adhesion molecule) | A 40 kDa type I transmembrane glycoprotein, contributes to a variety of biological processes, including cell adhesion, signaling, migration, and proliferation | NCT03013712 (phase1, 2) | CAR-T cells can be engineered to target EpCAM and show toxicity to antigen-positive tumor cells [79] | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, J.; Yang, D.; Ding, Y. Advances in Promoting the Efficacy of Chimeric Antigen Receptor T Cells in the Treatment of Hepatocellular Carcinoma. Cancers 2022, 14, 5018. https://doi.org/10.3390/cancers14205018
Shen J, Yang D, Ding Y. Advances in Promoting the Efficacy of Chimeric Antigen Receptor T Cells in the Treatment of Hepatocellular Carcinoma. Cancers. 2022; 14(20):5018. https://doi.org/10.3390/cancers14205018
Chicago/Turabian StyleShen, Jie, Dashuai Yang, and Youming Ding. 2022. "Advances in Promoting the Efficacy of Chimeric Antigen Receptor T Cells in the Treatment of Hepatocellular Carcinoma" Cancers 14, no. 20: 5018. https://doi.org/10.3390/cancers14205018
APA StyleShen, J., Yang, D., & Ding, Y. (2022). Advances in Promoting the Efficacy of Chimeric Antigen Receptor T Cells in the Treatment of Hepatocellular Carcinoma. Cancers, 14(20), 5018. https://doi.org/10.3390/cancers14205018