
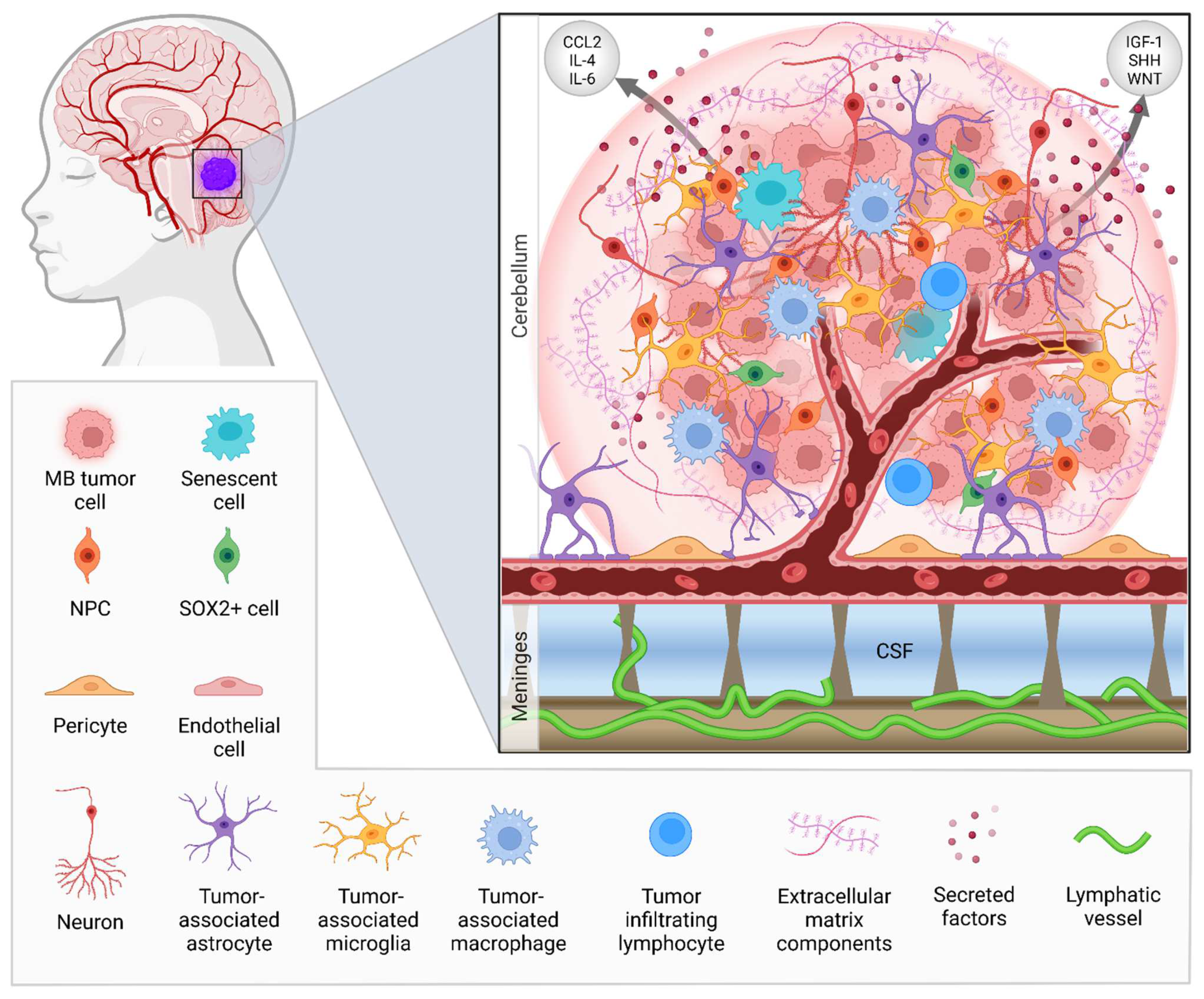
The Tumor Microenvironment of Medulloblastoma: An Intricate Multicellular Network with Therapeutic Potential


Abstract
Simple Summary
Abstract
1. Introduction
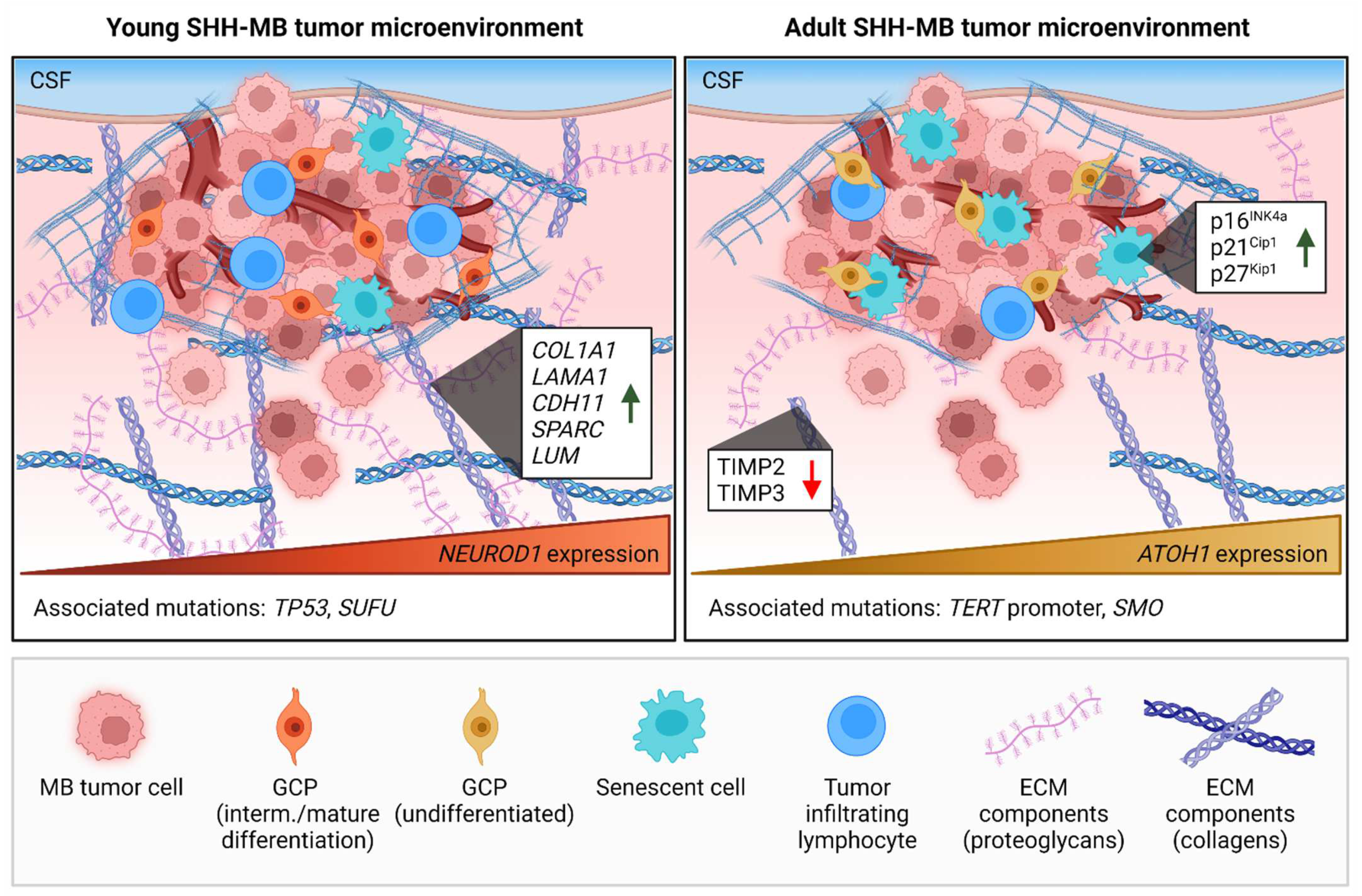
2. The Impact of Age on the Tumor Microenvironment of Brain Malignancies
Insights in TME Age-Related Differences in MB with Transcriptomics
3. The Extracellular Matrix Composition of Medulloblastoma
4. The Immune Cell Landscape of Medulloblastoma
Immunotherapy against Medulloblastoma
5. Involvement of Non-Hematopoietic CNS Cells in Medulloblastoma
5.1. Astrocytes in MB Tumor Progression and Relapse
5.2. Tumorigenic Activity of Neurons
6. Targeting the Brain Tumor Vasculature in Medulloblastoma
6.1. The Blood–Brain Barrier
6.2. Angiogenesis in MB
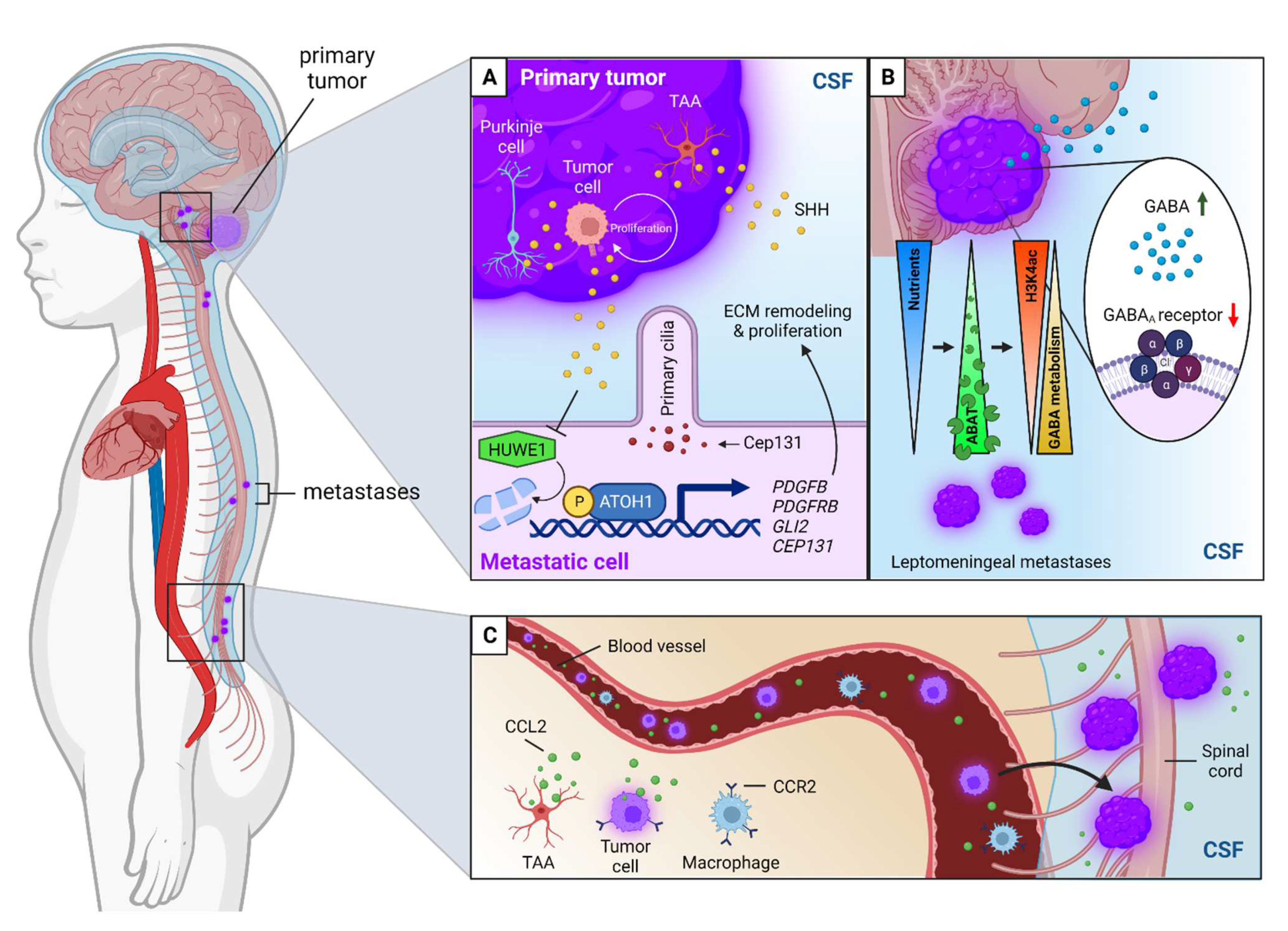
6.3. Cerebrospinal Fluid Circulation and the Lymphatic System
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Majd, N.; Penas-Prado, M. Updates on Management of Adult Medulloblastoma. Curr Treat. Options Oncol. 2019, 20, 64. [Google Scholar] [CrossRef]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754 e736. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Giangaspero, F.; Gyorffy, B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) medulloblastomas. J. Hematol. Oncol. 2019, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Riemondy, K.A.; Venkataraman, S.; Willard, N.; Nellan, A.; Sanford, B.; Griesinger, A.M.; Amani, V.; Mitra, S.; Hankinson, T.C.; Handler, M.H.; et al. Neoplastic and immune single-cell transcriptomics define subgroup-specific intra-tumoral heterogeneity of childhood medulloblastoma. Neuro. Oncol. 2022, 24, 273–286. [Google Scholar] [CrossRef]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.D.; Kumar, S.; Mack, S.C.; et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Schuller, U.; Heine, V.M.; Mao, J.; Kho, A.T.; Dillon, A.K.; Han, Y.G.; Huillard, E.; Sun, T.; Ligon, A.H.; Qian, Y.; et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 2008, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Ellis, T.; Markant, S.L.; Read, T.A.; Kessler, J.D.; Bourboulas, M.; Schuller, U.; Machold, R.; Fishell, G.; Rowitch, D.H.; et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 2008, 14, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Lin, G.N.; Song, W.; Zhang, Y.; Lai, H.; Zhang, M.; Miao, J.; Cheng, X.; Wang, Y.; Li, W.; et al. Single-cell spatial transcriptomic analysis reveals common and divergent features of developing postnatal granule cerebellar cells and medulloblastoma. BMC Biol. 2021, 19, 135. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.; Schwalbe, E.C.; Hicks, D.; Aldinger, K.A.; Lindsey, J.C.; Crosier, S.; Richardson, S.; Goddard, J.; Hill, R.M.; Castle, J.; et al. Medulloblastoma group 3 and 4 tumors comprise a clinically and biologically significant expression continuum reflecting human cerebellar development. Cell Rep. 2022, 40, 111162. [Google Scholar] [CrossRef]
- Hovestadt, V.; Smith, K.S.; Bihannic, L.; Filbin, M.G.; Shaw, M.L.; Baumgartner, A.; DeWitt, J.C.; Groves, A.; Mayr, L.; Weisman, H.R.; et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 2019, 572, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Adamski, J.; Bartels, U.; Tabori, U.; Wang, X.; Huang, A.; Hawkins, C.; Mabbott, D.; Laperriere, N.; et al. Medulloblastoma subgroup-specific outcomes in irradiated children: Who are the true high-risk patients? Neuro-oncology 2016, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Chevignard, M.; Camara-Costa, H.; Doz, F.; Dellatolas, G. Core deficits and quality of survival after childhood medulloblastoma: A review. Neurooncol Pract. 2017, 4, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.M.; Plasschaert, S.L.A.; Timmermann, B.; Dufour, C.; Aquilina, K.; Avula, S.; Donovan, L.; Lequin, M.; Pietsch, T.; Thomale, U.; et al. Relapsed Medulloblastoma in Pre-Irradiated Patients: Current Practice for Diagnostics and Treatment. Cancers 2021, 14, 126. [Google Scholar] [CrossRef]
- Hill, R.M.; Richardson, S.; Schwalbe, E.C.; Hicks, D.; Lindsey, J.C.; Crosier, S.; Rafiee, G.; Grabovska, Y.; Wharton, S.B.; Jacques, T.S.; et al. Time, pattern, and outcome of medulloblastoma relapse and their association with tumour biology at diagnosis and therapy: A multicentre cohort study. Lancet Child. Adolesc. Health 2020, 4, 865–874. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schuller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 2018, 7, e1462430. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, Y.; O’Brien, J.A.; Franco-Barraza, J.; Qi, X.; Yuan, H.; Jin, W.; Zhang, J.; Gu, C.; Zhao, Z.; et al. Necroptotic astrocytes contribute to maintaining stemness of disseminated medulloblastoma through CCL2 secretion. Neuro-oncology 2020, 22, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Grabovska, Y.; Mackay, A.; O’Hare, P.; Crosier, S.; Finetti, M.; Schwalbe, E.C.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Cockle, J.; et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 2020, 11, 4324. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, L.D.; Wetzel, M.D.; Marchetti, D. Heparanase Modulates Shh and Wnt3a Signaling in Human Medulloblastoma Cells. Exp. Ther. Med. 2011, 2, 229–238. [Google Scholar] [CrossRef]
- Yao, M.; Ventura, P.B.; Jiang, Y.; Rodriguez, F.J.; Wang, L.; Perry, J.S.A.; Yang, Y.; Wahl, K.; Crittenden, R.B.; Bennett, M.L.; et al. Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell 2020, 180, 502–520 e519. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Buhl, J.L.; Selt, F.; Hielscher, T.; Guiho, R.; Ecker, J.; Sahm, F.; Ridinger, J.; Riehl, D.; Usta, D.; Ismer, B.; et al. The Senescence-associated Secretory Phenotype Mediates Oncogene-induced Senescence in Pediatric Pilocytic Astrocytoma. Clin. Cancer Res. 2019, 25, 1851–1866. [Google Scholar] [CrossRef] [PubMed]
- Tamayo-Orrego, L.; Wu, C.L.; Bouchard, N.; Khedher, A.; Swikert, S.M.; Remke, M.; Skowron, P.; Taylor, M.D.; Charron, F. Evasion of Cell Senescence Leads to Medulloblastoma Progression. Cell Rep. 2016, 14, 2925–2937. [Google Scholar] [CrossRef] [PubMed]
- Pallavicini, G.; Sgro, F.; Garello, F.; Falcone, M.; Bitonto, V.; Berto, G.E.; Bianchi, F.T.; Gai, M.; Chiotto, A.M.A.; Filippi, M.; et al. Inactivation of Citron Kinase Inhibits Medulloblastoma Progression by Inducing Apoptosis and Cell Senescence. Cancer Res. 2018, 78, 4599–4612. [Google Scholar] [CrossRef] [PubMed]
- Smitherman, A.B.; Wood, W.A.; Mitin, N.; Ayer Miller, V.L.; Deal, A.M.; Davis, I.J.; Blatt, J.; Gold, S.H.; Muss, H.B. Accelerated aging among childhood, adolescent, and young adult cancer survivors is evidenced by increased expression of p16(INK4a) and frailty. Cancer 2020, 126, 4975–4983. [Google Scholar] [CrossRef]
- Lázničková, P.; Bendíčková, K.; Kepák, T.; Frič, J. Immunosenescence in Childhood Cancer Survivors and in Elderly: A Comparison and Implication for Risk Stratification. Front. Aging 2021, 2, 708788. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Parrinello, S.; Coppe, J.P.; Krtolica, A.; Campisi, J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef]
- Muhlisch, J.; Bajanowski, T.; Rickert, C.H.; Roggendorf, W.; Wurthwein, G.; Jurgens, H.; Fruhwald, M.C. Frequent but borderline methylation of p16 (INK4a) and TIMP3 in medulloblastoma and sPNET revealed by quantitative analyses. J. Neurooncol. 2007, 83, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Hielscher, T.; Dubuc, A.; Mack, S.; Shih, D.; Remke, M.; Al-Halabi, H.; Albrecht, S.; Jabado, N.; Eberhart, C.G.; et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011, 122, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Ozen, O.; Krebs, B.; Hemmerlein, B.; Pekrun, A.; Kretzschmar, H.; Herms, J. Expression of matrix metalloproteinases and their inhibitors in medulloblastomas and their prognostic relevance. Clin. Cancer Res. 2004, 10, 4746–4753. [Google Scholar] [CrossRef] [PubMed]
- Tamayo-Orrego, L.; Charron, F. Recent advances in SHH medulloblastoma progression: Tumor suppressor mechanisms and the tumor microenvironment. F1000Research 2019, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Accardi, G.; Monastero, R.; Nicoletti, F.; Libra, M. Ageing: From inflammation to cancer. Immun. Ageing 2018, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Immunosenescence and cancer. Biogerontology 2017, 18, 717–721. [Google Scholar] [CrossRef]
- Ocasio, J.; Babcock, B.; Malawsky, D.; Weir, S.J.; Loo, L.; Simon, J.M.; Zylka, M.J.; Hwang, D.; Dismuke, T.; Sokolsky, M.; et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 2019, 10, 5829. [Google Scholar] [CrossRef]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Skowron, P.; Farooq, H.; Cavalli, F.M.G.; Morrissy, A.S.; Ly, M.; Hendrikse, L.D.; Wang, E.Y.; Djambazian, H.; Zhu, H.; Mungall, K.L.; et al. The transcriptional landscape of Shh medulloblastoma. Nat. Commun. 2021, 12, 1749. [Google Scholar] [CrossRef]
- Kool, M.; Jones, D.T.; Jager, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.; Munoz, A.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Z.; Ma, Q.; Xu, Q.; Liu, H.; Duan, W.; Lei, J.; Ma, J.; Wang, X.; Lv, S.; et al. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin. Cancer Res. 2014, 20, 4326–4338. [Google Scholar] [CrossRef]
- Lam, D.; Enright, H.A.; Cadena, J.; Peters, S.K.G.; Sales, A.P.; Osburn, J.J.; Soscia, D.A.; Kulp, K.S.; Wheeler, E.K.; Fischer, N.O. Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Sci. Rep. 2019, 9, 4159. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, A.; Kundu, S.; Haseeb, L.; Yu, D.; Olofsson, T.; Dredge, K.; Hammond, E.; Barash, U.; Vlodavsky, I.; Forsberg-Nilsson, K. Inhibition of Heparanase in Pediatric Brain Tumor Cells Attenuates their Proliferation, Invasive Capacity, and In Vivo Tumor Growth. Mol. Cancer Ther. 2017, 16, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef] [PubMed]
- Clavreul, A.; Etcheverry, A.; Chassevent, A.; Quillien, V.; Avril, T.; Jourdan, M.L.; Michalak, S.; Francois, P.; Carre, J.L.; Mosser, J.; et al. Isolation of a new cell population in the glioblastoma microenvironment. J. Neurooncol. 2012, 106, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Rick, J.W.; Joshi, R.; Beniwal, A.; Spatz, J.; Chang, A.C.-C.; Nguyen, A.T.; Sudhir, S.; Chandra, A.; Haddad, A.; et al. Identification of Cancer-Associated Fibroblasts in Glioblastoma and Defining Their Pro-tumoral Effects. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, Z.; Zhuo, S.; He, G.; Tang, J.; Hao, W.; Gao, W.Q.; Yang, K.; Xu, H. Prognosis and Immunotherapy Significances of a Cancer-Associated Fibroblasts-Related Gene Signature in Gliomas. Front. Cell Dev. Biol. 2021, 9, 721897. [Google Scholar] [CrossRef]
- Trombetta-Lima, M.; Rosa-Fernandes, L.; Angeli, C.B.; Moretti, I.F.; Franco, Y.M.; Mousessian, A.S.; Wakamatsu, A.; Lerario, A.M.; Oba-Shinjo, S.M.; Pasqualucci, C.A.; et al. Extracellular Matrix Proteome Remodeling in Human Glioblastoma and Medulloblastoma. J. Proteome Res. 2021, 20, 4693–4707. [Google Scholar] [CrossRef]
- Larsen, A.M.H.; Kuczek, D.E.; Kalvisa, A.; Siersbaek, M.S.; Thorseth, M.L.; Johansen, A.Z.; Carretta, M.; Grontved, L.; Vang, O.; Madsen, D.H. Collagen Density Modulates the Immunosuppressive Functions of Macrophages. J. Immunol. 2020, 205, 1461–1472. [Google Scholar] [CrossRef]
- Pinto, M.L.; Rios, E.; Silva, A.C.; Neves, S.C.; Caires, H.R.; Pinto, A.T.; Duraes, C.; Carvalho, F.A.; Cardoso, A.P.; Santos, N.C.; et al. Decellularized human colorectal cancer matrices polarize macrophages towards an anti-inflammatory phenotype promoting cancer cell invasion via CCL18. Biomaterials 2017, 124, 211–224. [Google Scholar] [CrossRef]
- Stahl, M.; Schupp, J.; Jager, B.; Schmid, M.; Zissel, G.; Muller-Quernheim, J.; Prasse, A. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS ONE 2013, 8, e81382. [Google Scholar] [CrossRef]
- Bougherara, H.; Mansuet-Lupo, A.; Alifano, M.; Ngo, C.; Damotte, D.; Le Frere-Belda, M.A.; Donnadieu, E.; Peranzoni, E. Real-Time Imaging of Resident T Cells in Human Lung and Ovarian Carcinomas Reveals How Different Tumor Microenvironments Control T Lymphocyte Migration. Front. Immunol. 2015, 6, 500. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; de Fougerolles, A.R. Reprograming T cells: The role of extracellular matrix in coordination of T cell activation and migration. Curr. Opin. Immunol. 2001, 13, 286–290. [Google Scholar] [CrossRef]
- Gunzer, M.; Schäfer, A.; Borgmann, S.; Grabbe, S.; Zänker, K.S.; Bröcker, E.-B.; Kämpgen, E.; Friedl, P. Antigen Presentation in Extracellular Matrix: Interactions of T Cells with Dendritic Cells Are Dynamic, Short Lived, and Sequential. Immunity 2000, 13, 323–332. [Google Scholar] [CrossRef]
- Romer, A.M.A.; Thorseth, M.L.; Madsen, D.H. Immune Modulatory Properties of Collagen in Cancer. Front. Immunol. 2021, 12, 791453. [Google Scholar] [CrossRef]
- Linke, F.; Aldighieri, M.; Lourdusamy, A.; Grabowska, A.M.; Stolnik, S.; Kerr, I.D.; Merry, C.L.; Coyle, B. 3D hydrogels reveal medulloblastoma subgroup differences and identify extracellular matrix subtypes that predict patient outcome. J. Pathol. 2021, 253, 326–338. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T. Immune cell landscape and immunotherapy of medulloblastoma. Pediatr. Investig. 2021, 5, 299–309. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Pham, C.D.; Flores, C.; Yang, C.; Pinheiro, E.M.; Yearley, J.H.; Sayour, E.J.; Pei, Y.; Moore, C.; McLendon, R.E.; Huang, J.; et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin. Cancer Res. 2016, 22, 582–595. [Google Scholar] [CrossRef]
- Diao, S.; Gu, C.; Zhang, H.; Yu, C. Immune cell infiltration and cytokine secretion analysis reveal a non-inflammatory microenvironment of medulloblastoma. Oncol. Lett. 2020, 20, 397. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Margol, A.S.; Robison, N.J.; Gnanachandran, J.; Hung, L.T.; Kennedy, R.J.; Vali, M.; Dhall, G.; Finlay, J.L.; Erdreich-Epstein, A.; Krieger, M.D.; et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin. Cancer Res. 2015, 21, 1457–1465. [Google Scholar] [CrossRef]
- Dang, M.T.; Gonzalez, M.V.; Gaonkar, K.S.; Rathi, K.S.; Young, P.; Arif, S.; Zhai, L.; Alam, Z.; Devalaraja, S.; To, T.K.J.; et al. Macrophages in SHH subgroup medulloblastoma display dynamic heterogeneity that varies with treatment modality. Cell Rep. 2021, 34, 108917. [Google Scholar] [CrossRef] [PubMed]
- Maximov, V.; Chen, Z.; Wei, Y.; Robinson, M.H.; Herting, C.J.; Shanmugam, N.S.; Rudneva, V.A.; Goldsmith, K.C.; MacDonald, T.J.; Northcott, P.A.; et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat. Commun. 2019, 10, 2410. [Google Scholar] [CrossRef] [PubMed]
- Crotty, E.E.; Smith, S.M.C.; Brasel, K.; Pakiam, F.; Girard, E.J.; Connor, Y.D.; Zindy, F.; Mhyre, A.J.; Roussel, M.F.; Olson, J.M. Medulloblastoma recurrence and metastatic spread are independent of colony-stimulating factor 1 receptor signaling and macrophage survival. J. Neurooncol. 2021, 153, 225–237. [Google Scholar] [CrossRef]
- Tan, I.L.; Arifa, R.D.N.; Rallapalli, H.; Kana, V.; Lao, Z.; Sanghrajka, R.M.; Sumru Bayin, N.; Tanne, A.; Wojcinski, A.; Korshunov, A.; et al. CSF1R inhibition depletes tumor-associated macrophages and attenuates tumor progression in a mouse sonic Hedgehog-Medulloblastoma model. Oncogene 2021, 40, 396–407. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, Y.; Li, H.; Xu, L.; You, H.; Liu, Y.; Liu, Z.; Liu, X.; Zheng, D.; Bie, J.; et al. Exosomal microRNAs induce tumor-associated macrophages via PPARgamma during tumor progression in SHH medulloblastoma. Cancer Lett. 2022, 535, 215630. [Google Scholar] [CrossRef]
- Blaeschke, F.; Paul, M.C.; Schuhmann, M.U.; Rabsteyn, A.; Schroeder, C.; Casadei, N.; Matthes, J.; Mohr, C.; Lotfi, R.; Wagner, B.; et al. Low mutational load in pediatric medulloblastoma still translates into neoantigens as targets for specific T-cell immunotherapy. Cytotherapy 2019, 21, 973–986. [Google Scholar] [CrossRef]
- Hwang, E.I.; Sayour, E.J.; Flores, C.T.; Grant, G.; Wechsler-Reya, R.; Hoang-Minh, L.B.; Kieran, M.W.; Salcido, J.; Prins, R.M.; Figg, J.W.; et al. The current landscape of immunotherapy for pediatric brain tumors. Nat. Cancer 2022, 3, 11–24. [Google Scholar] [CrossRef]
- Vugmeyster, Y.; Grisic, A.M.; Brockhaus, B.; Rueckert, P.; Ruisi, M.; Dai, H.; Khandelwal, A. Avelumab Dose Selection for Clinical Studies in Pediatric Patients with Solid Tumors. Clin. Pharm. 2022, 61, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Loeb, D.M.; Lee, J.W.; Morgenstern, D.A.; Samson, Y.; Uyttebroeck, A.; Lyu, C.J.; Van Damme, A.; Nysom, K.; Macy, M.E.; Zorzi, A.P.; et al. Avelumab in paediatric patients with refractory or relapsed solid tumours: Dose-escalation results from an open-label, single-arm, phase 1/2 trial. Cancer Immunol. Immunother. 2022, 71, 2485–2495. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, I.J.; Cohen, K.; Foreman, N.K.; Hargrave, D.; Lassaletta, A.; André, N.; Hansford, J.R.; Hassall, T.; Eyrich, M.; Gururangan, S.; et al. IMMU-08. Nivolumab with or without ipilimumab in pediatric patients with high-grade CNS malignancies: Efficacy, safety, biomarker, and pharmacokinetic results from Checkmate 908. Neuro-Oncology 2022, 24, i82–i83. [Google Scholar] [CrossRef]
- Johnson, T.S.; Aguilera, D.; Al-Basheer, A.; Cooksey, R.M.; Eaton, B.R.; Esiashvili, N.; Firat, S.; Fiveash, J.B.; Foreman, N.; Fridlyand, D.; et al. PDCT-06. Radio-immunotherapy using the IDO-inhibitor indoximod in combination with re-irradiation for children with progressive brain tumors in the phase 1 setting: An updated report of safety and tolerability (NCT02502708). Neuro-Oncology 2017, 19, vi185. [Google Scholar] [CrossRef][Green Version]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Ode, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro-Oncology 2021, 23, 999–1011. [Google Scholar] [CrossRef]
- Thompson, E.; Landi, D.; Archer, G.; Lipp, E.; Walter, A.; Archambault, B.; Balajonda, B.; Flahiff, C.; Jaggers, D.; Herndon, J.; et al. EPCT-01. A novel peptide vaccine directed to CMV pp65 for treatment of recurrent malignant glioma and medulloblastoma in children and young adults: Preliminary results of a phase 1 trial. Neuro-Oncology 2021, 23, i46. [Google Scholar] [CrossRef]
- Yang, W.Q.; Senger, D.; Muzik, H.; Shi, Z.Q.; Johnson, D.; Brasher, P.M.A.; Rewcastle, N.B.; Hamilton, M.; Rutka, J.; Wolff, J.; et al. Reovirus Prolongs Survival and Reduces the Frequency of Spinal and Leptomeningeal Metastases from Medulloblastoma1. Cancer Res. 2003, 63, 3162–3172. [Google Scholar]
- Stolarek, R.; Gomez-Manzano, C.; Jiang, H.; Suttle, G.; Lemoine, M.G.; Fueyo, J. Robust infectivity and replication of Delta-24 adenovirus induce cell death in human medulloblastoma. Cancer Gene Ther. 2004, 11, 713–720. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Kreofsky, C.R.; Pierson, C.R.; Russell, S.J.; Galanis, E.; Raffel, C. Treatment of medulloblastoma with a modified measles virus. Neuro-oncology 2010, 12, 1034–1042. [Google Scholar] [CrossRef]
- Friedman, G.K.; Moore, B.P.; Nan, L.; Kelly, V.M.; Etminan, T.; Langford, C.P.; Xu, H.; Han, X.; Markert, J.M.; Beierle, E.A.; et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro-oncology 2016, 18, 227–235. [Google Scholar] [CrossRef]
- Schuelke, M.R.; Gundelach, J.H.; Coffey, M.; West, E.; Scott, K.; Johnson, D.R.; Samson, A.; Melcher, A.; Vile, R.G.; Bram, R.J. Phase I trial of sargramostim/pelareorep therapy in pediatric patients with recurrent or refractory high-grade brain tumors. Neurooncol. Adv. 2022, 4, vdac085. [Google Scholar] [CrossRef] [PubMed]
- Matyash, V.; Kettenmann, H. Heterogeneity in astrocyte morphology and physiology. Brain Res. Rev. 2010, 63, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Guo, D.; Zheng, C.; Ma, Z.; Zhang, J.; Qu, Y.; Li, X.; Li, G.; Zhang, L.; Wang, Y. Complement C3a activates astrocytes to promote medulloblastoma progression through TNF-alpha. J. Neuroinflamm. 2022, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yuelling, L.W.; Wang, Y.; Du, F.; Gordon, R.E.; O’Brien, J.A.; Ng, J.M.Y.; Robins, S.; Lee, E.H.; Liu, H.; et al. Astrocytes Promote Medulloblastoma Progression through Hedgehog Secretion. Cancer Res. 2017, 77, 6692–6703. [Google Scholar] [CrossRef]
- Cheng, Y.; Franco-Barraza, J.; Wang, Y.; Zheng, C.; Zhang, L.; Qu, Y.; Long, Y.; Cukierman, E.; Yang, Z.J. Sustained hedgehog signaling in medulloblastoma tumoroids is attributed to stromal astrocytes and astrocyte-derived extracellular matrix. Lab. Investig. 2020, 100, 1208–1222. [Google Scholar] [CrossRef]
- Cherry, J.D.; Meng, G.; Daley, S.; Xia, W.; Svirsky, S.; Alvarez, V.E.; Nicks, R.; Pothast, M.; Kelley, H.; Huber, B.; et al. CCL2 is associated with microglia and macrophage recruitment in chronic traumatic encephalopathy. J. Neuroinflamm. 2020, 17, 370. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Sun, P.; Ma, Z.; Sun, P. BRD4 promotes tumor progression and NF-kappaB/CCL2-dependent tumor-associated macrophage recruitment in GIST. Cell Death Dis. 2019, 10, 935. [Google Scholar] [CrossRef] [PubMed]
- Seike, T.; Fujita, K.; Yamakawa, Y.; Kido, M.A.; Takiguchi, S.; Teramoto, N.; Iguchi, H.; Noda, M. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin. Exp. Metastasis 2011, 28, 13–25. [Google Scholar] [CrossRef]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Doheny, D.; Zhu, D.; Aguayo, N.R.; Xing, F.; Chan, M.; Ruiz, J.; Metheny-Barlow, L.J.; et al. TGLI1 transcription factor mediates breast cancer brain metastasis via activating metastasis-initiating cancer stem cells and astrocytes in the tumor microenvironment. Oncogene 2020, 39, 64–78. [Google Scholar] [CrossRef]
- Gong, X.; Hou, Z.; Endsley, M.P.; Gronseth, E.I.; Rarick, K.R.; Jorns, J.M.; Yang, Q.; Du, Z.; Yan, K.; Bordas, M.L.; et al. Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-beta2/ANGPTL4 axes. NPJ Precis. Oncol. 2019, 3, 24. [Google Scholar] [CrossRef]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Swiderska-Syn, M.; Mir-Pedrol, J.; Oles, A.; Schleuger, O.; Salvador, A.D.; Greiner, S.M.; Seward, C.; Yang, F.; Babcock, B.R.; Shen, C.; et al. Noncanonical activation of GLI signaling in SOX2(+) cells drives medulloblastoma relapse. Sci. Adv. 2022, 8, eabj9138. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Murad, N.; Xu, Z.; Zhang, P.; Okonechnikov, K.; Kool, M.; Rivero-Hinojosa, S.; Lazarski, C.; Zheng, P.; Liu, Y.; et al. MYC Drives Group 3 Medulloblastoma through Transformation of Sox2(+) Astrocyte Progenitor Cells. Cancer Res. 2019, 79, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Wang, Y.; Cheng, Y.; Liao, S.; Hu, J.; Du, F.; Xu, G.; Liu, Y.; Cai, K.Q.; Cheung, M.; et al. Tumor cells generate astrocyte-like cells that contribute to SHH-driven medulloblastoma relapse. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Boue, S.; Belmonte, J.C.I. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef]
- Svalina, M.N.; Kikuchi, K.; Abraham, J.; Lal, S.; Davare, M.A.; Settelmeyer, T.P.; Young, M.C.; Peckham, J.L.; Cho, Y.J.; Michalek, J.E.; et al. IGF1R as a Key Target in High Risk, Metastatic Medulloblastoma. Sci. Rep. 2016, 6, 27012. [Google Scholar] [CrossRef]
- Srinivasan, E.S.; Deshpande, K.; Neman, J.; Winkler, F.; Khasraw, M. The microenvironment of brain metastases from solid tumors. Neurooncol. Adv. 2021, 3, v121–v132. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Korber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wissmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e31. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Hysinger, J.D.; Barron, T.; Schindler, N.F.; Cobb, O.; Guo, X.; Yalcin, B.; Anastasaki, C.; Mulinyawe, S.B.; Ponnuswami, A.; et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 2021, 594, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Lin, C.J.; Hatcher, A.; Lozzi, B.; Kong, K.; Huang-Hobbs, E.; Cheng, Y.T.; Beechar, V.B.; Zhu, W.; Zhang, Y.; et al. PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature 2020, 578, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Niesen, J.; Ohli, J.; Sedlacik, J.; Duhrsen, L.; Hellwig, M.; Spohn, M.; Holsten, T.; Schuller, U. Pik3ca mutations significantly enhance the growth of SHH medulloblastoma and lead to metastatic tumour growth in a novel mouse model. Cancer Lett. 2020, 477, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.C.; Li, K.K.; Wang, W.W.; Liu, A.P.; Huang, Q.J.; Chan, A.K.; Poon, M.F.; Chung, N.Y.; Wong, Q.H.; Chen, H.; et al. Clinical and mutational profiles of adult medulloblastoma groups. Acta Neuropathol. Commun. 2020, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef]
- Landry, J.P.; Schertz, K.L.; Chiang, Y.J.; Bhalla, A.D.; Yi, M.; Keung, E.Z.; Scally, C.P.; Feig, B.W.; Hunt, K.K.; Roland, C.L.; et al. Comparison of Cancer Prevalence in Patients With Neurofibromatosis Type 1 at an Academic Cancer Center vs in the General Population From 1985 to 2020. JAMA Netw. Open 2021, 4, e210945. [Google Scholar] [CrossRef]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef]
- O’Keeffe, E.; Campbell, M. Modulating the paracellular pathway at the blood-brain barrier: Current and future approaches for drug delivery to the CNS. Drug Discov. Today Technol. 2016, 20, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lee, H.; Fang, Z.; Velalopoulou, A.; Kim, J.; Thomas, M.B.; Liu, J.; Abramowitz, R.G.; Kim, Y.; Coskun, A.F.; et al. Single-cell analysis reveals effective siRNA delivery in brain tumors with microbubble-enhanced ultrasound and cationic nanoparticles. Sci. Adv. 2021, 7, eabf7390. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.J.; Liu, J.; Yu, B.; Malhotra, A.; Munson, J.; Park, J.C.; Wang, K.; Fei, B.; Bellamkonda, R.; Arbiser, J. Liposome-Imipramine Blue Inhibits Sonic Hedgehog Medulloblastoma In Vivo. Cancers 2021, 13, 1220. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Dismuke, T.; Malawsky, D.; Ramsey, J.D.; Hwang, D.; Godfrey, V.L.; Kabanov, A.V.; Gershon, T.R.; Sokolsky-Papkov, M. Enhancing CDK4/6 inhibitor therapy for medulloblastoma using nanoparticle delivery and scRNA-seq-guided combination with sapanisertib. Sci. Adv. 2022, 8, eabl5838. [Google Scholar] [CrossRef]
- Hwang, D.; Dismuke, T.; Tikunov, A.; Rosen, E.P.; Kagel, J.R.; Ramsey, J.D.; Lim, C.; Zamboni, W.; Kabanov, A.V.; Gershon, T.R.; et al. Poly(2-oxazoline) nanoparticle delivery enhances the therapeutic potential of vismodegib for medulloblastoma by improving CNS pharmacokinetics and reducing systemic toxicity. Nanomedicine 2021, 32, 102345. [Google Scholar] [CrossRef]
- Kumar, V.; Wang, Q.; Sethi, B.; Lin, F.; Kumar, V.; Coulter, D.W.; Dong, Y.; Mahato, R.I. Polymeric nanomedicine for overcoming resistance mechanisms in hedgehog and Myc-amplified medulloblastoma. Biomaterials 2021, 278, 121138. [Google Scholar] [CrossRef]
- Sajesh, B.V.; On, N.H.; Omar, R.; Alrushaid, S.; Kopec, B.M.; Wang, W.G.; Sun, H.D.; Lillico, R.; Lakowski, T.M.; Siahaan, T.J.; et al. Validation of Cadherin HAV6 Peptide in the Transient Modulation of the Blood-Brain Barrier for the Treatment of Brain Tumors. Pharmaceutics 2019, 11, 481. [Google Scholar] [CrossRef]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef]
- Pagnuzzi-Boncompagni, M.; Picco, V.; Vial, V.; Planas-Bielsa, V.; Vandenberghe, A.; Daubon, T.; Derieppe, M.A.; Montemagno, C.; Durivault, J.; Grepin, R.; et al. Antiangiogenic Compound Axitinib Demonstrates Low Toxicity and Antitumoral Effects against Medulloblastoma. Cancers 2021, 14, 70. [Google Scholar] [CrossRef]
- Schwinn, S.; Mokhtari, Z.; Thusek, S.; Schneider, T.; Siren, A.L.; Tiemeyer, N.; Caruana, I.; Miele, E.; Schlegel, P.G.; Beilhack, A.; et al. Cytotoxic effects and tolerability of gemcitabine and axitinib in a xenograft model for c-myc amplified medulloblastoma. Sci. Rep. 2021, 11, 14062. [Google Scholar] [CrossRef]
- Bai, R.Y.; Staedtke, V.; Rudin, C.M.; Bunz, F.; Riggins, G.J. Effective treatment of diverse medulloblastoma models with mebendazole and its impact on tumor angiogenesis. Neuro-oncology 2015, 17, 545–554. [Google Scholar] [CrossRef]
- Chan, T.S.Y.; Picard, D.; Hawkins, C.E.; Lu, M.; Pfister, S.; Korshunov, A.; Roussel, M.F.; Wechsler-Reya, R.J.; Henkin, J.; Bouffet, E.; et al. Thrombospondin-1 mimetics are promising novel therapeutics for MYC-associated medulloblastoma. Neurooncol. Adv. 2021, 3, vdab002. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, D.; Mazewski, C.; Fangusaro, J.; MacDonald, T.J.; McNall-Knapp, R.Y.; Hayes, L.L.; Kim, S.; Castellino, R.C. Response to bevacizumab, irinotecan, and temozolomide in children with relapsed medulloblastoma: A multi-institutional experience. Childs Nerv. Syst 2013, 29, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Piha-Paul, S.A.; Shin, S.J.; Vats, T.; Guha-Thakurta, N.; Aaron, J.; Rytting, M.; Kleinerman, E.; Kurzrock, R. Pediatric patients with refractory central nervous system tumors: Experiences of a clinical trial combining bevacizumab and temsirolimus. Anticancer Res. 2014, 34, 1939–1945. [Google Scholar] [PubMed]
- Chen, Z.; Tian, F.; Chen, X. Cost-Effectiveness Analysis of a Three-Drug Regimen Containing Bevacizumab for the Treatment of Recurrent Pediatric Medulloblastoma in China: Based on a COG Randomized Phase II Screening Trial. Front. Public Health 2022, 10, 914536. [Google Scholar] [CrossRef] [PubMed]
- Packer, R.J.; Rood, B.R.; Turner, D.C.; Stewart, C.F.; Fisher, M.; Smith, C.; Young-Pouissant, T.; Goldman, S.; Lulla, R.; Banerjee, A.; et al. Phase I and pharmacokinetic trial of PTC299 in pediatric patients with refractory or recurrent central nervous system tumors: A PBTC study. J. Neurooncol. 2015, 121, 217–224. [Google Scholar] [CrossRef][Green Version]
- Fults, D.W.; Taylor, M.D.; Garzia, L. Leptomeningeal dissemination: A sinister pattern of medulloblastoma growth. J. Neurosurg. Pediatr. 2019, 23, 613–621. [Google Scholar] [CrossRef]
- Zapotocky, M.; Mata-Mbemba, D.; Sumerauer, D.; Liby, P.; Lassaletta, A.; Zamecnik, J.; Krskova, L.; Kyncl, M.; Stary, J.; Laughlin, S.; et al. Differential patterns of metastatic dissemination across medulloblastoma subgroups. J. Neurosurg. Pediatr. 2018, 21, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Deng, Y.; Zhang, W. Molecular Determinants of Medulloblastoma Metastasis and Leptomeningeal Dissemination. Mol. Cancer Res. 2021, 19, 743–752. [Google Scholar] [CrossRef]
- Grausam, K.B.; Dooyema, S.D.R.; Bihannic, L.; Premathilake, H.; Morrissy, A.S.; Forget, A.; Schaefer, A.M.; Gundelach, J.H.; Macura, S.; Maher, D.M.; et al. ATOH1 Promotes Leptomeningeal Dissemination and Metastasis of Sonic Hedgehog Subgroup Medulloblastomas. Cancer Res. 2017, 77, 3766–3777. [Google Scholar] [CrossRef]
- Ayrault, O.; Zhao, H.; Zindy, F.; Qu, C.; Sherr, C.J.; Roussel, M.F. Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma-initiating cells. Cancer Res. 2010, 70, 5618–5627. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Zanini, M.; Shirvani, H.; Cheng, J.S.; Yu, H.; Feng, C.H.; Mercier, A.L.; Hung, S.Y.; Forget, A.; Wang, C.H.; et al. Atoh1 Controls Primary Cilia Formation to Allow for SHH-Triggered Granule Neuron Progenitor Proliferation. Dev. Cell 2019, 48, 184–199.e5. [Google Scholar] [CrossRef] [PubMed]
- Forget, A.; Bihannic, L.; Cigna, S.M.; Lefevre, C.; Remke, M.; Barnat, M.; Dodier, S.; Shirvani, H.; Mercier, A.; Mensah, A.; et al. Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Dev. Cell 2014, 29, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Martirosian, V.; Deshpande, K.; Zhou, H.; Shen, K.; Smith, K.; Northcott, P.; Lin, M.; Stepanosyan, V.; Das, D.; Remsik, J.; et al. Medulloblastoma uses GABA transaminase to survive in the cerebrospinal fluid microenvironment and promote leptomeningeal dissemination. Cell Rep. 2021, 35, 109302. [Google Scholar] [CrossRef]
- Kaushik, I.; Srivastava, S.K. GABAA receptor agonist suppresses pediatric medulloblastoma progression by inhibiting PKA-Gli1 signaling axis. Molecules 2022, 30, 2584–2602. [Google Scholar] [CrossRef]
- Garzia, L.; Kijima, N.; Morrissy, A.S.; De Antonellis, P.; Guerreiro-Stucklin, A.; Holgado, B.L.; Wu, X.; Wang, X.; Parsons, M.; Zayne, K.; et al. A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell 2018, 172, 1050–1062.e1014. [Google Scholar] [CrossRef]
- Low, S.Y.Y.; Bte Syed Sulaiman, N.; Tan, E.E.K.; Ng, L.P.; Kuick, C.H.; Chang, K.T.E.; Tang, P.H.; Wong, R.X.; Looi, W.S.; Low, D.C.Y.; et al. Cerebrospinal fluid cytokines in metastatic group 3 and 4 medulloblastoma. BMC Cancer 2020, 20, 554. [Google Scholar] [CrossRef]
- Lee, B.; Mahmud, I.; Pokhrel, R.; Murad, R.; Yuan, M.; Stapleton, S.; Bettegowda, C.; Jallo, G.; Eberhart, C.G.; Garrett, T.; et al. Medulloblastoma cerebrospinal fluid reveals metabolites and lipids indicative of hypoxia and cancer-specific RNAs. Acta Neuropathol. Commun. 2022, 10, 25. [Google Scholar] [CrossRef]
- Reichl, B.; Niederstaetter, L.; Boegl, T.; Neuditschko, B.; Bileck, A.; Gojo, J.; Buchberger, W.; Peyrl, A.; Gerner, C. Determination of a Tumor-Promoting Microenvironment in Recurrent Medulloblastoma: A Multi-Omics Study of Cerebrospinal Fluid. Cancers 2020, 12, 1350. [Google Scholar] [CrossRef]
- Engelhard, H.H.; Willis, A.J.; Hussain, S.I.; Papavasiliou, G.; Banner, D.J.; Kwasnicki, A.; Lakka, S.S.; Hwang, S.; Shokuhfar, T.; Morris, S.C.; et al. Etoposide-Bound Magnetic Nanoparticles Designed for Remote Targeting of Cancer Cells Disseminated Within Cerebrospinal Fluid Pathways. Front. Neurol. 2020, 11, 596632. [Google Scholar] [CrossRef]
- Hogan, B.M.; Bower, N.I. Lymphatics and the Brain: It’s Time to Go Fishing. Circ. Res. 2021, 128, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Cho, H.; Kim, J.H.; Kim, S.H.; Ham, J.S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.H.; Hong, Y.K.; et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 2019, 572, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Penco-Campillo, M.; Comoglio, Y.; Feliz Morel, A.J.; Hanna, R.; Durivault, J.; Leloire, M.; Mejias, B.; Pagnuzzi, M.; Morot, A.; Burel-Vandenbos, F.; et al. VEGFC negatively regulates the growth and aggressiveness of medulloblastoma cells. Commun. Biol. 2020, 3, 579. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| NCT | Phase | Treatment Modality | Drug | Target | Patient Enrollment | Age Group | Primary Outcome |
|---|---|---|---|---|---|---|---|
| NCT02359565 | 1 | ICI | Pembrolizumab | PD-1 | MB, EP, HGG, DIPG, HypBT | Pediatric, AYA | Safety, ORR, PD-1+ T-cell change |
| NCT02813135 | 1–2 | ICI | Nivolumab lirilumab | PD-1 and KIR2DL1/ KIR2L3 | MB, EP, HGG, DIPG | Pediatric | ORR, TTP |
| NCT03173950 | 2 | ICI | Nivolumab | PD-1 | MB, EP, CPC, A/MM, PRT | Adult | 6-month PFS, ORR |
| NCT02793466 | 1 | ICI | Durvalumab | PD-L1 | MB, EP, HGG, DIPG | Pediatric, AYA | Safety, MTD |
| NCT04049669 | 2 | ICI | Indoximod with radiation and chemotherapy | IDO | MB, EP, HGG, DIPG | Pediatric | 8-month PFS, 12-month OS |
| NCT05106296 | 1 | ICI and SMI | Indoximod with ibrutinib and chemotherapy | IDO and BTK (respectively) | MB, EP, HGG, PNET | Pediatric | ORR, toxicity |
| NCT03389802 | 1 | ICI | Agonistic monoclonal antibody APX005M | CD40 | MB, EP, HGG, DIPG | Pediatric | Safety, RP2D |
| NCT03500991 | 1 | ACT | Anti-HER2 CAR T-cells | HER2+ | MB, EP, HGG, CPC, PNET, ATRT | Pediatric, AYA | Safety and feasibility |
| NCT03638167 | 1 | ACT | Anti-EGFR806 CAR T-cells | EGFR+ | MB, EP, HGG, CPC, PNET, ATRT | Pediatric, AYA | Safety and feasibility |
| NCT03652545 | 1 | ACT | Tumor multi-antigen associated T-cells | PRAME+, WT1+ and/or BIRC5+ | MB, EP, HGG, CPC, DIPG | All | Safety and feasibility, MTD |
| NCT04099797 | 1 | ACT | Anti-GD2 CAR T-cells | GD2+ | MB, EP, HGG, DIPG | Pediatric | MTD |
| NCT05298995 | 1 | ACT | Anti-GD2 CAR T-cells | GD2+ | MB, HGG, DIPG | Pediatric, AYA | Safety, MTD |
| NCT04185038 | 1 | ACT | Anti-B7-H3 CAR T-cells | B7-H3+ | MB, EP, DIPG, PB, CPC, PNET, ATRT | Pediatric, AYA | Safety and feasibility |
| NCT04510051 | 1 | ACT | Anti-IL13Rα2 CAR T-cells | IL13Rα2+ | Recurrent/refractory brain tumors | Pediatric, AYA | Safety and feasibility |
| NCT04661384 | 1 | ACT | Anti-IL13Rα2 CAR T-cells | IL13Rα2+ | MB, EP, HGG | Adult | Safety and feasibility, 3-month OS |
| NCT05131763 | 1 | ACT | Anti-NKG2D CAR T-cells | NKG2DL+ | MB, HGG | Adult | Safety/toxicity |
| NCT01326104 | 2 | ACT | Chemotherapy followed by treatment with total tumor RNA-loaded dendritic cells | Patient- specific antigens | MB, PNET | Pediatric, AYA | 12-month PFS |
| NCT03299309 | 1 | Vaccine | PEP-CMV vaccine | CMV pp65 | MB, HGG | Pediatric, AYA | Safety |
| NCT05096481 | 2 | Vaccine | PEP-CMV vaccine | CMV pp65 | MB, HGG, DIPG | Pediatric | 4-month PFS |
| NCT04978727 | 1 | Vaccine | SurVaxM vaccine | Survivin (BIRC5) | MB, EP, HGG, DIPG, AA, AOD | Pediatric | Safety/toxicity |
| NCT02962167 | 1 | Oncolytic virus | Modified measles virus (MV-NIS) | Na+/I− symporter | MB, ATRT | Pediatric, AYA | Safety, RP2D |
| NCT03911388 | 1 | Oncolytic virus | Engineered HSV G207 | Cytopathic effect | MB, EP, HGG, PNET | Pediatric | Safety |
| NCT03043391 | 1 | Oncolytic virus | Recombinant polio/rhinovirus (PVSRIPO) | CD155 | MB, EP, HGG, ATRT, AA, AOA, AOD | AYA | Safety/toxicity |
| NCT02444546 | 1 | Oncolytic virus | Engineered wild-type reovirus with GM-CSF | Cytopathic effect | MB, HGG, DIPG, AA, AOD, ATRT, PNET | AYA | MTD |
| NCT04758533 | 1–2 | Oncolytic virus | Optimized adenovirus (ICOVIR-5) | pRB pathway | MB, DIPG | Pediatric | Safety, efficacy, MTD |
| NCT01356290 | 2 | MAB | Bevacizumab with chemotherapy | VEGF | MB, EP, ATRT | Pediatric | Efficacy |
| NCT04743661 | 2 | MAB | Bevacizumab with omburtamab and chemotherapy | VEGFB7-H3+ | MB, EP | Pediatric | 2-year EFS |
| NCT04501718 | 2 | SMI | Apatinib with chemotherapy | VEGFR-2 | Recurrent MB | Pediatric | ORR, PFS, OS |
| NCT03155620 | 2 | SMI | Erdafitinib | FGFR | Recurrent/refractory pediatric tumors with mutations | Pediatric | ORR |
| NCT03257631 | 2 | IMiD | Pomalidomide | COX-2 and cereblon | MB, EP, HGG, DIPG | Pediatric, AYA | ORR or long-term SD |
| NCT01661400 | 1 | IMiD | Thalidomide | COX-2 and cereblon | MB, EP, HGG, DIPG | Pediatric, AYA post-transplant | Safety, stem-cell transplant-related toxicity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Bree, N.F.H.N.; Wilhelm, M. The Tumor Microenvironment of Medulloblastoma: An Intricate Multicellular Network with Therapeutic Potential. Cancers 2022, 14, 5009. https://doi.org/10.3390/cancers14205009
van Bree NFHN, Wilhelm M. The Tumor Microenvironment of Medulloblastoma: An Intricate Multicellular Network with Therapeutic Potential. Cancers. 2022; 14(20):5009. https://doi.org/10.3390/cancers14205009
Chicago/Turabian Stylevan Bree, Niek F. H. N., and Margareta Wilhelm. 2022. "The Tumor Microenvironment of Medulloblastoma: An Intricate Multicellular Network with Therapeutic Potential" Cancers 14, no. 20: 5009. https://doi.org/10.3390/cancers14205009
APA Stylevan Bree, N. F. H. N., & Wilhelm, M. (2022). The Tumor Microenvironment of Medulloblastoma: An Intricate Multicellular Network with Therapeutic Potential. Cancers, 14(20), 5009. https://doi.org/10.3390/cancers14205009