
Sentinel Lymph Node Gene Expression Signature Predicts Recurrence-Free Survival in Cutaneous Melanoma

 , ,
, , 

Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Data Source and Study Population
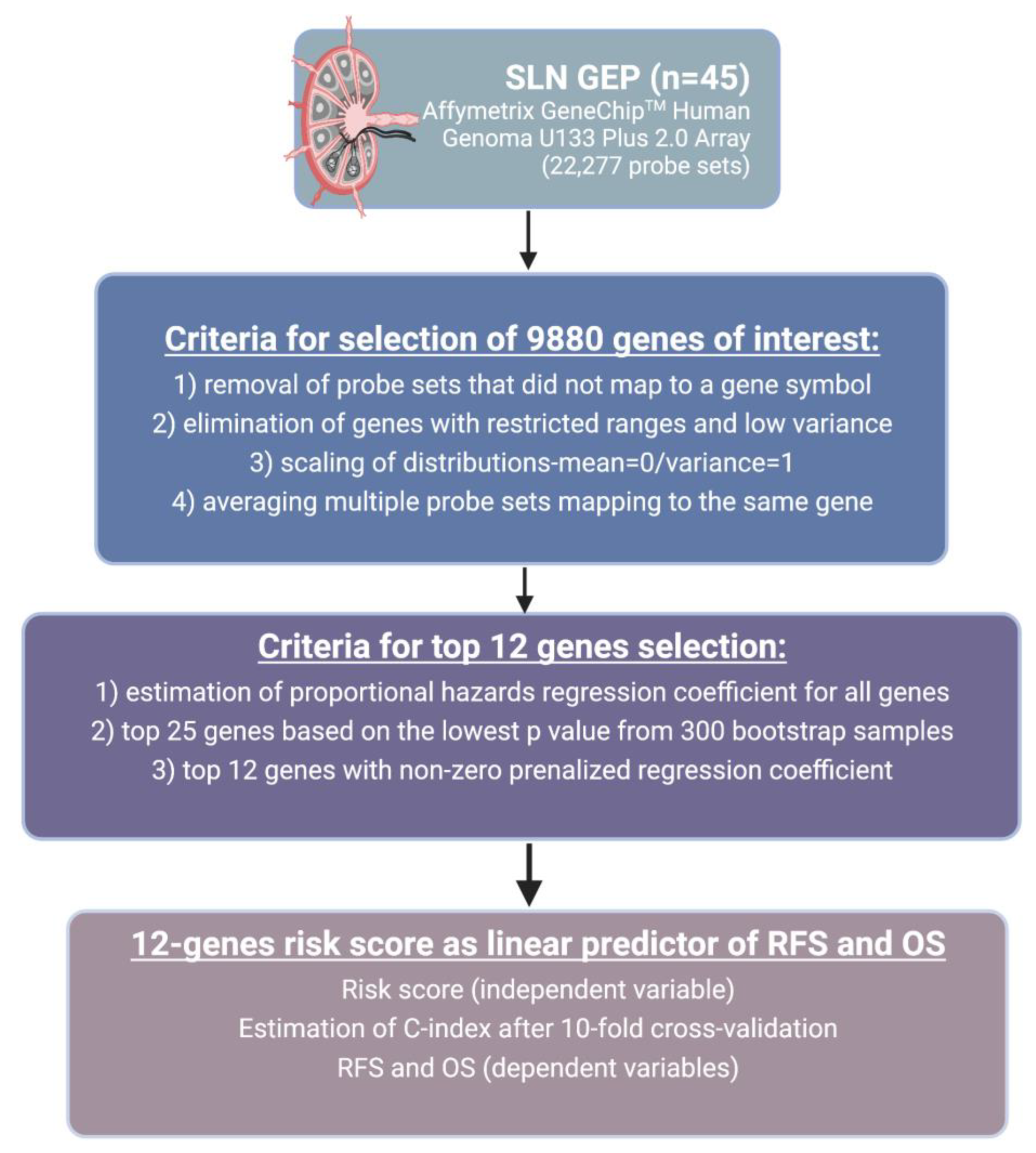
2.2. Data Analysis
2.3. Prognostic Role of 12-Gene Signature Score Using TCGA SKCM Cohort
2.4. Functional Enrichment Analysis
3. Results
3.1. Patients
3.2. Gene Expression Analysis
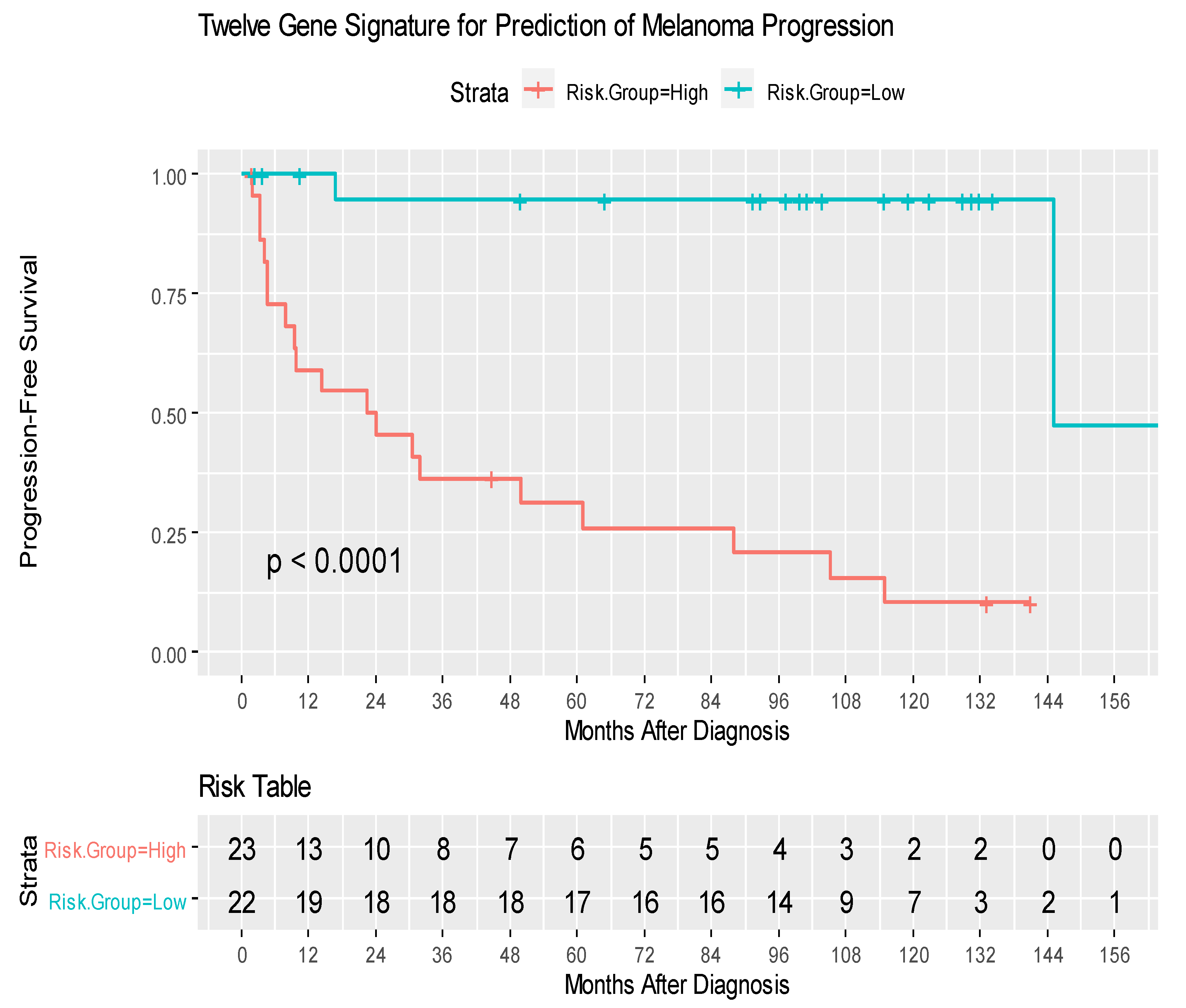
3.3. Prognostic Role of 12-Gene Signature Score Using TCGA SKCM Cohort
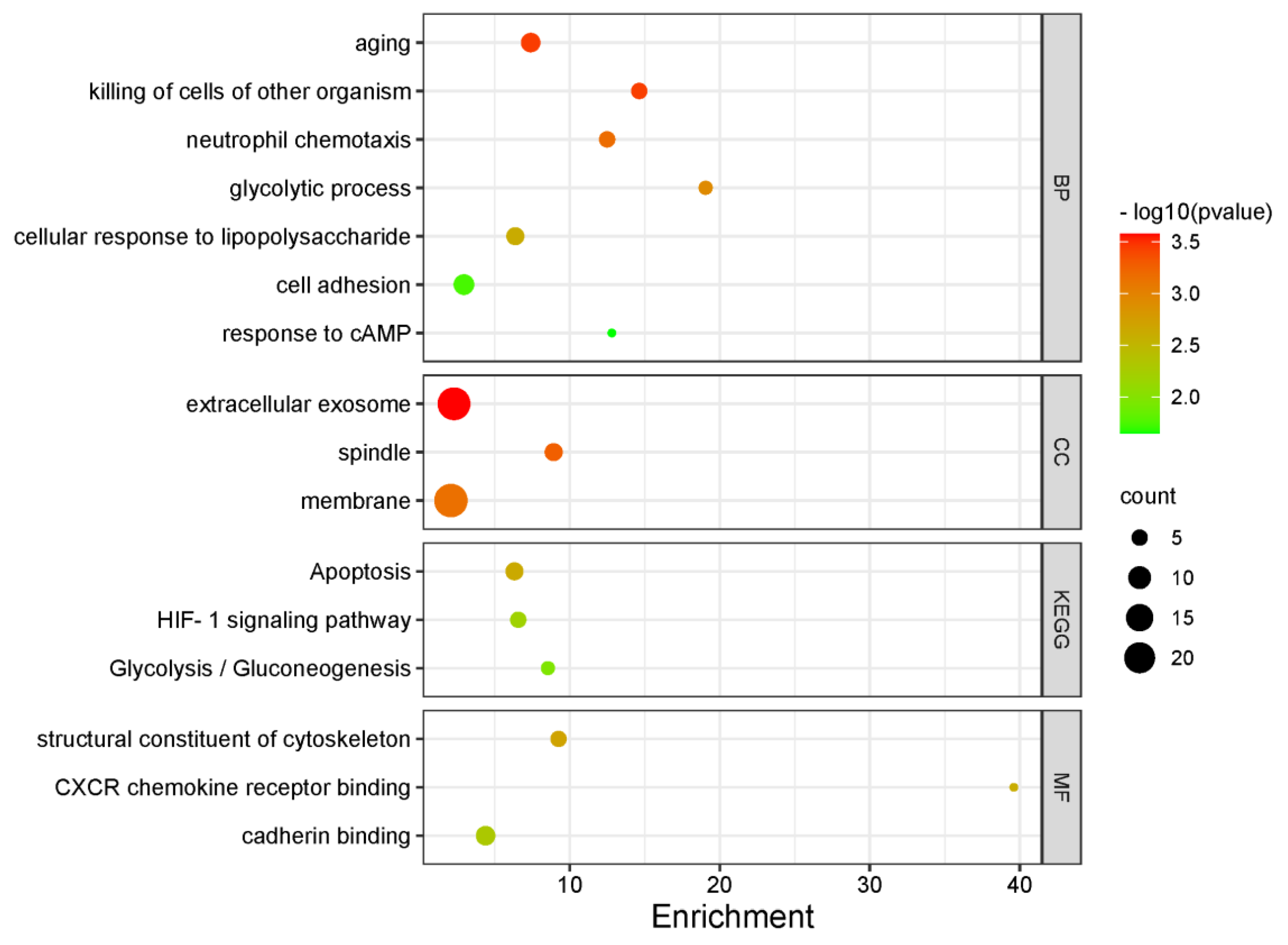
3.4. Functional Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SLN | sentinel lymph node |
| SLNB | sentinel lymph node biopsy |
| GEP | gene expression profiling |
| DEG | differentially expressed genes |
| RFS | recurrence-free survival |
| MSLT-II | Multicenter Selective Lymphadenectomy Trial-II |
| TIL | tumor infiltrating lymphocytes |
| ROC | Receiver Operating Characteristic |
| TCGA | The Cancer Genomic Atlas |
| RLN | Regional Lymph Nodes |
| OS | Overall Survival |
| GO | Gene Ontology |
| BP | Biological Processes |
| CC | Cellular Components |
| MF | Molecular Function |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef]
- Lee, J.H.; Torisu-Itakara, H.; Cochran, A.J.; Kadison, A.; Huynh, Y.; Morton, D.L.; Essner, R. Quantitative analysis of melanoma-induced cytokine-mediated immunosuppression in melanoma sentinel nodes. Clin. Cancer Res. 2005, 11, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Chen, Y.; Chan, J.L.; Qian, Y.W.; Goydos, J.S. Molecular analysis of melanoma-induced sentinel lymph node immune dysfunction. Cancer Immunol. Immunother. 2011, 60, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.L.; Thompson, J.F.; Cochran, A.J.; Mozzillo, N.; Elashoff, R.; Essner, R.; Nieweg, O.E.; Roses, D.F.; Hoekstra, H.J.; Karakousis, C.P.; et al. Sentinel-node biopsy or nodal observation in melanoma. N. Engl. J. Med. 2006, 355, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.L.; Wen, D.-R.; Wong, J.H.; Economou, J.S.; Cagle, L.A.; Storm, F.K.; Foshag, L.J.; Cochran, A.J. Technical details of intraoperative lymphatic mapping for early stage melanoma. Arch. Surg. 1992, 127, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.B.; Dueck, A.C.; Gray, R.J.; Wasif, N.; Swanson, D.L.; Sekulic, A.; Pockaj, B.A. Malignant melanoma in the elderly: Different regional disease and poorer prognosis. J. Cancer 2011, 2, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Ecker, B.L.; Kaur, A.; Douglass, S.M.; Webster, M.R.; Almeida, F.V.; Marino, G.E.; Sinnamon, A.J.; Neuwirth, M.G.; Alicea, G.M.; Ndoye, A.; et al. Age-related changes in HAPLN1 increase lymphatic permeability and affect routes of melanoma metastasis. Cancer Discov. 2019, 9, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Wong, S.L.; Edwards, M.J.; Ross, M.I.; Reintgen, D.S.; Noyes, R.D.; Stadelmann, W.K.; Lentsch, E.; McMasters, K.M.; for the Sunbelt Melanoma Trial Group. Sentinel lymph node biopsy for head and neck melanomas. Ann. Surg. Oncol. 2003, 10, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Fadaki, N.; Li, R.; Parrett, B.; Sanders, G.; Thummala, S.; Martineau, L.; Cardona-Huerta, S.; Miranda, S.; Cheng, S.T.; Miller, J.R., III; et al. Is head and neck melanoma different from trunk and extremity melanomas with respect to sentinel lymph node status and clinical outcome? Ann. Surg. Oncol. 2013, 20, 3089–3097. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.J.; Huang, R.-R.; Lee, J.; Itakura, E.; Leong, S.P.L.; Essner, R. Tumour—Induced immune modulation of sentinel lymph nodes. Nat. Rev. Immunol. 2006, 6, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Grotz, T.E.; Jakub, J.W.; Mansfield, A.S.; Goldenstein, R.; Enninga, E.A.; Nevala, W.K.; Leontovich, A.A.; Markovic, S.N. Evidence of Th2 polarization of the sentinel lymph node (SLN) in melanoma. Oncoimmunology 2015, 4, e1026504. [Google Scholar] [CrossRef] [PubMed]
- Mozzillo, N.; Pennacchioli, E.; Gandini, S.; Caracò, C.; Crispo, A.; Botti, G.; Lastoria, S.; Barberis, M.; Verrecchia, F.; Testori, A. Sentinel node biopsy in thin and thick melanoma. Ann. Surg. Oncol. 2013, 20, 2780–2786. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Long, G.V.; Khattak, M.A.; de La Cruz Merino, L.; Del Vecchio, M.; Spagnolo, F. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): A randomised, double-blind, phase 3 trial. Lancet 2021, 399, 1718–1729. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Floros, T.; Lin, H.M.; Lin, Y.; Rahman, Z.; Ashraf, M.; Vallabhaneni, P.; Sander, C.; Rao, U.N.M.; Panelli, M.; et al. A unique gene expression signature is significantly differentially expressed in tumor-positive or tumor-negative sentinel lymph nodes in patients with melanoma. Melanoma Res. 2017, 27, 429–438. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216. [Google Scholar] [CrossRef]
- Schroer, T.A. Dynactin. Annu. Rev. Cell Dev. Biol. 2004, 20, 759–779. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, X.; Liang, Q.; Wang, S.; Liao, X.; Li, D.; Pan, F. Prognostic value of dynactin mRNA expression in cutaneous melanoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 3752. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, Y.; Zhu, X.; Mo, Z. Prediction of survival outcome in lower-grade glioma using a prognostic signature with 33 immune-related gene pairs. Int. J. Gen. Med. 2021, 14, 8149. [Google Scholar] [CrossRef] [PubMed]
- Iyevleva, A.G.; Raskin, G.A.; Tiurin, V.I.; Sokolenko, A.P.; Mitiushkina, N.V.; Aleksakhina, S.N.; Garifullina, A.R.; Strelkova, T.N.; Merkulov, V.O.; Ivantsov, A.O. Novel ALK fusion partners in lung cancer. Cancer Lett. 2015, 362, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Ross, O.A.; Teive, H.A.; Sławek, J.; Dickson, D.W.; Wszolek, Z.K. DCTN1-related neurodegeneration: Perry syndrome and beyond. Park. Relat. Disord. 2017, 41, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Cresswell, J.; Löffler, M.G.; Bogan, J.S. The glucose transporter 4-regulating protein TUG is essential for highly insulin-responsive glucose uptake in 3T3-L1 adipocytes. J. Biol. Chem. 2007, 282, 7710–7722. [Google Scholar] [CrossRef]
- Kobos, R.; Nagai, M.; Tsuda, M.; Merl, M.Y.; Saito, T.; Laé, M.; Mo, Q.; Olshen, A.; Lianoglou, S.; Leslie, C. Combining integrated genomics and functional genomics to dissect the biology of a cancer-associated, aberrant transcription factor, the ASPSCR1–TFE3 fusion oncoprotein. J. Pathol. 2013, 229, 743–754. [Google Scholar] [CrossRef]
- Butler, M.; Sanmugalingam, D.; Burton, V.J.; Wilson, T.; Pearson, R.; Watson, R.P.; Smith, P.; Parkinson, S.J. Impairment of adenosine A3 receptor activity disrupts neutrophil migratory capacity and impacts innate immune function in vivo. Eur. J. Immunol. 2012, 42, 3358–3368. [Google Scholar] [CrossRef]
- Merighi, S.; Varani, K.; Gessi, S.; Cattabriga, E.; Iannotta, V.; Ulouglu, C.; Leung, E.; Borea, P.A. Pharmacological and biochemical characterization of adenosine receptors in the human malignant melanoma A375 cell line. Br. J. Pharmacol. 2001, 134, 1215–1226. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K. A3 adenosine receptors as modulators of inflammation: From medicinal chemistry to therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef]
- Madi, L.; Ochaion, A.; Rath-Wolfson, L.; Bar-Yehuda, S.; Erlanger, A.; Ohana, G.; Harish, A.; Merimski, O.; Barer, F.; Fishman, P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin. Cancer Res. 2004, 10, 4472–4479. [Google Scholar] [CrossRef]
- Antonioli, L.; Csóka, B.; Fornai, M.; Colucci, R.; Kókai, E.; Blandizzi, C.; Haskó, G. Adenosine and inflammation: What’s new on the horizon? Drug Discov. Today 2014, 19, 1051–1068. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Varani, K.; Merighi, S.; Cattabriga, E.; Avitabile, A.; Gavioli, R.; Fortini, C.; Leung, E.; Mac Lennan, S.; Borea, P.A. Expression of A3 adenosine receptors in human lymphocytes: Up-regulation in T cell activation. Mol. Pharmacol. 2004, 65, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; MacLennan, S.; Baraldi, P.G.; Borea, P.A. A3 adenosine receptors modulate hypoxia-inducible factor-1a expression in human a375 melanoma cells. Neoplasia 2005, 7, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Wang, D.; Xu, S.; Lee, C.A.; Zhen, E.; Yoon, C.H.; Abarzua, P.; Wang, S.; Frank, N.Y.; Wu, X. ATF-3 expression inhibits melanoma growth by downregulating ERK and AKT pathways. Lab. Investig. 2021, 101, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, S.; Mou, Z.; Zhou, Q.; Zhang, Z.; Chen, Y.; Ou, Y.; Chen, X.; Dai, X.; Xu, C. Transcriptomic analysis identified ARHGAP family as a novel biomarker associated with tumor-promoting immune infiltration and nanomechanical characteristics in bladder cancer. Front. Cell Dev. Biol. 2021, 9, 657219. [Google Scholar] [CrossRef]
- Johnstone, C.N.; Castellví-Bel, S.; Chang, L.M.; Bessa, X.; Nakagawa, H.; Harada, H.; Sung, R.K.; Piqué, J.M.; Castells, A.; Rustgi, A.K. ARHGAP8 is a novel member of the RHOGAP family related to ARHGAP1/CDC42GAP/p50RHOGAP: Mutation and expression analyses in colorectal and breast cancers. Gene 2004, 336, 59–71. [Google Scholar] [CrossRef]
- Song, J.; Lee, J.; Lee, N.; Jung, H.; Kim, S.; Lee, K. Microarray analysis of normal cervix, carcinoma in situ, and invasive cervical cancer: Identification of candidate genes in pathogenesis of invasion in cervical cancer. Int. J. Gynecol. Cancer 2008, 18, 1051–1059. [Google Scholar] [CrossRef]
- Ikawa, M.; Wada, I.; Kominami, K.; Watanabe, D.; Toshimori, K.; Nishimune, Y.; Okabe, M. The putative chaperone calmegin is required for sperm fertility. Nature 1997, 387, 607–611. [Google Scholar] [CrossRef]
- Dugué, P.A.; Dowty, J.G.; Joo, J.E.; Wong, E.M.; Makalic, E.; Schmidt, D.F.; English, D.R.; Hopper, J.L.; Pedersen, J.; Severi, G. Heritable methylation marks associated with breast and prostate cancer risk. Prostate 2018, 78, 962–969. [Google Scholar] [CrossRef]
- Li, Y.; Wright, G.L.; Peterson, J.M. C1q/TNF-related protein 3 (CTRP3) function and regulation. Compr. Physiol. 2017, 7, 863. [Google Scholar]
- Schäffler, A.; Ehling, A.; Neumann, E.; Herfarth, H.; Paul, G.; Tarner, I.; Gay, S.; Schölmerich, J.; Müller-Ladner, U. Genomic organization, promoter, amino acid sequence, chromosomal localization, and expression of the human gene for CORS-26 (collagenous repeat-containing sequence of 26-kDa protein). Biochim. Biophys. Acta BBA-Gene Struct. Expr. 2003, 1630, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; de Souza, J.V.; Ahmad, A.; Bronowska, A.K. Structure, dynamics, and ligand recognition of human-specific CHRFAM7A (Dupα7) nicotinic receptor linked to neuropsychiatric disorders. Int. J. Mol. Sci. 2021, 22, 5466. [Google Scholar] [CrossRef] [PubMed]
- Bordas, A.; Cedillo, J.L.; Arnalich, F.; Esteban-Rodriguez, I.; Guerra-Pastrián, L.; de Castro, J.; Martín-Sánchez, C.; Atienza, G.; Fernández-Capitan, C.; Rios, J.J. Expression patterns for nicotinic acetylcholine receptor subunit genes in smoking-related lung cancers. Oncotarget 2017, 8, 67878. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Chao, S.-H.; Lane, D. HEXIM1 and the control of transcription elongation: From cancer and inflammation to AIDS and cardiac hypertrophy. Cell Cycle 2007, 6, 1856–1863. [Google Scholar] [CrossRef]
- Ok Atılgan, A.; Özdemir, B.H.; Akçay, E.Y.; Ataol Demirkan, Ö.; Tekindal, M.A.; Özkardeş, H. Role of tumor-associated macrophages in the Hexim1 and TGFβ/SMAD pathway, and their influence on progression of prostatic adenocarcinoma. Pathol.-Res. Pract. 2016, 212, 83–92. [Google Scholar] [CrossRef]
- Smebye, M.L.; Sveen, A.; Haugom, L.; Davidson, B.; Trope, C.G.; Lothe, R.A.; Heim, S.; Skotheim, R.I.; Micci, F. Chromosome 19 rearrangements in ovarian carcinomas: Zinc finger genes are particularly targeted. Genes Chromosomes Cancer 2014, 53, 558–567. [Google Scholar] [CrossRef]
- Dvir, L.; Srour, G.; Abu-Ras, R.; Miller, B.; Shalev, S.A.; Ben-Yosef, T. Autosomal-recessive early-onset retinitis pigmentosa caused by a mutation in PDE6G, the gene encoding the gamma subunit of rod cGMP phosphodiesterase. Am. J. Hum. Genet. 2010, 87, 258–264. [Google Scholar] [CrossRef]
- López-Lago, M.A.; Posner, S.; Thodima, V.J.; Molina, A.M.; Motzer, R.J.; Chaganti, R.S. Neutrophil chemokines secreted by tumor cells mount a lung antimetastatic response during renal cell carcinoma progression. Oncogene 2013, 32, 1752–1760. [Google Scholar] [CrossRef]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- Panelli, M.C.; Wang, E.; Phan, G.; Puhlmann, M.; Miller, L.; Ohnmacht, G.A.; Klein, H.G.; Marincola, F.M. Gene-expression profiling of the response of peripheral blood mononuclear cells and melanoma metastases to systemic IL-2 administration. Genome Biol. 2002, 3, research0035.1. [Google Scholar] [CrossRef]
- Watts, C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat. Immunol. 2004, 5, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Blair, S.L.; Burstein, H.J.; Dang, C.; Elias, A.D.; et al. NCCN guidelines® insights: Breast cancer, version 4.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Sivendran, S.; Chang, R.; Pham, L.; Phelps, R.G.; Harcharik, S.T.; Hall, L.D.; Bernardo, S.G.; Moskalenko, M.M.; Sivendran, M.; Fu, Y.; et al. Dissection of immune gene networks in primary melanoma tumors critical for antitumor surveillance of patients with stage II–III resectable disease. J. Investig. Dermatol. 2014, 134, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Greenhaw, B.N.; Covington, K.R.; Kurley, S.J.; Yeniay, Y.; Cao, N.A.; Plasseraud, K.M.; Cook, R.W.; Hsueh, E.C.; Gastman, B.R.; Wei, M.L. Molecular risk prediction in cutaneous melanoma: A meta-analysis of the 31-gene expression profile prognostic test in 1479 patients. J. Am. Acad. Dermatol. 2020, 83, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Gastman, B.R.; Gerami, P.; Kurley, S.J.; Cook, R.W.; Leachman, S.; Vetto, J.T. Identification of patients at risk of metastasis using a prognostic 31-gene expression profile in subpopulations of melanoma patients with favorable outcomes by standard criteria. J. Am. Acad. Dermatol. 2019, 80, 149–157.e144. [Google Scholar] [CrossRef]
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; van der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin. Cancer Res. 2016, 22, 1234–1242. [Google Scholar] [CrossRef]
- Farrow, N.E.; Holl, E.K.; Jung, J.; Gao, J.; Jung, S.H.; Al-Rohil, R.N.; Selim, M.A.; Mosca, P.J.; Ollila, D.W.; Antonia, S.J.; et al. Characterization of sentinel lymph node immune signatures and implications for risk stratification for adjuvant therapy in melanoma. Ann. Surg. Oncol. 2021, 28, 3501–3510. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Number (%) |
|---|---|
| Median Age at Diagnosis (range) | 56 (16–81) |
| Gender | |
| Male | 27 (60%) |
| Female | 18 (40%) |
| Breslow Score median (IQR) | 4.1 (3.3–6.0) |
| Ulceration | |
| Yes | 29 (67%) |
| No | 14 (33%) |
| TIL | |
| Absent | 7 (16%) |
| Brisk | 5 (11%) |
| Non-Brisk | 20 (44%) |
| Unknown | 13 (29%) |
| Sentinel Lymph Node Status | |
| Positive | 23 (51%) |
| Negative | 22 (49%) |
| Number of positive SLN | |
| 1 | 19 (83%) |
| 2 | 4 (17%) |
| Extracapsular Extension | |
| Yes | 7 (30) |
| No | 16 (70) |
| Location | |
| Head and Neck | 5 (11%) |
| Trunk | 18 (40%) |
| Upper Extremities | 12 (27%) |
| Lower Extremities | 10 (22%) |
| Histology | |
| Acral Lentiginous | 4 (9%) |
| Nodular | 25 (56%) |
| Superficial Spreading | 8 (18%) |
| NOS | 8 (18%) |
| Molecular Testing | |
| BRAF mutant | 10(50%) |
| NRAS mutant | 6 (30%) |
| NF mutant | 0 (0) |
| Triple wild type | 4 (20%) |
| Gene | HR | L95 | U95 | Penalized Coefficient |
|---|---|---|---|---|
| CLGN | 0.25 | 0.13 | 0.50 | −0.211 |
| C1QTNF3 | 0.27 | 0.14 | 0.52 | −0.236 |
| ADORA3 | 0.19 | 0.08 | 0.45 | −0.160 |
| ARHGAP8 | 0.27 | 0.14 | 0.54 | −0.067 |
| DCTN1 | 4.24 | 1.86 | 9.68 | 0.112 |
| ASPSCR1 | 2.30 | 1.42 | 3.74 | 0.149 |
| CHRFAM7A | 0.37 | 0.20 | 0.66 | −0.112 |
| ZNF223 | 0.35 | 0.19 | 0.66 | −0.004 |
| PDE6G | 0.21 | 0.08 | 0.53 | −0.058 |
| CXCL3 | 0.45 | 0.27 | 0.73 | −0.096 |
| HEXIM1 | 2.97 | 1.45 | 6.09 | 0.187 |
| HLA-DRB | 0.60 | 0.42 | 0.86 | −0.124 |
| Covariate | Reference | Hazard Ratio | 95 % CI | p Value |
|---|---|---|---|---|
| Gene Signature | −0.72 to + 0.48 * | 30.07 | 8.12–111.24 | <0.0001 |
| Breslow thickness | 3.3–6.0 | 1.41 | 1.14–1.74 | 0.0014 |
| Number of positive SLNs | 0–2 | 6.01 | 1.52–23.80 | 0.0106 |
| Histology | Nodular | 0.2549 | ||
| Acral lentiginous | 4.07 | 1.00–16.47 | ||
| NOS | 1.24 | 0.38–4.04 | ||
| Superficial spreading | 1.02 | 0.31–3.33 | ||
| Ulceration | No | 3.61 | 1.05–12.41 | 0.0419 |
| SLN status | Negative | 2.5 | 1.01–6.21 | 0.0485 |
| TIL | Absent | 0.3093 | ||
| Brisk | 0.64 | 0.11–3.86 | ||
| Non brisk | 1.61 | 0.45–5.76 | ||
| Unknown | 0.61 | 0.14–2.74 | ||
| Location | Trunk | 0.0764 | ||
| HN | 2.64 | 0.65–10.62 | ||
| LE | 2.32 | 0.74–7.32 | ||
| UE | 1.38 | 0.42–4.54 | ||
| Sex | Male | 1.21 | 0.49–2.96 | 0.6817 |
| Age at Diagnosis | 49–66 | 1.93 | 0.97–3.81 | 0.0592 |
| Class | Terms | Genes | Enrichment | p Value |
|---|---|---|---|---|
| BP | aging | IL10, VCAM1, ELAVL4, TSPO, TIMP1, PAX5, CD68 | 7.39 | 0.0004 |
| killing of cells of other organism | CXCL1, CXCL3, GAPDH, CXCL2, LTF | 14.63 | 0.0004 | |
| neutrophil chemotaxis | CXADR, BSG, CXCL1, CXCL3, CXCL2 | 12.49 | 0.0007 | |
| glycolytic process | LDHA, ALDOA, GAPDH, PFKM | 19.06 | 0.0012 | |
| cellular response to lipopolysaccharide | IL10, TSPO, CXCL1, CD68, CXCL3, CXCL2 | 6.37 | 0.0024 | |
| cell adhesion | LGALS3BP, COL1A1, CLDN10, VCAM1, SPECC1L, BSG, FOLR2, SIGLEC7 | 2.94 | 0.0182 | |
| response to cAMP | COL1A1, LDHA, BSG | 12.80 | 0.0225 | |
| CC | extracellular exosome | LGALS3BP, PLVAP, VCAM1, GOT2, HSPB1, LIFR, SNF8, LTBP3, C1QTNF3, LDHA, TUBA1A, TUBB2A, FXYD2, BSG, TSPO, MYH9, TIMP1, ALDOA, GAPDH, EPHB1, HLA-DRB1, EIF3B, LTF | 2.28 | 0.0003 |
| spindle | DCTN1, SPECC1L, MYH9, HSPB1, KATNB1, AURKC | 8.92 | 0.0005 | |
| membrane | LGALS3BP, GRAMD1B, VCAM1, DCTN1, PRKDC, ELAVL4, ILK, KATNB1, SNF8, PCSK6, C1QTNF3, LDHA, BSG, IL3RA, MLEC, MYH9, SLC39A7, ALDOA, CD68, HRAS, GAPDH, YKT6, PFKM, HLA-DRB1 | 2.07 | 0.0007 | |
| MF | structural constituent of cytoskeleton | TUBA1B, TUBB2A, TUBA1A, HLA-DRB1, LMNB2 | 9.25 | 0.0020 |
| CXCR chemokine receptor binding | CXCL1, CXCL3, CXCL2 | 39.60 | 0.0025 | |
| cadherin binding | GOLGA2, LDHA, BSG, MYH9, ALDOA, YKT6, CDH18 | 4.39 | 0.0051 | |
| KEGG | Apoptosis | TUBA1B, TUBA1A, CTSK, IL3RA, HRAS, LMNB2 | 6.31 | 0.0023 |
| HIF-1 signaling pathway | LDHA, TIMP1, ALDOA, GAPDH, PFKM | 6.56 | 0.0065 | |
| Glycolysis/Gluconeogenesis | LDHA, ALDOA, GAPDH, PFKM | 8.54 | 0.0108 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karapetyan, L.; Gooding, W.; Li, A.; Yang, X.; Knight, A.; Abushukair, H.M.; Vargas De Stefano, D.; Sander, C.; Karunamurthy, A.; Panelli, M.; et al. Sentinel Lymph Node Gene Expression Signature Predicts Recurrence-Free Survival in Cutaneous Melanoma. Cancers 2022, 14, 4973. https://doi.org/10.3390/cancers14204973
Karapetyan L, Gooding W, Li A, Yang X, Knight A, Abushukair HM, Vargas De Stefano D, Sander C, Karunamurthy A, Panelli M, et al. Sentinel Lymph Node Gene Expression Signature Predicts Recurrence-Free Survival in Cutaneous Melanoma. Cancers. 2022; 14(20):4973. https://doi.org/10.3390/cancers14204973
Chicago/Turabian StyleKarapetyan, Lilit, William Gooding, Aofei Li, Xi Yang, Andrew Knight, Hassan M. Abushukair, Danielle Vargas De Stefano, Cindy Sander, Arivarasan Karunamurthy, Monica Panelli, and et al. 2022. "Sentinel Lymph Node Gene Expression Signature Predicts Recurrence-Free Survival in Cutaneous Melanoma" Cancers 14, no. 20: 4973. https://doi.org/10.3390/cancers14204973
APA StyleKarapetyan, L., Gooding, W., Li, A., Yang, X., Knight, A., Abushukair, H. M., Vargas De Stefano, D., Sander, C., Karunamurthy, A., Panelli, M., Storkus, W. J., Tarhini, A. A., & Kirkwood, J. M. (2022). Sentinel Lymph Node Gene Expression Signature Predicts Recurrence-Free Survival in Cutaneous Melanoma. Cancers, 14(20), 4973. https://doi.org/10.3390/cancers14204973