
Clinical Significance and Regulation of ERK5 Expression and Function in Cancer

 , , , , , ,
, , , , , ,  and
and 
Simple Summary
Abstract
1. Introduction
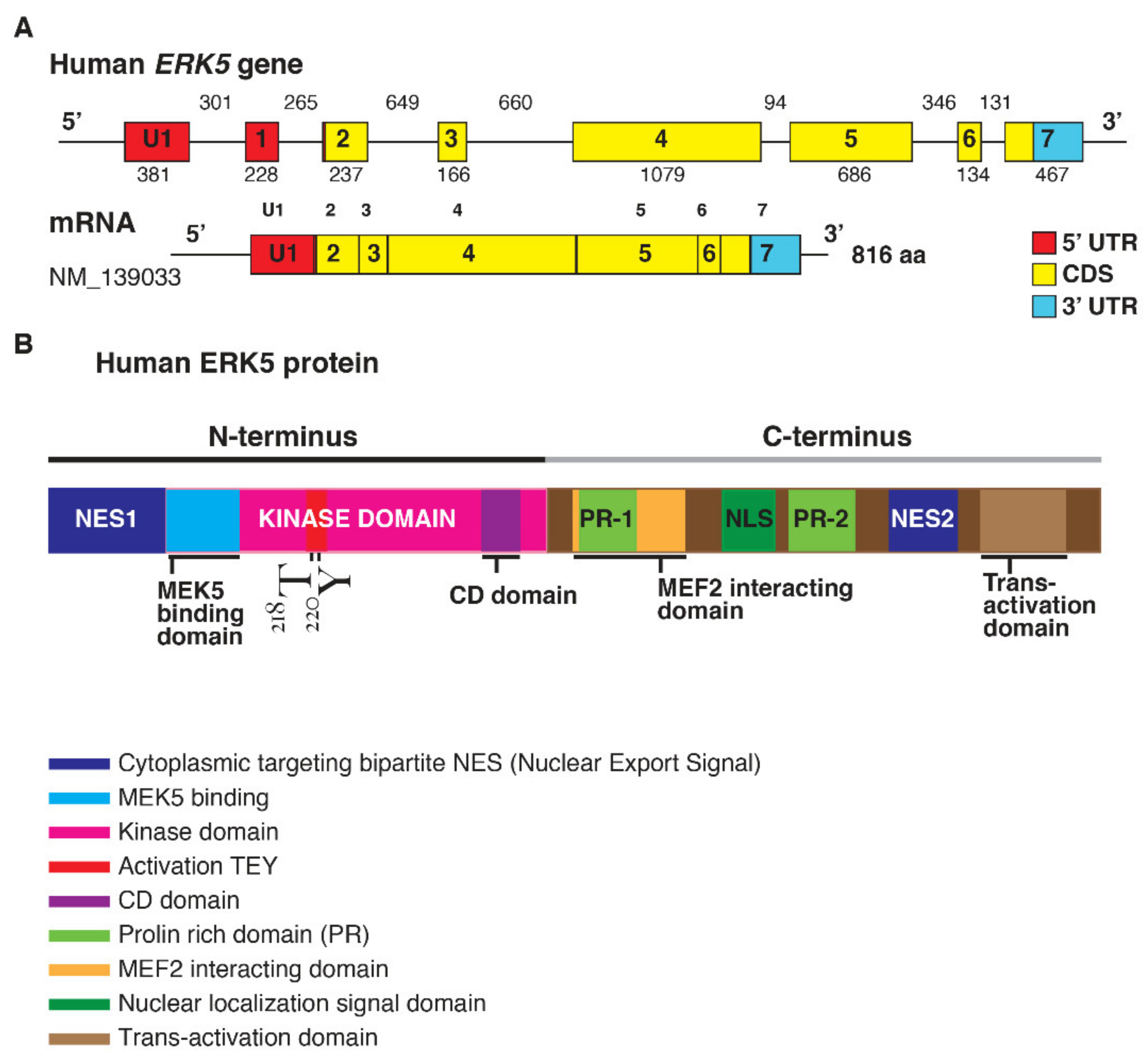
1.1. ERK5 Structure
1.2. Regulation of ERK5 Activation
1.3. ERK5 and Cancer Proliferation
1.4. Role of ERK5 in Cancer Migration and Metastasis
1.5. ERK5 and Cancer Stemness
1.6. ERK5 and Cancer Cell Metabolism
2. ERK5 Gene Expression among Human Cancers
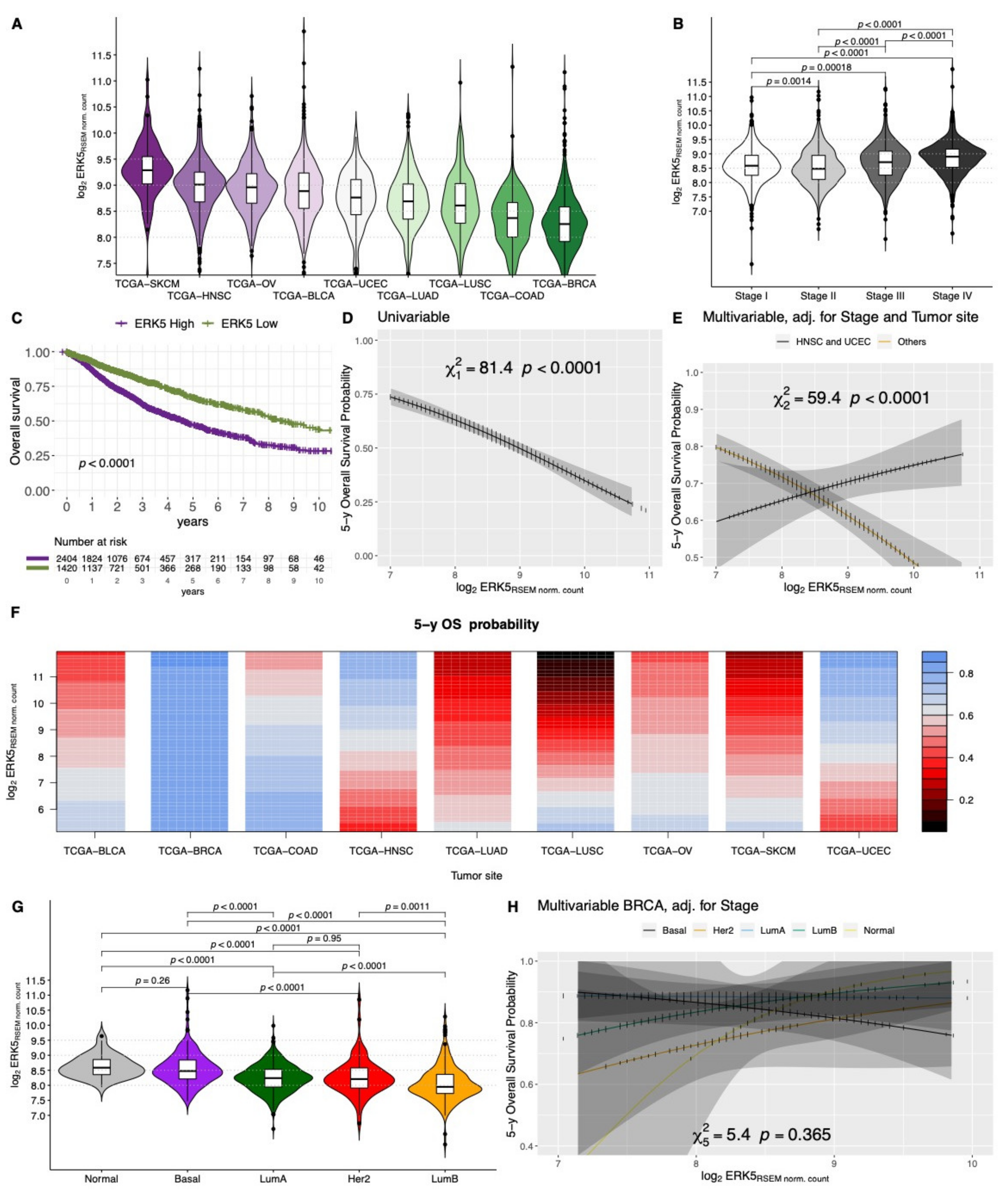
2.1. Clinical Significance of ERK5 Gene Expression in Solid Tumors
2.2. Localization and Clinical Significance of ERK5 Protein Expression in Human Cancers

{kind=link}
{kind=link}
{kind=link}
| TUMOR TYPE | PATIENTS N° | TECHNIQUE | REAGENT/BIOINFORMATIC TOOL | OUTCOME * | REFERENCE |
|---|---|---|---|---|---|
| SKCM | 479 | TCGA (genomic alterations) | cBioportal | poor DFS | [87] |
| 66 | IHC–TMA | anti-ERK5 (mMs, C-7; SCB) | |||
| LGG and HGG | 524 | REMBRANDT (mRNA) | poor | [138] | |
| 162 | IHC–TMA | anti-ERK5 (mMs, C-7; SCB) | T+ | ||
| APC | 1 | IHC | anti-ERK5 (pRb; CST) | - | [153] |
| APC | 11 | IHC | anti-ERK5 | - | [154] |
| 11 | RT-PCR | ||||
| 11 | WB | anti-ERK5 (CST) | |||
| CCA | 104 | GEO (mRNA) | Portal invasion | [155] | |
| IHC–TMA | anti-ERK5 (polyclonal) | T+ | |||
| OSCC | 35 | whole-transcriptome chip | [96] | ||
| RT-qPCR | |||||
| 306 | IHC–TMA | anti-pERK5 (Thr218/Tyr220, mRb; CST) | T+; LN+ | ||
| BC | 252 | mRNA | R2: Genomics Analysis and Visualization Platform | poor | [156] |
| BC | 84 | IHC | anti-ERK5 (791–805 C-term, Rb; hm) | poor DFS | [145] |
| 23 | WB | anti-ERK5 (791–805 aa C-term, Rb; hm) and anti-pERK5 (213–225 aa Erk5 C-term, Rb; hm) | |||
| BC | 39 | IHC–TMA | anti-pERK5 | - | [146] |
| OC | IHC–TMA | anti-pERK5 | |||
| BC | 1803 | KM Plotter | poor DMFS; M+ | [95] | |
| 514 | TCGA | poor | |||
| IHC | anti-ERK5 (pRb; Abcam) | ||||
| BC | 1809 | KM Plotter (mRNA) | poor | [82] | |
| 14 | WB | anti-ERK5 (C-20) | |||
| BC | 384 | Kinex™ antibody arrays (mRNA) | poor RFS and DMFS | [139] | |
| LC (NSCLC) | 74 | IHC | anti-ERK5 (mRb, D23E9; CST) | - | [89] |
| LC | 23 | IHC | anti-pERK5 | - | [147] |
| 23 | WB | anti-pERK5 | |||
| LC (LUAD; LUSC) | 36 | IHC–TMA | anti-ERK5 and anti-pERK5 (CST) | T+ | [140] |
| 8 | RT-qPCR | poor OS | |||
| LC (LUAD; LUSC) | 1925 | KM Plotter (mRNA) | poor | [39] | |
| 972 | TCGA (mRNA and CNA) | cBioportal | poor | ||
| MESO | 73 | IHC–TMA | anti-ERK5 and pERK5 (pRb; CST) | - | [149] |
| MESO | 15 | IF–TMA | anti-pERK5 | - | [148] |
| HCC | 43 | IHC | anti-ERK5 (polyclonal; Sigma-Aldrich) | - | [142] |
| 66 | RT-qPCR (CNA) | ||||
| HCC | 16 | IHC | anti-ERK5 (polyclonal) | - | [9] |
| 13 | WB | anti-pERK5 | |||
| CC | 228 | IHC | anti-ERK5 (pRb; CST) | [58] | |
| WB | anti-ERK5 (pRb; CST) | T+; LN+; M+ | |||
| CC | 151 | TCGA (RNA-Seq) | SurvExpress web resource | poor OS | [84] |
| 482 | GEO (microarray) | ||||
| CRC | 2 | DNA methylation | - | [157] | |
| CRC | 9 | WB | anti-ERK5 (pRb; Abcam) and anti-pERK5 (Thr732, pRb; CST) | - | [56] |
| RCC | 50 | WB | anti-ERK5 (CST) | T+; M+ | [116] |
| RCC | 19 | WB | anti-ERK5 (hm or CST) | T+; M+ | [152] |
| PCa | 19 | IHC | anti-ERK5 (pGt; SCB) | - | [150] |
| PCa | 81 | IHC | anti-ERK5 (pSp; hm) | poor; M+ and extracapsular disease | [83] |
| PCa | 112 | IHC–TMA | anti-ERK5 | M+ | [151] |
| OS | 30 | RT-qPCR | poor OS; T+; M+ | [143] | |
| OS | 19 | PCR (gDNA) | - | [141] | |
| CSCC † | 12 | IHC | anti-ERK5 SCB (pRb, C-20) | - | [158] |
| Mixed types † | 7 | IHC | anti-ERK5 SCB (pRb, C-20) | ||
| Mixed types † | 2 | ICC | anti-ERK5 SCB (pRb, C-20) | - | [159] |
| 6 | IHC | anti-ERK5 SCB (pRb, C-20) |
3. Role of ERK5 in Tumor Microenvironment
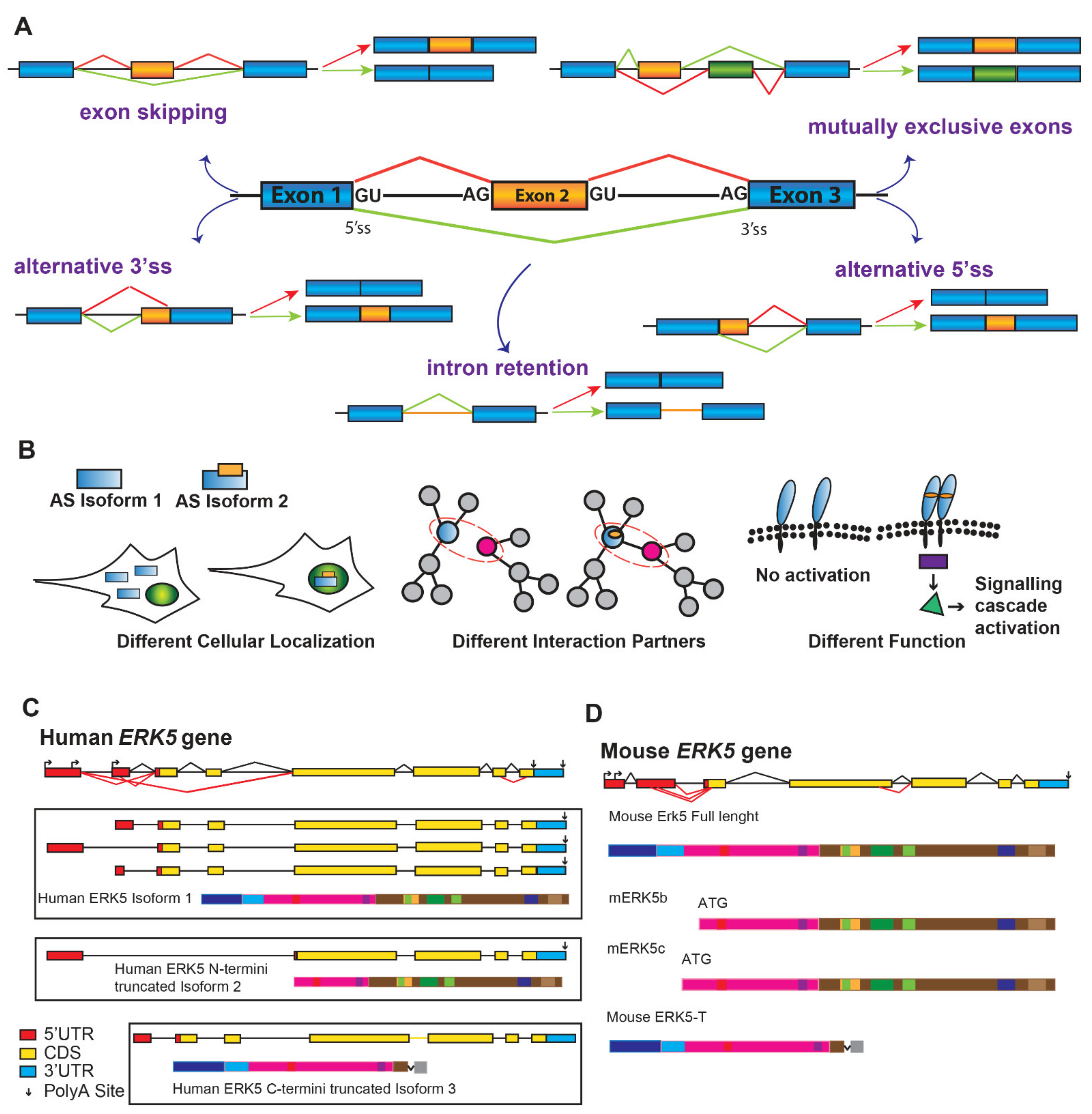
4. Alternatively Spliced Variants of ERK5
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Morimoto, H.; Kondoh, K.; Nishimoto, S.; Terasawa, K.; Nishida, E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J. Biol. Chem. 2007, 282, 35449–35456. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Tapping, R.I.; Chao, T.H.; Lo, J.F.; King, C.C.; Yang, Y.; Lee, J.D. BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J. Biol. Chem. 2001, 276, 8631–8634. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, R.; Salinas, S.; Philips, A.; Hipskind, R.A. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene 2003, 22, 5387–5398. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Fearns, C.; Eliceiri, B.; Yang, Y.; Lee, J.-D. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005, 65, 7699–7706. [Google Scholar] [CrossRef]
- Garaude, J.; Cherni, S.; Kaminski, S.; Delepine, E.; Chable-Bessia, C.; Benkirane, M.; Borges, J.; Pandiella, A.; Iñiguez, M.A.; Fresno, M.; et al. ERK5 activates NF-kappaB in leukemic T cells and is essential for their growth in vivo. J. Immunol. 2006, 177, 7607–7617. [Google Scholar] [CrossRef]
- Cude, K.; Wang, Y.; Choi, H.-J.; Hsuan, S.-L.; Zhang, H.; Wang, C.-Y.; Xia, Z. Regulation of the G2-M cell cycle progression by the ERK5-NFkappaB signaling pathway. J. Cell Biol. 2007, 177, 253–264. [Google Scholar] [CrossRef]
- Umapathy, G.; El Wakil, A.; Witek, B.; Chesler, L.; Danielson, L.; Deng, X.; Gray, N.S.; Johansson, M.; Kvarnbrink, S.; Ruuth, K.; et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci. Signal. 2014, 7, ra102. [Google Scholar] [CrossRef]
- Kato, Y.; Kravchenko, V.V.; Tapping, R.I.; Han, J.; Ulevitch, R.J.; Lee, J.D. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997, 16, 7054–7066. [Google Scholar] [CrossRef]
- Rovida, E.; Di Maira, G.; Tusa, I.; Cannito, S.; Paternostro, C.; Navari, N.; Vivoli, E.; Deng, X.; Gray, N.S.; Esparís-Ogando, A.; et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut 2015, 64, 1454–1465. [Google Scholar] [CrossRef]
- Liu, L.; Cavanaugh, J.E.; Wang, Y.; Sakagami, H.; Mao, Z.; Xia, Z. ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc. Natl. Acad. Sci. USA 2003, 100, 8532–8537. [Google Scholar] [CrossRef]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R.; Gray, N.S.; Lee, J.-D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef]
- Yang, Q.; Lee, J.-D. Targeting the BMK1 MAP kinase pathway in cancer therapy. Clin. Cancer Res. 2011, 17, 3527–3532. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Gilley, R.; Cook, S.J. ERK5 and its role in tumour development. Biochem. Soc. Trans. 2012, 40, 251–256. [Google Scholar] [CrossRef]
- Simões, A.E.S.; Rodrigues, C.M.P.; Borralho, P.M. The MEK5/ERK5 signalling pathway in cancer: A promising novel therapeutic target. Drug Discov. Today 2016, 21, 1654–1663. [Google Scholar] [CrossRef]
- Stecca, B.; Rovida, E. Impact of ERK5 on the hallmarks of cancer. Int. J. Mol. Sci. 2019, 20, 1426. [Google Scholar] [CrossRef]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signalling pathway. Cell. Signal. 2006, 18, 753–760. [Google Scholar] [CrossRef]
- Yang, S.-H.; Sharrocks, A.D.; Whitmarsh, A.J. Transcriptional regulation by the MAP kinase signaling cascades. Gene 2003, 320, 3–21. [Google Scholar] [CrossRef]
- Zhou, G.; Bao, Z.Q.; Dixon, J.E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [Google Scholar] [CrossRef]
- Yan, C.; Luo, H.; Lee, J.D.; Abe, J.; Berk, B.C. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J. Biol. Chem. 2001, 276, 10870–10878. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Mu, D.-W.; Guo, H.-Q.; Zhou, G.-B.; Li, J.-Y.; Su, B. Oleanolic acid suppresses the proliferation of human bladder cancer by Akt/mTOR/S6K and ERK1/2 signaling. Int. J. Clin. Exp. Pathol. 2015, 8, 13864–13870. [Google Scholar]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713–716. [Google Scholar] [CrossRef]
- Kesavan, K.; Lobel-Rice, K.; Sun, W.; Lapadat, R.; Webb, S.; Johnson, G.L.; Garrington, T.P. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J. Cell Physiol. 2004, 199, 140–148. [Google Scholar] [CrossRef]
- Hayashi, M.; Kim, S.-W.; Imanaka-Yoshida, K.; Yoshida, T.; Abel, E.D.; Eliceiri, B.; Yang, Y.; Ulevitch, R.J.; Lee, J.-D. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Investig. 2004, 113, 1138–1148. [Google Scholar] [CrossRef]
- Carvajal-Vergara, X.; Tabera, S.; Montero, J.C.; Esparís-Ogando, A.; López-Pérez, R.; Mateo, G.; Gutiérrez, N.; Parmo-Cabañas, M.; Teixidó, J.; San Miguel, J.F.; et al. Multifunctional role of Erk5 in multiple myeloma. Blood 2005, 105, 4492–4499. [Google Scholar] [CrossRef]
- Abe, J.; Kusuhara, M.; Ulevitch, R.J.; Berk, B.C.; Lee, J.D. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J. Biol. Chem. 1996, 271, 16586–16590. [Google Scholar] [CrossRef]
- Chao, T.H.; Hayashi, M.; Tapping, R.I.; Kato, Y.; Lee, J.D. MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J. Biol. Chem. 1999, 274, 36035–36038. [Google Scholar] [CrossRef]
- Nakamura, K.; Johnson, G.L. PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J. Biol. Chem. 2003, 278, 36989–36992. [Google Scholar] [CrossRef]
- Nakamura, K.; Johnson, G.L. Noncanonical function of MEKK2 and MEK5 PB1 domains for coordinated extracellular signal-regulated kinase 5 and c-Jun N-terminal kinase signaling. Mol. Cell. Biol. 2007, 27, 4566–4577. [Google Scholar] [CrossRef][Green Version]
- Barros, J.C.; Marshall, C.J. Activation of either ERK1/2 or ERK5 MAP kinase pathways can lead to disruption of the actin cytoskeleton. J. Cell Sci. 2005, 118, 1663–1671. [Google Scholar] [CrossRef]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.P.; Müller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell. Signal. 2012, 24, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Nino, M.E.; Blumen, S.R.; Sabo-Attwood, T.; Pass, H.; Carbone, M.; Testa, J.R.; Altomare, D.A.; Mossman, B.T. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra-1 pathway. Am. J. Respir. Cell Mol. Biol. 2008, 38, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, H.; Song, H. Upregulation of MEK5 by Stat3 promotes breast cancer cell invasion and metastasis. Oncol. Rep. 2017, 37, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Browne, J.A.; Pearson, A.L.; Zahr, R.A.; Niculescu-Duvaz, I.; Baines, D.L.; Dockrell, M.E.C. TGF-beta activates ERK5 in human renal epithelial cells. Biochem. Biophys. Res. Commun. 2008, 373, 440–444. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, M.; Wang, L.; Fan, X.; Wang, P.; Wang, D.; Fu, X.; Wang, J. Overexpression of MEKK2 is associated with colorectal carcinogenesis. Oncol. Lett. 2013, 6, 1333–1337. [Google Scholar] [CrossRef]
- Lu, H.; Cao, X.; Chen, Q.; Chen, L.; Chen, L.; Gan, M. The expression and role of MEKK3 in renal clear cell carcinoma. Anat. Rec. 2015, 298, 727–734. [Google Scholar] [CrossRef]
- Samanta, A.K.; Huang, H.J.; Bast, R.C.; Liao, W.S.-L. Overexpression of MEKK3 confers resistance to apoptosis through activation of NFkappaB. J. Biol. Chem. 2004, 279, 7576–7583. [Google Scholar] [CrossRef]
- Hasan, R.; Sharma, R.; Saraya, A.; Chattopadhyay, T.K.; DattaGupta, S.; Walfish, P.G.; Chauhan, S.S.; Ralhan, R. Mitogen activated protein kinase kinase kinase 3 (MAP3K3/MEKK3) overexpression is an early event in esophageal tumorigenesis and is a predictor of poor disease prognosis. BMC Cancer 2014, 14, 2. [Google Scholar] [CrossRef]
- Sánchez-Fdez, A.; Re-Louhau, M.F.; Rodríguez-Núñez, P.; Ludeña, D.; Matilla-Almazán, S.; Pandiella, A.; Esparís-Ogando, A. Clinical, genetic and pharmacological data support targeting the MEK5/ERK5 module in lung cancer. NPJ Precis. Oncol. 2021, 5, 78. [Google Scholar] [CrossRef]
- Dong, F.; Gutkind, J.S.; Larner, A.C. Granulocyte colony-stimulating factor induces ERK5 activation, which is differentially regulated by protein-tyrosine kinases and protein kinase C. Regulation of cell proliferation and survival. J. Biol. Chem. 2001, 276, 10811–10816. [Google Scholar] [CrossRef]
- Buschbeck, M.; Eickhoff, J.; Sommer, M.N.; Ullrich, A. Phosphotyrosine-specific phosphatase PTP-SL regulates the ERK5 signaling pathway. J. Biol. Chem. 2002, 277, 29503–29509. [Google Scholar] [CrossRef]
- Sarközi, R.; Miller, B.; Pollack, V.; Feifel, E.; Mayer, G.; Sorokin, A.; Schramek, H. ERK1/2-driven and MKP-mediated inhibition of EGF-induced ERK5 signaling in human proximal tubular cells. J. Cell Physiol. 2007, 211, 88–100. [Google Scholar] [CrossRef]
- Adam, C.; Fusi, L.; Weiss, N.; Goller, S.G.; Meder, K.; Frings, V.G.; Kneitz, H.; Goebeler, M.; Houben, R.; Schrama, D.; et al. Efficient Suppression of NRAS-Driven Melanoma by Co-Inhibition of ERK1/2 and ERK5 MAPK Pathways. J. Investig. Dermatol. 2020, 140, 2455–2465.e10. [Google Scholar] [CrossRef]
- De Jong, P.R.; Taniguchi, K.; Harris, A.R.; Bertin, S.; Takahashi, N.; Duong, J.; Campos, A.D.; Powis, G.; Corr, M.; Karin, M.; et al. ERK5 signalling rescues intestinal epithelial turnover and tumour cell proliferation upon ERK1/2 abrogation. Nat. Commun. 2016, 7, 11551. [Google Scholar] [CrossRef]
- Lee, B.; Sahoo, A.; Sawada, J.; Marchica, J.; Sahoo, S.; Layng, F.I.A.L.; Finlay, D.; Mazar, J.; Joshi, P.; Komatsu, M.; et al. MicroRNA-211 Modulates the DUSP6-ERK5 Signaling Axis to Promote BRAFV600E-Driven Melanoma Growth In Vivo and BRAF/MEK Inhibitor Resistance. J. Investig. Dermatol. 2021, 141, 385–394. [Google Scholar] [CrossRef]
- Moncho-Amor, V.; Pintado-Berninches, L.; Ibañez de Cáceres, I.; Martín-Villar, E.; Quintanilla, M.; Chakravarty, P.; Cortes-Sempere, M.; Fernández-Varas, B.; Rodriguez-Antolín, C.; de Castro, J.; et al. Role of Dusp6 Phosphatase as a Tumor Suppressor in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2019, 20, 2036. [Google Scholar] [CrossRef]
- Garcia, L.; Garcia, F.; Llorens, F.; Unzeta, M.; Itarte, E.; Gómez, N. PP1/PP2A phosphatases inhibitors okadaic acid and calyculin A block ERK5 activation by growth factors and oxidative stress. FEBS Lett. 2002, 523, 90–94. [Google Scholar] [CrossRef]
- Buschbeck, M.; Ullrich, A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J. Biol. Chem. 2005, 280, 2659–2667. [Google Scholar] [CrossRef]
- Erazo, T.; Moreno, A.; Ruiz-Babot, G.; Rodríguez-Asiain, A.; Morrice, N.A.; Espadamala, J.; Bayascas, J.R.; Gómez, N.; Lizcano, J.M. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol. Cell. Biol. 2013, 33, 1671–1686. [Google Scholar] [CrossRef]
- Erazo, T.; Espinosa-Gil, S.; Diéguez-Martínez, N.; Gómez, N.; Lizcano, J.M. SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 2203. [Google Scholar] [CrossRef]
- Kamakura, S.; Moriguchi, T.; Nishida, E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J. Biol. Chem. 1999, 274, 26563–26571. [Google Scholar] [CrossRef]
- Kato, Y.; Chao, T.H.; Hayashi, M.; Tapping, R.I.; Lee, J.D. Role of BMK1 in regulation of growth factor-induced cellular responses. Immunol. Res. 2000, 21, 233–237. [Google Scholar] [CrossRef]
- Terasawa, K.; Okazaki, K.; Nishida, E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells 2003, 8, 263–273. [Google Scholar] [CrossRef]
- Yang, S.H.; Whitmarsh, A.J.; Davis, R.J.; Sharrocks, A.D. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 1998, 17, 1740–1749. [Google Scholar] [CrossRef]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef]
- Zhuang, K.; Zhang, J.; Xiong, M.; Wang, X.; Luo, X.; Han, L.; Meng, Y.; Zhang, Y.; Liao, W.; Liu, S. CDK5 functions as a tumor promoter in human colorectal cancer via modulating the ERK5-AP-1 axis. Cell Death Dis. 2016, 7, e2415. [Google Scholar] [CrossRef]
- Luiz, J.P.M.; Toller-Kawahisa, J.E.; Viacava, P.R.; Nascimento, D.C.; Pereira, P.T.; Saraiva, A.L.; Prado, D.S.; LeBert, M.; Giurisato, E.; Tournier, C.; et al. MEK5/ERK5 signaling mediates IL-4-induced M2 macrophage differentiation through regulation of c-Myc expression. J. Leukoc. Biol. 2020, 108, 1215–1223. [Google Scholar] [CrossRef]
- Simões, A.E.S.; Pereira, D.M.; Gomes, S.E.; Brito, H.; Carvalho, T.; French, A.; Castro, R.E.; Steer, C.J.; Thibodeau, S.N.; Rodrigues, C.M.P.; et al. Aberrant MEK5/ERK5 signalling contributes to human colon cancer progression via NF-κB activation. Cell Death Dis. 2015, 6, e1718. [Google Scholar] [CrossRef]
- Samanta, A.K.; Huang, H.J.; Le, X.-F.; Mao, W.; Lu, K.H.; Bast, R.C.; Liao, W.S.-L. MEKK3 expression correlates with nuclear factor kappa B activity and with expression of antiapoptotic genes in serous ovarian carcinoma. Cancer 2009, 115, 3897–3908. [Google Scholar] [CrossRef]
- Mody, N.; Campbell, D.G.; Morrice, N.; Peggie, M.; Cohen, P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem. J. 2003, 372, 567–575. [Google Scholar] [CrossRef]
- Pearson, A.J.; Fullwood, P.; Toro Tapia, G.; Prise, I.; Smith, M.P.; Xu, Q.; Jordan, A.; Giurisato, E.; Whitmarsh, A.J.; Francavilla, C.; et al. Discovery of a Gatekeeper Residue in the C-Terminal Tail of the Extracellular Signal-Regulated Protein Kinase 5 (ERK5). Int. J. Mol. Sci. 2020, 21, 929. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Rodríguez, E.; Pandiella, A. Multisite phosphorylation of Erk5 in mitosis. J. Cell Sci. 2010, 123, 3146–3156. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Obara, Y.; Yamauchi, A.; Couvillon, A.D.; Mason, J.J.; Ishii, K.; Nakahata, N. Phosphorylation of ERK5 on Thr732 is associated with ERK5 nuclear localization and ERK5-dependent transcription. PLoS ONE 2015, 10, e0117914. [Google Scholar] [CrossRef] [PubMed]
- Clapé, C.; Fritz, V.; Henriquet, C.; Apparailly, F.; Fernandez, P.L.; Iborra, F.; Avancès, C.; Villalba, M.; Culine, S.; Fajas, L. miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PLoS ONE 2009, 4, e7542. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Singh, L.B.; Yang, Z.H.; Kalna, G.; Fleming, J.; Fisher, G.; Cooper, C.; Cuzick, J.; Berney, D.M.; Møller, H.; et al. Mir143 expression inversely correlates with nuclear ERK5 immunoreactivity in clinical prostate cancer. Br. J. Cancer 2013, 108, 149–154. [Google Scholar] [CrossRef]
- Noguchi, S.; Mori, T.; Hoshino, Y.; Maruo, K.; Yamada, N.; Kitade, Y.; Naoe, T.; Akao, Y. MicroRNA-143 functions as a tumor suppressor in human bladder cancer T24 cells. Cancer Lett. 2011, 307, 211–220. [Google Scholar] [CrossRef]
- Zhou, L.L.; Dong, J.L.; Huang, G.; Sun, Z.L.; Wu, J. MicroRNA-143 inhibits cell growth by targeting ERK5 and MAP3K7 in breast cancer. Braz. J. Med. Biol. Res. 2017, 50, e5891. [Google Scholar] [CrossRef]
- Zhai, L.; Ma, C.; Li, W.; Yang, S.; Liu, Z. miR-143 suppresses epithelial-mesenchymal transition and inhibits tumor growth of breast cancer through down-regulation of ERK5. Mol. Carcinog. 2016, 55, 1990–2000. [Google Scholar] [CrossRef]
- Ni, Y.; Meng, L.; Wang, L.; Dong, W.; Shen, H.; Wang, G.; Liu, Q.; Du, J. MicroRNA-143 functions as a tumor suppressor in human esophageal squamous cell carcinoma. Gene 2013, 517, 197–204. [Google Scholar] [CrossRef]
- Chen, J.-H.; Yang, R.; Zhang, W.; Wang, Y.-P. Functions of microRNA-143 in the apoptosis, invasion and migration of nasopharyngeal carcinoma. Exp. Ther. Med. 2016, 12, 3749–3755. [Google Scholar] [CrossRef]
- Hartmann, J.-U.; Bräuer-Hartmann, D.; Kardosova, M.; Wurm, A.A.; Wilke, F.; Schödel, C.; Gerloff, D.; Katzerke, C.; Krakowsky, R.; Namasu, C.Y.; et al. MicroRNA-143 targets ERK5 in granulopoiesis and predicts outcome of patients with acute myeloid leukemia. Cell Death Dis. 2018, 9, 814. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Iio, A.; Naoe, T. Role of microRNA-143 in Fas-mediated apoptosis in human T-cell leukemia Jurkat cells. Leuk. Res. 2009, 33, 1530–1538. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Kitade, Y.; Kinoshita, T.; Naoe, T. Downregulation of microRNAs-143 and -145 in B-cell malignancies. Cancer Sci. 2007, 98, 1914–1920. [Google Scholar] [CrossRef]
- Wu, J.; Cui, H.; Zhu, Z.; Wang, L. MicroRNA-200b-3p suppresses epithelial-mesenchymal transition and inhibits tumor growth of glioma through down-regulation of ERK5. Biochem. Biophys. Res. Commun. 2016, 478, 1158–1164. [Google Scholar] [CrossRef]
- Hoang, V.T.; Yan, T.J.; Cavanaugh, J.E.; Flaherty, P.T.; Beckman, B.S.; Burow, M.E. Oncogenic signaling of MEK5-ERK5. Cancer Lett. 2017, 392, 51–59. [Google Scholar] [CrossRef]
- Paudel, R.; Fusi, L.; Schmidt, M. The MEK5/ERK5 pathway in health and disease. Int. J. Mol. Sci. 2021, 22, 7594. [Google Scholar] [CrossRef]
- Rovida, E.; Spinelli, E.; Sdelci, S.; Barbetti, V.; Morandi, A.; Giuntoli, S.; Dello Sbarba, P. ERK5/BMK1 is indispensable for optimal colony-stimulating factor 1 (CSF-1)-induced proliferation in macrophages in a Src-dependent fashion. J. Immunol. 2008, 180, 4166–4172. [Google Scholar] [CrossRef]
- Buse, P.; Tran, S.H.; Luther, E.; Phu, P.T.; Aponte, G.W.; Firestone, G.L. Cell cycle and hormonal control of nuclear-cytoplasmic localization of the serum- and glucocorticoid-inducible protein kinase, Sgk, in mammary tumor cells. A novel convergence point of anti-proliferative and proliferative cell signaling pathways. J. Biol. Chem. 1999, 274, 7253–7263. [Google Scholar] [CrossRef]
- Perez-Madrigal, D.; Finegan, K.G.; Paramo, B.; Tournier, C. The extracellular-regulated protein kinase 5 (ERK5) promotes cell proliferation through the down-regulation of inhibitors of cyclin dependent protein kinases (CDKs). Cell. Signal. 2012, 24, 2360–2368. [Google Scholar] [CrossRef]
- Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Menconi, A.; Lulli, M.; Dello Sbarba, P.; Stecca, B.; Rovida, E. The Hedgehog-GLI Pathway Regulates MEK5-ERK5 Expression and Activation in Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 11259. [Google Scholar] [CrossRef]
- Shukla, A.; Miller, J.M.; Cason, C.; Sayan, M.; MacPherson, M.B.; Beuschel, S.L.; Hillegass, J.; Vacek, P.M.; Pass, H.I.; Mossman, B.T. Extracellular signal-regulated kinase 5: A potential therapeutic target for malignant mesotheliomas. Clin. Cancer Res. 2013, 19, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Ruiz, M.J.; Álvarez-Fernández, S.; Parrott, T.; Zaknoen, S.; Burrows, F.J.; Ocaña, A.; Pandiella, A.; Esparís-Ogando, A. Therapeutic potential of ERK5 targeting in triple negative breast cancer. Oncotarget 2014, 5, 11308–11318. [Google Scholar] [CrossRef] [PubMed]
- McCracken, S.R.C.; Ramsay, A.; Heer, R.; Mathers, M.E.; Jenkins, B.L.; Edwards, J.; Robson, C.N.; Marquez, R.; Cohen, P.; Leung, H.Y. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene 2008, 27, 2978–2988. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Simões, A.E.S.; Gomes, S.E.; Castro, R.E.; Carvalho, T.; Rodrigues, C.M.P.; Borralho, P.M. MEK5/ERK5 signaling inhibition increases colon cancer cell sensitivity to 5-fluorouracil through a p53-dependent mechanism. Oncotarget 2016, 7, 34322–34340. [Google Scholar] [CrossRef] [PubMed]
- Giurisato, E.; Tournier, C. Can tumor cells proliferate without ERK5? Cell Cycle 2016, 15, 619–620. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Clark, J.; Wang, L.-Z.; Gilmour, L.; Squires, M.; Gilley, R.; Foxton, C.; Newell, D.R.; Wedge, S.R.; Cook, S.J. Tumor cells with KRAS or BRAF mutations or ERK5/MAPK7 amplification are not addicted to ERK5 activity for cell proliferation. Cell Cycle 2016, 15, 506–518. [Google Scholar] [CrossRef]
- Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Urso, C.; Borgognoni, L.; Wang, J.; Deng, X.; Gray, N.S.; Stecca, B.; et al. ERK5 is activated by oncogenic BRAF and promotes melanoma growth. Oncogene 2018, 37, 2601–2614. [Google Scholar] [CrossRef]
- Tubita, A.; Lombardi, Z.; Tusa, I.; Lazzeretti, A.; Sgrignani, G.; Papini, D.; Menconi, A.; Gagliardi, S.; Lulli, M.; Dello Sbarba, P.; et al. Inhibition of ERK5 elicits cellular senescence in melanoma via the cyclin-dependent kinase inhibitor p21. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Gavine, P.R.; Wang, M.; Yu, D.; Hu, E.; Huang, C.; Xia, J.; Su, X.; Fan, J.; Zhang, T.; Ye, Q.; et al. Identification and validation of dysregulated MAPK7 (ERK5) as a novel oncogenic target in squamous cell lung and esophageal carcinoma. BMC Cancer 2015, 15, 454. [Google Scholar] [CrossRef]
- Ranganathan, A.; Pearson, G.W.; Chrestensen, C.A.; Sturgill, T.W.; Cobb, M.H. The MAP kinase ERK5 binds to and phosphorylates p90 RSK. Arch. Biochem. Biophys. 2006, 449, 8–16. [Google Scholar] [CrossRef]
- Lennartsson, J.; Burovic, F.; Witek, B.; Jurek, A.; Heldin, C.-H. Erk 5 is necessary for sustained PDGF-induced Akt phosphorylation and inhibition of apoptosis. Cell. Signal. 2010, 22, 955–960. [Google Scholar] [CrossRef]
- Villa-Moruzzi, E. Targeting of FAK Ser910 by ERK5 and PP1delta in non-stimulated and phorbol ester-stimulated cells. Biochem. J. 2007, 408, 7–18. [Google Scholar] [CrossRef]
- Sawhney, R.S.; Liu, W.; Brattain, M.G. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J. Cell. Physiol. 2009, 219, 152–161. [Google Scholar] [CrossRef]
- Ali, M.; Mutahir, Z.; Riaz, A. CRISPR/Cas9 engineering of ERK5 identifies its FAK/PYK2 dependent role in adhesion-mediated cell survival. Biochem. Biophys. Res. Commun. 2019, 513, 179–185. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, J.; Telfer, B.A.; Zhang, H.; Ali, N.; Chen, F.; Risa, B.; Pearson, A.J.; Zhang, W.; Finegan, K.G.; et al. The extracellular-regulated protein kinase 5 (ERK5) enhances metastatic burden in triple-negative breast cancer through focal adhesion protein kinase (FAK)-mediated regulation of cell adhesion. Oncogene 2021, 40, 3929–3941. [Google Scholar] [CrossRef]
- Sticht, C.; Freier, K.; Knöpfle, K.; Flechtenmacher, C.; Pungs, S.; Hofele, C.; Hahn, M.; Joos, S.; Lichter, P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC). Neoplasia 2008, 10, 462–470. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Ventrella, R.; Petry, L.; Katzenellenbogen, B.S. Novel roles for ERK5 and cofilin as critical mediators linking ERα-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol. Cancer Res. 2014, 12, 714–727. [Google Scholar] [CrossRef]
- Pavan, S.; Meyer-Schaller, N.; Diepenbruck, M.; Kalathur, R.K.R.; Saxena, M.; Christofori, G. A kinome-wide high-content siRNA screen identifies MEK5-ERK5 signaling as critical for breast cancer cell EMT and metastasis. Oncogene 2018, 37, 4197–4213. [Google Scholar] [CrossRef]
- Castro, N.E.; Lange, C.A. Breast tumor kinase and extracellular signal-regulated kinase 5 mediate Met receptor signaling to cell migration in breast cancer cells. Breast Cancer Res. 2010, 12, R60. [Google Scholar] [CrossRef]
- Zuo, Y.; Wu, Y.; Wehrli, B.; Chakrabarti, S.; Chakraborty, C. Modulation of ERK5 is a novel mechanism by which Cdc42 regulates migration of breast cancer cells. J. Cell. Biochem. 2015, 116, 124–132. [Google Scholar] [CrossRef]
- Locatelli, A.; Lange, C.A. Met receptors induce Sam68-dependent cell migration by activation of alternate extracellular signal-regulated kinase family members. J. Biol. Chem. 2011, 286, 21062–21072. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.A.; Kahle, M.P.; Ameka, M.; Campbell, E.M.; Cuevas, B.D. MEKK2 regulates focal adhesion stability and motility in invasive breast cancer cells. Biochim. Biophys. Acta 2014, 1843, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Cronan, M.R.; Nakamura, K.; Johnson, N.L.; Granger, D.A.; Cuevas, B.D.; Wang, J.G.; Mackman, N.; Scott, J.E.; Dohlman, H.G.; Johnson, G.L. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene 2012, 31, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Nitschke, A.M.; Xiong, W.; Zhang, Q.; Tang, Y.; Bloch, M.; Elliott, S.; Zhu, Y.; Bazzone, L.; Yu, D.; et al. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008, 10, R105. [Google Scholar] [CrossRef] [PubMed]
- Matossian, M.D.; Hoang, V.T.; Burks, H.E.; La, J.; Elliott, S.; Brock, C.; Rusch, D.B.; Buechlein, A.; Nephew, K.P.; Bhatt, A.; et al. Constitutive activation of MEK5 promotes a mesenchymal and migratory cell phenotype in triple negative breast cancer. Oncoscience 2021, 8, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Hoang, V.T.; Matossian, M.D.; Ucar, D.A.; Elliott, S.; La, J.; Wright, M.K.; Burks, H.E.; Perles, A.; Hossain, F.; King, C.T.; et al. ERK5 is required for tumor growth and maintenance through regulation of the extracellular matrix in triple negative breast cancer. Front. Oncol. 2020, 10, 1164. [Google Scholar] [CrossRef]
- Javaid, S.; Zhang, J.; Smolen, G.A.; Yu, M.; Wittner, B.S.; Singh, A.; Arora, K.S.; Madden, M.W.; Desai, R.; Zubrowski, M.J.; et al. MAPK7 regulates EMT features and modulates the generation of ctcs. Mol. Cancer Res. 2015, 13, 934–943. [Google Scholar] [CrossRef]
- Yue, B.; Ren, Q.X.; Su, T.; Wang, L.N.; Zhang, L. ERK5 silencing inhibits invasion of human osteosarcoma cell via modulating the Slug/MMP-9 pathway. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2640–2647. [Google Scholar]
- Huang, Y.; Yao, J.; Zhu, B.; Zhang, J.; Sun, T. Mitogen-activated protein kinase 7 promotes cell proliferation, migration and invasion in SOSP-M human osteosarcoma cell line. Tumori 2017, 103, 483–488. [Google Scholar] [CrossRef]
- Tesser-Gamba, F.; da Silva Lopes, L.J.; Petrilli, A.S.; Toledo, S.R.C. MAPK7 gene controls proliferation, migration and cell invasion in osteosarcoma. Mol. Carcinog. 2016, 55, 1700–1713. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Xu, S.; Wang, S.; Tu, Y.; Xiong, Y.; Mei, J.; Wang, C. The role of MAPK signaling pathway in the Her-2-positive meningiomas. Oncol. Rep. 2016, 36, 685–695. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, T.; Deng, Q.; Zhou, Q.; Sun, X.; Li, E.; Yu, D.; Zhong, C. Benzidine Induces Epithelial-Mesenchymal Transition of Human Bladder Cancer Cells through Activation of ERK5 Pathway. Mol. Cells 2018, 41, 188–197. [Google Scholar]
- Schramp, M.; Ying, O.; Kim, T.Y.; Martin, G.S. ERK5 promotes Src-induced podosome formation by limiting Rho activation. J. Cell Biol. 2008, 181, 1195–1210. [Google Scholar] [CrossRef]
- Dai, J.; Wang, T.; Wang, W.; Zhang, S.; Liao, Y.; Chen, J. Role of MAPK7 in cell proliferation and metastasis in ovarian cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 10444–10451. [Google Scholar]
- Park, S.J.; Choi, Y.S.; Lee, S.; Lee, Y.J.; Hong, S.; Han, S.; Kim, B.-C. BIX02189 inhibits TGF-β1-induced lung cancer cell metastasis by directly targeting TGF-β type I receptor. Cancer Lett. 2016, 381, 314–322. [Google Scholar] [CrossRef]
- Serrano-Oviedo, L.; Giménez-Bachs, J.M.; Nam-Cha, S.Y.; Cimas, F.J.; García-Cano, J.; Sánchez-Prieto, R.; Salinas-Sánchez, A.S. Implication of VHL, ERK5, and HIF-1alpha in clear cell renal cell carcinoma: Molecular basis. Urol. Oncol. 2017, 35, 114.e15–114.e22. [Google Scholar]
- Im, J.-Y.; Yoon, S.-H.; Kim, B.-K.; Ban, H.S.; Won, K.-J.; Chung, K.-S.; Jung, K.E.; Won, M. DNA damage induced apoptosis suppressor (DDIAS) is upregulated via ERK5/MEF2B signaling and promotes β-catenin-mediated invasion. Biochim. Biophys. Acta 2016, 1859, 1449–1458. [Google Scholar] [CrossRef]
- Zheng, Y.; Asara, J.M.; Tyner, A.L. Protein-tyrosine kinase 6 promotes peripheral adhesion complex formation and cell migration by phosphorylating p130 CRK-associated substrate. J. Biol. Chem. 2012, 287, 148–158. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, B.; Guo, W.; Gao, L.; Shi, L.; Li, H.; Lu, S.; Liu, Y.; Li, X. miR-429 inhibits glioma invasion through BMK1 suppression. J. Neurooncol. 2015, 125, 43–54. [Google Scholar] [CrossRef]
- Xia, C.; Yang, Y.; Kong, F.; Kong, Q.; Shan, C. MiR-143-3p inhibits the proliferation, cell migration and invasion of human breast cancer cells by modulating the expression of MAPK7. Biochimie 2018, 147, 98–104. [Google Scholar] [CrossRef]
- Chen, R.; Yang, Q.; Lee, J.-D. BMK1 kinase suppresses epithelial-mesenchymal transition through the Akt/GSK3β signaling pathway. Cancer Res. 2012, 72, 1579–1587. [Google Scholar] [CrossRef]
- Williams, C.A.C.; Fernandez-Alonso, R.; Wang, J.; Toth, R.; Gray, N.S.; Findlay, G.M. Erk5 Is a Key Regulator of Naive-Primed Transition and Embryonic Stem Cell Identity. Cell Rep. 2016, 16, 1820–1828. [Google Scholar] [CrossRef]
- Brown, H.A.; Williams, C.A.; Zhou, H.; Rios-Szwed, D.; Fernandez-Alonso, R.; Mansoor, S.; McMulkin, L.; Toth, R.; Gourlay, R.; Peltier, J.; et al. An ERK5-KLF2 signalling module regulates early embryonic gene expression and telomere rejuvenation in stem cells. Biochem. J. 2021, 478, 4119–4136. [Google Scholar] [CrossRef]
- Lai, V.K.; Ashraf, M.; Jiang, S.; Haider, K. MicroRNA-143 is a critical regulator of cell cycle activity in stem cells with co-overexpression of Akt and angiopoietin-1 via transcriptional regulation of Erk5/cyclin D1 signaling. Cell Cycle 2012, 11, 767–777. [Google Scholar]
- Tusa, I.; Cheloni, G.; Poteti, M.; Gozzini, A.; DeSouza, N.H.; Shan, Y.; Deng, X.; Gray, N.S.; Li, S.; Rovida, E.; et al. Targeting the Extracellular Signal-Regulated Kinase 5 Pathway to Suppress Human Chronic Myeloid Leukemia Stem Cells. Stem Cell Rep. 2018, 11, 929–943. [Google Scholar] [CrossRef]
- Pereira, D.M.; Gomes, S.E.; Borralho, P.M.; Rodrigues, C.M.P. MEK5/ERK5 activation regulates colon cancer stem-like cell properties. Cell Death Discov. 2019, 5, 68. [Google Scholar] [CrossRef]
- Kedika, S.R.; Shukla, S.P.; Udugamasooriya, D.G. Design of a dual ERK5 kinase activation and autophosphorylation inhibitor to block cancer stem cell activity. Bioorg. Med. Chem. Lett. 2020, 30, 127552. [Google Scholar] [CrossRef]
- Lopez-Royuela, N.; Rathore, M.G.; Allende-Vega, N.; Annicotte, J.-S.; Fajas, L.; Ramachandran, B.; Gulick, T.; Villalba, M. Extracellular-signal-regulated kinase 5 modulates the antioxidant response by transcriptionally controlling Sirtuin 1 expression in leukemic cells. Int. J. Biochem. Cell Biol. 2014, 53, 253–261. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Rathore, M.G.; Allende-Vega, N.; Vo, D.-N.; Belkhala, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; Lecellier, C.-H.; et al. Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen species (ROS) Production. EBioMedicine 2016, 3, 43–53. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Gerbal-Chaloin, S.; Gondeau, C.; Vo, D.-N.; Belkahla, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; et al. The PDK1 inhibitor dichloroacetate controls cholesterol homeostasis through the ERK5/MEF2 pathway. Sci. Rep. 2017, 7, 10654. [Google Scholar] [CrossRef]
- Charni, S.; de Bettignies, G.; Rathore, M.G.; Aguiló, J.I.; van den Elsen, P.J.; Haouzi, D.; Hipskind, R.A.; Enriquez, J.A.; Sanchez-Beato, M.; Pardo, J.; et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. J. Immunol. 2010, 185, 3498–3503. [Google Scholar] [CrossRef] [PubMed]
- Rathore, M.G.; Saumet, A.; Rossi, J.-F.; de Bettignies, C.; Tempé, D.; Lecellier, C.-H.; Villalba, M. The NF-κB member p65 controls glutamine metabolism through miR-23a. Int. J. Biochem. Cell Biol. 2012, 44, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Villalba, M.; Rathore, M.G.; Lopez-Royuela, N.; Krzywinska, E.; Garaude, J.; Allende-Vega, N. From tumor cell metabolism to tumor immune escape. Int. J. Biochem. Cell Biol. 2013, 45, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, R.E.; Cheung, T.H.; Ahn, N.G. Global gene expression analysis of ERK5 and ERK1/2 signaling reveals a role for HIF-1 in ERK5-mediated responses. J. Biol. Chem. 2006, 281, 20993–21003. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.K.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822.e7. [Google Scholar] [CrossRef]
- Cristea, S.; Coles, G.L.; Hornburg, D.; Gershkovitz, M.; Arand, J.; Cao, S.; Sen, T.; Williamson, S.C.; Kim, J.W.; Drainas, A.P.; et al. The MEK5-ERK5 Kinase Axis Controls Lipid Metabolism in Small-Cell Lung Cancer. Cancer Res. 2020, 80, 1293–1303. [Google Scholar] [CrossRef]
- Belkahla, S.; Haq Khan, A.U.; Gitenay, D.; Alexia, C.; Gondeau, C.; Vo, D.-N.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; et al. Changes in metabolism affect expression of ABC transporters through ERK5 and depending on p53 status. Oncotarget 2018, 9, 1114–1129. [Google Scholar] [CrossRef]
- Carmell, N.; Rominiyi, O.; Myers, K.N.; McGarrity-Cottrell, C.; Vanderlinden, A.; Lad, N.; Perroux-David, E.; El-Khamisy, S.F.; Fernando, M.; Finegan, K.G.; et al. Identification and validation of ERK5 as a DNA damage modulating drug target in glioblastoma. Cancers 2021, 13, 944. [Google Scholar] [CrossRef]
- Miranda, M.; Rozali, E.; Khanna, K.K.; Al-Ejeh, F. MEK5-ERK5 pathway associates with poor survival of breast cancer patients after systemic treatments. Oncoscience 2015, 2, 99–101. [Google Scholar] [CrossRef]
- Jiang, W.; Cai, F.; Xu, H.; Lu, Y.; Chen, J.; Liu, J.; Cao, N.; Zhang, X.; Chen, X.; Huang, Q.; et al. Extracellular signal regulated kinase 5 promotes cell migration, invasion and lung metastasis in a FAK-dependent manner. Protein Cell 2020, 11, 825–845. [Google Scholar] [CrossRef]
- Van Dartel, M.; Cornelissen, P.W.A.; Redeker, S.; Tarkkanen, M.; Knuutila, S.; Hogendoorn, P.C.W.; Westerveld, A.; Gomes, I.; Bras, J.; Hulsebos, T.J.M. Amplification of 17p11.2∼p12, including PMP22, TOP3A, and MAPK7, in high-grade osteosarcoma. Cancer Genet. Cytogenet. 2002, 139, 91–96. [Google Scholar] [CrossRef]
- Zen, K.; Yasui, K.; Nakajima, T.; Zen, Y.; Zen, K.; Gen, Y.; Mitsuyoshi, H.; Minami, M.; Mitsufuji, S.; Tanaka, S.; et al. ERK5 is a target for gene amplification at 17p11 and promotes cell growth in hepatocellular carcinoma by regulating mitotic entry. Genes Chromosomes Cancer 2009, 48, 109–120. [Google Scholar] [CrossRef]
- Tesser-Gamba, F.; Petrilli, A.S.; de Seixas Alves, M.T.; Filho, R.J.G.; Juliano, Y.; Toledo, S.R.C. MAPK7 and MAP2K4 as prognostic markers in osteosarcoma. Hum. Pathol. 2012, 43, 994–1002. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Montero, J.C.; Ocaña, A.; Abad, M.; Ortiz-Ruiz, M.J.; Pandiella, A.; Esparís-Ogando, A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS ONE 2009, 4, e5565. [Google Scholar] [CrossRef]
- Antoon, J.W.; Martin, E.C.; Lai, R.; Salvo, V.A.; Tang, Y.; Nitzchke, A.M.; Elliott, S.; Nam, S.Y.; Xiong, W.; Rhodes, L.V.; et al. MEK5/ERK5 signaling suppresses estrogen receptor expression and promotes hormone-independent tumorigenesis. PLoS ONE 2013, 8, e69291. [Google Scholar] [CrossRef]
- Sánchez-Fdez, A.; Ortiz-Ruiz, M.J.; Re-Louhau, M.F.; Ramos, I.; Blanco-Múñez, Ó.; Ludeña, D.; Abad, M.; Sánchez-Martín, M.; Pandiella, A.; Esparís-Ogando, A. MEK5 promotes lung adenocarcinoma. Eur. Respir. J. 2019, 53, 1801327. [Google Scholar] [CrossRef]
- Thompson, J.K.; Shukla, A.; Leggett, A.L.; Munson, P.B.; Miller, J.M.; MacPherson, M.B.; Beuschel, S.L.; Pass, H.I.; Shukla, A. Extracellular signal regulated kinase 5 and inflammasome in progression of mesothelioma. Oncotarget 2018, 9, 293–305. [Google Scholar] [CrossRef]
- Wang, H.; Dai, Y.-Y.; Zhang, W.-Q.; Hsu, P.-C.; Yang, Y.-L.; Wang, Y.-C.; Chan, G.; Au, A.; Xu, Z.-D.; Jiang, S.-J.; et al. DCLK1 is correlated with MET and ERK5 expression, and associated with prognosis in malignant pleural mesothelioma. Int. J. Oncol. 2017, 51, 91–103. [Google Scholar] [CrossRef][Green Version]
- Mehta, P.B.; Jenkins, B.L.; McCarthy, L.; Thilak, L.; Robson, C.N.; Neal, D.E.; Leung, H.Y. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene 2003, 22, 1381–1389. [Google Scholar] [CrossRef]
- Ramsay, A.K.; McCracken, S.R.C.; Soofi, M.; Fleming, J.; Yu, A.X.; Ahmad, I.; Morland, R.; Machesky, L.; Nixon, C.; Edwards, D.R.; et al. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br. J. Cancer 2011, 104, 664–672. [Google Scholar] [CrossRef]
- Arias-González, L.; Moreno-Gimeno, I.; del Campo, A.R.; Serrano-Oviedo, L.; Valero, M.L.; Esparís-Ogando, A.; de la Cruz-Morcillo, M.Á.; Melgar-Rojas, P.; García-Cano, J.; Cimas, F.J.; et al. ERK5/BMK1 is a novel target of the tumor suppressor VHL: Implication in clear cell renal carcinoma. Neoplasia 2013, 15, 649–659. [Google Scholar] [CrossRef]
- Suenaga, S.; Ichiyanagi, O.; Ito, H.; Naito, S.; Kato, T.; Nagaoka, A.; Kato, T.; Yamakawa, M.; Obara, Y.; Tsuchiya, N. Expression of Extracellular Signal-regulated Kinase 5 and Ankyrin Repeat Domain 1 in Composite Pheochromocytoma and Ganglioneuroblastoma Detected Incidentally in the Adult Adrenal Gland. Intern. Med. 2016, 55, 3611–3621. [Google Scholar] [CrossRef]
- Obara, Y.; Nagasawa, R.; Nemoto, W.; Pellegrino, M.J.; Takahashi, M.; Habecker, B.A.; Stork, P.J.S.; Ichiyanagi, O.; Ito, H.; Tomita, Y.; et al. ERK5 induces ankrd1 for catecholamine biosynthesis and homeostasis in adrenal medullary cells. Cell. Signal. 2016, 28, 177–189. [Google Scholar] [CrossRef]
- Gentilini, A.; Lori, G.; Caligiuri, A.; Raggi, C.; Di Maira, G.; Pastore, M.; Piombanti, B.; Lottini, T.; Arcangeli, A.; Madiai, S.; et al. Extracellular Signal-Regulated Kinase 5 Regulates the Malignant Phenotype of Cholangiocarcinoma Cells. Hepatology 2021, 74, 2007–2020. [Google Scholar] [CrossRef]
- Bhatt, A.B.; Wright, T.D.; Barnes, V.; Chakrabarty, S.; Matossian, M.D.; Lexner, E.; Ucar, D.A.; Miele, L.; Flaherty, P.T.; Burow, M.E.; et al. Diverse and converging roles of ERK1/2 and ERK5 pathways on mesenchymal to epithelial transition in breast cancer. Transl. Oncol. 2021, 14, 101046. [Google Scholar] [CrossRef]
- Tian, X.; Sun, D.; Zhao, S.; Xiong, H.; Fang, J. Screening of potential diagnostic markers and therapeutic targets against colorectal cancer. Onco. Targets Ther. 2015, 8, 1691–1699. [Google Scholar]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef]
- Giurisato, E.; Lonardi, S.; Telfer, B.; Lussoso, S.; Risa-Ebrí, B.; Zhang, J.; Russo, I.; Wang, J.; Santucci, A.; Finegan, K.G.; et al. Extracellular-Regulated Protein Kinase 5-Mediated Control of p21 Expression Promotes Macrophage Proliferation Associated with Tumor Growth and Metastasis. Cancer Res. 2020, 80, 3319–3330. [Google Scholar] [CrossRef]
- Regan, C.P.; Li, W.; Boucher, D.M.; Spatz, S.; Su, M.S.; Kuida, K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. USA 2002, 99, 9248–9253. [Google Scholar] [CrossRef]
- Sohn, S.J.; Sarvis, B.K.; Cado, D.; Winoto, A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 2002, 277, 43344–43351. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Garin, G.; Xie, L.; Zheng, Q.; Wei, H.; Abe, J.; Yan, C.; Berk, B.C. BMK1/ERK5 is a novel regulator of angiogenesis by destabilizing hypoxia inducible factor 1alpha. Circ. Res. 2005, 96, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Finegan, K.G.; Perez-Madrigal, D.; Hitchin, J.R.; Davies, C.C.; Jordan, A.M.; Tournier, C. ERK5 is a critical mediator of inflammation-driven cancer. Cancer Res. 2015, 75, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Riegel, K.; Yurugi, H.; Schlöder, J.; Jonuleit, H.; Kaulich, M.; Kirschner, F.; Arnold-Schild, D.; Tenzer, S.; Schild, H.; Rajalingam, K. ERK5 modulates IL-6 secretion and contributes to tumor-induced immune suppression. Cell Death Dis. 2021, 12, 969. [Google Scholar] [CrossRef]
- Ren, J.; He, J.; Zhang, H.; Xia, Y.; Hu, Z.; Loughran, P.; Billiar, T.; Huang, H.; Tsung, A. Platelet TLR4-ERK5 Axis Facilitates NET-Mediated Capturing of Circulating Tumor Cells and Distant Metastasis after Surgical Stress. Cancer Res. 2021, 81, 2373–2385. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, R.; Guo, Z.; He, J. Cancer-associated fibroblasts promote cell growth by activating ERK5/PD-L1 signaling axis in colorectal cancer. Pathol. Res. Pract. 2020, 216, 152884. [Google Scholar] [CrossRef]
- Charni, S.; Aguilo, J.I.; Garaude, J.; de Bettignies, G.; Jacquet, C.; Hipskind, R.A.; Singer, D.; Anel, A.; Villalba, M. ERK5 knockdown generates mouse leukemia cells with low MHC class I levels that activate NK cells and block tumorigenesis. J. Immunol. 2009, 182, 3398–3405. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, P.; Li, X.; Wang, Q.; Deng, Z.-B.; Zhuang, X.; Mu, J.; Zhang, L.; Wang, B.; Yan, J.; et al. Restoration of miR17/20a in solid tumor cells enhances the natural killer cell antitumor activity by targeting Mekk2. Cancer Immunol. Res. 2014, 2, 789–799. [Google Scholar] [CrossRef]
- Loveridge, C.J.; Mui, E.J.; Patel, R.; Tan, E.H.; Ahmad, I.; Welsh, M.; Galbraith, J.; Hedley, A.; Nixon, C.; Blyth, K.; et al. Increased T-cell Infiltration Elicited by Erk5 Deletion in a Pten-Deficient Mouse Model of Prostate Carcinogenesis. Cancer Res. 2017, 77, 3158–3168. [Google Scholar] [CrossRef]
- Lin, E.C.K.; Amantea, C.M.; Nomanbhoy, T.K.; Weissig, H.; Ishiyama, J.; Hu, Y.; Sidique, S.; Li, B.; Kozarich, J.W.; Rosenblum, J.S. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 11865–11870. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Stolc, V.; Gauhar, Z.; Mason, C.; Halasz, G.; van Batenburg, M.F.; Rifkin, S.A.; Hua, S.; Herreman, T.; Tongprasit, W.; Barbano, P.E.; et al. A gene expression map for the euchromatic genome of Drosophila melanogaster. Science 2004, 306, 655–660. [Google Scholar] [CrossRef]
- Hillier, L.W.; Reinke, V.; Green, P.; Hirst, M.; Marra, M.A.; Waterston, R.H. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res. 2009, 19, 657–666. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef]
- Ellis, J.D.; Barrios-Rodiles, M.; Colak, R.; Irimia, M.; Kim, T.; Calarco, J.A.; Wang, X.; Pan, Q.; O’Hanlon, D.; Kim, P.M.; et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell 2012, 46, 884–892. [Google Scholar] [CrossRef]
- Bonomi, S.; Gallo, S.; Catillo, M.; Pignataro, D.; Biamonti, G.; Ghigna, C. Oncogenic alternative splicing switches: Role in cancer progression and prospects for therapy. Int. J. Cell Biol. 2013, 2013, 962038. [Google Scholar] [CrossRef]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef]
- Di Matteo, A.; Belloni, E.; Pradella, D.; Cappelletto, A.; Volf, N.; Zacchigna, S.; Ghigna, C. Alternative splicing in endothelial cells: Novel therapeutic opportunities in cancer angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 275. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Anamika, K.; Garnier, N.; Srinivasan, N. Functional diversity of human protein kinase splice variants marks significant expansion of human kinome. BMC Genom. 2009, 10, 622. [Google Scholar] [CrossRef]
- Druillennec, S.; Dorard, C.; Eychène, A. Alternative splicing in oncogenic kinases: From physiological functions to cancer. J. Nucleic Acids 2012, 2012, 639062. [Google Scholar] [CrossRef] [PubMed]
- El Marabti, E.; Younis, I. The cancer spliceome: Reprograming of alternative splicing in cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- English, J.M.; Vanderbilt, C.A.; Xu, S.; Marcus, S.; Cobb, M.H. Isolation of MEK5 and differential expression of alternatively spliced forms. J. Biol. Chem. 1995, 270, 28897–28902. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.J.; Abe, J.; Malik, S.; Che, W.; Yang, J. Differential role of MEK5α and MEK5β in BMK1/ERK5 activation. J. Biol. 2004, 279, 1506–1512. [Google Scholar] [CrossRef]
- McCaw, B.J.; Chow, S.Y.; Wong, E.S.M.; Tan, K.L.; Guo, H.; Guy, G.R. Identification and characterization of mErk5-T, a novel Erk5/Bmk1 splice variant. Gene 2005, 345, 183–190. [Google Scholar] [CrossRef]
- Kawakami, T.; Park, S.W.; Kaku, R.; Yang, J. Extracellular-regulated-kinase 5-mediated renal protection against ischemia-reperfusion injury. Biochem. Biophys. Res. Commun. 2012, 418, 603–608. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Tucker, J.A.; Tatum, N.J.; Wang, J.; Oxley, D.; Kidger, A.M.; Johnson, V.P.; Cassidy, M.A.; Gray, N.S.; Noble, M.E.M.; et al. Paradoxical activation of the protein kinase-transcription factor ERK5 by ERK5 kinase inhibitors. Nat. Commun. 2020, 11, 1383. [Google Scholar] [CrossRef]
- Pereira, D.M.; Rodrigues, C.M.P. Targeted Avenues for Cancer Treatment: The MEK5-ERK5 Signaling Pathway. Trends Mol. Med. 2020, 26, 394–407. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monti, M.; Celli, J.; Missale, F.; Cersosimo, F.; Russo, M.; Belloni, E.; Di Matteo, A.; Lonardi, S.; Vermi, W.; Ghigna, C.; et al. Clinical Significance and Regulation of ERK5 Expression and Function in Cancer. Cancers 2022, 14, 348. https://doi.org/10.3390/cancers14020348
Monti M, Celli J, Missale F, Cersosimo F, Russo M, Belloni E, Di Matteo A, Lonardi S, Vermi W, Ghigna C, et al. Clinical Significance and Regulation of ERK5 Expression and Function in Cancer. Cancers. 2022; 14(2):348. https://doi.org/10.3390/cancers14020348
Chicago/Turabian StyleMonti, Matilde, Jacopo Celli, Francesco Missale, Francesca Cersosimo, Mariapia Russo, Elisa Belloni, Anna Di Matteo, Silvia Lonardi, William Vermi, Claudia Ghigna, and et al. 2022. "Clinical Significance and Regulation of ERK5 Expression and Function in Cancer" Cancers 14, no. 2: 348. https://doi.org/10.3390/cancers14020348
APA StyleMonti, M., Celli, J., Missale, F., Cersosimo, F., Russo, M., Belloni, E., Di Matteo, A., Lonardi, S., Vermi, W., Ghigna, C., & Giurisato, E. (2022). Clinical Significance and Regulation of ERK5 Expression and Function in Cancer. Cancers, 14(2), 348. https://doi.org/10.3390/cancers14020348