Simple Summary
Recently, despite the common application of various novel therapies (e.g., immunotherapy and stem cell transplantation) in hematologic tumors, hematologic malignancies remain suboptimal and have a worse prognosis due to the lack of donors and their high heterogeneity. Among them, epigenetic alterations (e.g., the abnormal modification of m6A) are essential to facilitate the progression of tumors and drug resistance. Our purpose in this study is to pinpoint the molecular targets of pathogenesis, as well as to analyze the oncogenic characteristics of m6A modifications. In this article, we, therefore, elaborate on the mechanisms of m6A modification and its role in normal hematopoietic regulation and malignant tumorigenesis, thus contributing to the refinement of molecularly targeted therapies.
Abstract
Epigenetics is identified as the study of heritable modifications in gene expression and regulation that do not involve DNA sequence alterations, such as DNA methylation, histone modifications, etc. Importantly, N6-methyladenosine (m6A) methylation modification is one of the most common epigenetic modifications of eukaryotic messenger RNA (mRNA), which plays a key role in various cellular processes. It can not only mediate various RNA metabolic processes such as RNA splicing, translation, and decay under the catalytic regulation of related enzymes but can also affect the normal development of bone marrow hematopoiesis by regulating the self-renewal, proliferation, and differentiation of pluripotent stem cells in the hematopoietic microenvironment of bone marrow. In recent years, numerous studies have demonstrated that m6A methylation modifications play an important role in the development and progression of hematologic malignancies (e.g., leukemia, lymphoma, myelodysplastic syndromes [MDS], multiple myeloma [MM], etc.). Targeting the inhibition of m6A-associated factors can contribute to increased susceptibility of patients with hematologic malignancies to therapeutic agents. Therefore, this review elaborates on the biological characteristics and normal hematopoietic regulatory functions of m6A methylation modifications and their role in the pathogenesis of hematologic malignancies.
1. Introduction
Hematologic malignancies are a wide group of malignant clonal disorders of hematopoietic cells. Importantly, they typically manifest as uncontrolled proliferation and differentiation disorders of the hematopoietic cells, with high heterogeneity and poor prognosis, and so far, there is the lack of an effective cure. Currently, emerging evidence has revealed that the incidence of hematological malignancies ranks 6th in the total incidence of cancer in the world and 1st in the mortality rate of malignant tumors in adolescents [1]. Hematologic tumors are the second most common cause of cancer death in the United States. The occurrence of hematologic tumors is a complex multi-step process, and the malignancies mainly include lymphoma, leukemia, and myeloma, among other hematologic malignancies. In recent decades, studies [2,3] have shown that the pathogenesis of such diseases is associated with a variety of factors, including genomic abnormalities, epigenetic alterations, abnormal regulation of the bone marrow hematopoietic microenvironment, and disorders of the immune system.
In addition to the increasing number of patients suffering from this malignancy each year [1], inappropriate treatment modalities also lead to an annual increase in the mortality rate of this disease, which has inspired doctors to look for better treatment strategies. Notably, the progress of gene editing technologies and their application in the field of hematological tumors over the past decades have led to significant developments in the diagnosis and treatment of these malignancies [2]. Meanwhile, these findings shed light on the different mechanisms involved in the pathogenesis of hematologic malignancies [3]. Nevertheless, while most previous studies have focused on the study of genetic mechanisms of hematologic malignancies [4], recently, more attention has been paid to the field of epigenetics, which involves many closely related mechanisms regulating changes in gene expression levels without involving alterations in the DNA sequence [5].
In recent decades, epigenetics has been identified as a novel concept that corresponds to genetics and encompasses many types of well-documented epigenetic modifications. In plain words, epigenetic modification is a form of gene expression regulation that affects gene transcription and translation without changes in the nucleotide sequence and can regulate and thus affect gene expression at the level of DNA and chromatin structural modifications, RNA stability, and transcriptional activity, including DNA methylation modifications, histone covalent modifications, chromatin remodeling, non-coding RNA regulation, RNA modifications, etc. [6,7,8]. Further, epitranscriptomics is one of the newly emerging hot fields, which mainly focuses on the effects of chemical modifications carried by RNAs and their correlated regulators on gene expression. To date, it has been reported that more than one hundred chemical modifications (e.g., m6A) have been identified on RNAs, which perform extremely significant biological functions in living organisms via their involvement in mediating epigenetic regulation. More importantly, in recent years, epigenetic modifications have played a highly important role in the development and progression of hematologic malignancies (Figure 1).
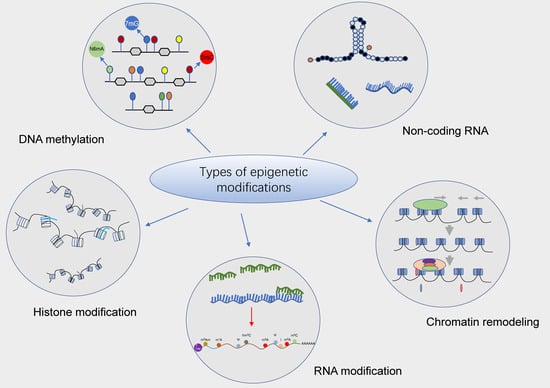
Figure 1.
Potential epigenetic regulatory mechanisms in hematologic malignancies. Epigenetic modifications are heritable alterations that can generate changes in gene activity independent of changes in gene nucleotide sequences, including DNA methylation, histone modifications, chromatin remodeling, non-coding RNA, and RNA and RNA modifications, etc., which primarily mediate changes in gene transcription as well as translation activity. Numerous studies have confirmed that epigenetic modifications (e.g., DNA methylation and histone modifications) play an essential role in the development and progression of hematologic malignancies and are considered to be a vital target for the treatment of different types of leukemias and other hematologic malignancies.
At present, epitranscriptomics, represented by N6-methyladenosine (m6A) modifications, has become a hot topic of research due to the current study of second-generation sequencing, i.e., targeting their epigenetic alterations at the transcriptome level [9,10,11,12]. In essence, m6A methylation modifications can mediate the post-transcriptional regulation of gene expression without altering base sequences. In addition, RNA m6A modification is reversible and dynamically modulated by m6A modifiers (e.g., writers, erasers, and readers), which have currently been proven to play an important role in regulating mRNA decay, stability, variable splicing, translation efficiency, and localization [11,13,14]. Moreover, m6A sites are also present in long non-coding RNAs as well as non-coding RNAs such as microRNAs.
With the rapid development of high-throughput m6A sequencing technology in recent years, accumulated evidence has supported that m6A and its related factors are involved in the hematopoietic development of bone marrow by regulating the self-renewal, proliferation and differentiation of pluripotent stem cells in the bone marrow hematopoietic microenvironment. Identically, emerging studies [4,7,15,16] have indicated that abnormal m6A modification is closely associated with the development and progression of hematologic malignancies. In this paper, we systematically review the progress of research on the biological characteristics of m6A methylation modifications, their effects on normal hematopoietic regulatory functions, and their role in hematologic malignancies. It is expected to provide a scientific basis for the development of novel molecularly targeted therapies based on the aberrant m6A modifications in related hematologic tumors.
2. Biological Features of m6A Methylation Modifications
2.1. Overview of m6A Methylation
RNA epigenetic modifications are an important part of RNA regulation [17]. Currently, more than 150 RNA modifications have been identified in eukaryotes, mainly occurring on mRNA, tRNA, rRNA, and other non-coding RNAs, which are regulated at the post-transcriptional level [18]. Of note, the m6A is a methylation modification located on the sixth nitrogen atom of RNA adenine, which is the most common type of modification in higher eukaryotic mRNAs [19,20], and the number of m6A modified adenines accounts for 0.2 to 0.5% of the total number of adenines [21]. Moreover, m6A modification is widely found not only in mammals but also in yeast, plants, protozoa, and various viruses [22,23]. With the rapid development of high-throughput sequencing technology, it has been recently found that m6A is sequentially distributed in the protein-coding sequence (CDS), near the stop codon and in the 3’ untranslated region (3’UTR) [24,25,26], i.e., mainly localized in the highly conserved sequence of mRNA RRACH motif (R = G/A; H = U/A/C) [27,28,29]. In addition, m6A methylation modifications are widely distributed in mRNA and non-coding RNA (ncRNA) and play important roles in the metabolism of many RNAs, including mRNA splicing, processing and maturation of microRNA (miRNA), and long non-coding RNA (lncRNA)-mediated transcriptional repression [30,31,32].
Precisely, m6A was first discovered in the mRNA of hepatocellular carcinoma cells in 1974 [20,33] and has since been detected in a variety of eukaryotic organisms such as mice and yeast whose biological functions involve cell differentiation, mitosis, immune homeostasis, and other aspects. As a dynamic and reversible epistatic modification, m6A possesses many recognition proteins, which can be classified into three major categories: “Writers”, “Erasers”, and “Readers”, according to their functions [11] (Figure 2). Importantly, the enzymatic reaction process is divided into three modification states in sequence according to the classification: (1) mRNA is methylated by the writer (m6A methyltransferase); (2) the process can be reversed by the eraser (m6A demethylase); and (3) the methylated mRNA is recognized by the reader (m6A binding protein) [32]. To sum up, the specific mechanism of m6A is illustrated as follows.
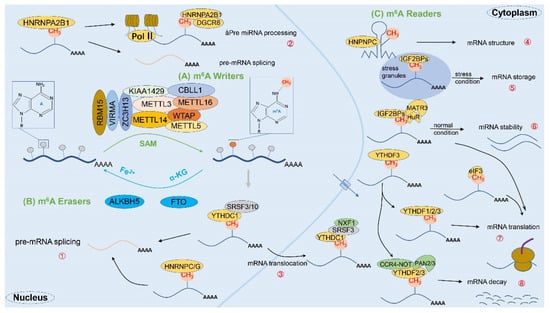
Figure 2.
The complete process of m6A methylation modification. The m6A methylation modification is a dynamic and reversible enzymatic process and is strictly modulated by writers, erasers, and readers. (A) First, mRNA methylation is catalyzed by writers (e.g., m6A methyltransferases METTL3, METTL14, WTAP, RBM15, VIRMA, and ZC3H13, etc.); (B) Second, the process can be reversed by erasers (e.g., m6A demethylases FTO and ALKBH5); (C) Finally, the methylated mRNA will be recognized by readers (e.g., m6A binding proteins YTHDF1/2/3, YTHDC1/2, HNRNPs, IGF2BP, eIF3, etc.), thereby fulfilling the corresponding physiological roles. Specifically: ① mRNA splicing: YTHDC1, HNRNPA2/B1, HNRNPC/G, etc.; ② miRNA processing: HNRNPA2/B1; ③ mRNA translocation: YTHDC1; ④ mRNA structure: HNPNPC; ⑤ mRNA storage: IGF2BPs; ⑥ mRNA stability: IGF2BPs; ⑦ mRNA translation: YTHDF1/2/3, YTHDC2, IGF2BPs, eIF3, etc.; ⑧ mRNA decay: YTHDF2/3, YTHDC2.
2.2. m6A-Related Enzymes
2.2.1. m6A Methyltransferase (Writers)
The m6A methyltransferase is also known as the writer [34]. The core components of the m6A methyltransferase complex, which performs m6A methylation, include three main species: METTL3 (methyltransferase-like 3), METTL14 (methyltransferase-like 14), and WTAP (Wilms’ tumor 1-associating protein) [35,36,37]. In fact, the m6A methyltransferase complex adds m6A methylation to the target mRNA through the methyl group on S-adenosylmethionine (SAM) transferase [38,39]. Among them, METTL3, with its N-terminal structural domain, methyltransferase structural domain, and two tandem zinc finger structural domains, is the main catalytic structure; on the other hand, METTL14 has a strong interaction with METTL3, but its methyl donor site is degraded and does not have catalytic activity, so it only plays a catalytic auxiliary role for METTL3 and maintains METTL3 activity [30,40]. METTL3–METTL14 heterodimer formation can induce m6A deposition on mammalian nuclear RNA [41,42]. Moreover, WTAP is not catalytically active, but it can help localize the complex to the nucleus and promote affinity between the complex and mRNA [43,44,45].
Indeed, METTL3 can catalyze the methylation modification of most mRNAs in vivo, and its homologue, METTL16 (methyltransferase-like protein 16), regulates the m6A modification of U6snRNA and a small fraction of mRNAs [46,47]. In addition, there are METTL5 (methyltransferase-like 5), ZCCHC4 (zinc finger CCHC-type containing 4), etc., which also have some catalytic functions for m6A on rRNAs [48]. In the methyltransferase complex, some new components including VIRMA (vir-Like m6A methyltransferase associated) [49,50,51], RBM15/15B(RNA binding motif protein 15/15B) [52], ZC3H13 (zinc finger CCCH-type containing 13) [53,54], etc. can interact with WTAP directly or indirectly as regulatory subunits, which together with WTAP form the MACOM complex and direct the localization of the whole complex in the cell [55,56]. Similarly, the activity of the methyltransferase complex is also susceptible to the influence of miRNAs [57].
Currently, it is generally known that m6A is co-transcriptionally modified on transcripts, and then METTL3 is recruited to chromatin in a transcription-dependent manner, mediating the methylation of nascent transcripts, and that the degree of methylation is strongly correlated with the activity of RNA polymerase II [58]. However, how the methyltransferase complex recognizes the substrate RNA and how the activity of the complex is regulated remain to be further investigated.
2.2.2. m6A Demethylases (Erasers)
The m6A demethylases are called erasers, and their role is to catalyze the removal of methyl from m6A that has been methylated. Mechanistically, m6A methyltransferases and demethylases affect post-transcriptional gene expression levels by regulating dynamic changes in m6A methylation modifications of mRNA sequences. In fact, the two types of m6A demethylases identified so far are FTO (fat mass and obesity-associated gene) [59] and ALKBH5 (alkB homolog 5) [60]. It has been reported that FTO is an obesity-associated protein with the role of catalyzing m6A demethylation [59,61]. FTO belongs to the α ketoglutaric acid (α-KG) and divalent iron ion Fe(II) dependent ALKB dioxygenase family, which is localized in both the nucleus and cytoplasm of cells [61]. It possesses two structural domains: the N-terminal domain, which is conserved with the dioxygenase family sequence, and the C-terminal domain, which mainly plays a scaffolding role. The mechanism by which FTO affects the demethylation of m6A is that m6A is first oxidized to hm6A (N6-hydroxymethyladenosine) and f6A (N6-formyladenosine), then formaldehyde and formic acid are removed to complete the demethylation. Ultimately, adenine is produced [40,62].
However, the substrate of FTO is not only m6A but also N6,2’-O-dimethyladenine (m6Am) and m1A on tRNA are its catalytic substrates [11]. For example, in 2019, Guifang Jia’s group obtained the crystal structures of the human FTO nucleic acid complexes and resolved the interaction mechanism of FTO with each substrate [63]. It was found that although the FTO active pocket can recognize molecules of multiple RNA modification substrates, it prefers to bind N6-methyladenine bases and has the same demethylation activity for internal m6A and m6Am with the same RNA sequence (N6-methyladenine bases), indicating that the demethylation activity of FTO is mainly dependent on the recognition of residues and bases in its catalytic pocket rather than on the ribose ring and that different RNA sequences and tertiary structures also affect the catalytic function of FTO. Thus, these findings provide an understanding of the catalytic mechanism of how FTO demethylates a variety of substrates and provide potential research directions for future selective chemistries for cancer therapy.
In recent studies, ALKBH5 was identified to be localized in the nucleus, and either silencing or overexpression of ALKBH5 could alter m6A levels [60]. More importantly, ALKBH5, similar to FTO, is also a member of the divalent iron ion, α-KG-dependent ALKB dioxygenase family. However, unlike FTO, the demethylation regarding the m6A modification site of target genes mediated by ALKBH5 does not require a multi-step reaction but is performed directly. At present, it has been indicated that ALKBH5 may regulate the out-of-nucleus transport of mRNAs [11], which is ultimately involved in the development of multiple diseases [64]. Therefore, as an m6A demethylase, it has been conclusively demonstrated that the knockdown of ALKBH5 in mouse models does not affect health status other than causing impaired spermatogenesis in mice, which makes ALKBH5 a potential therapeutic target in the future [65,66]. Collectively, the discovery of methyltransferases and demethylases confirms that m6A modification is dynamically reversible and that both work together to maintain a dynamic balance of cellular m6A methylation and demethylation.
2.2.3. m6A Binding Proteins (Readers)
Currently, the m6A-binding protein is recognized as the reader. Specifically, m6A-binding proteins can regulate the biological behavior and function of mRNA by reading m6A methylation, and the YTH domain family, which binds to RNA, is the first of the “Readers” found to directly recognize m6A [67]. YTH family proteins, as reading proteins of m6A, are mainly divided into two isoforms, i.e., a total of five family members: YTH domain family protein (YTHDF1/2/3) and the nucleus member YTH domain-containing (YTHDC1/2) in humans. At present, the mechanism of m6A recognition by the YTH domain is relatively well understood [67,68]. The YTH domain contains several conserved aromatic amino acids, and the benzene ring on it not only interacts with m6A adenine and methyl groups but also forms a cage-like tertiary structure, which firmly holds the methyl group of m6A, thus recognizing m6A [69]. In terms of functional studies, YTH domain family proteins play a rather important role in regulating RNA stability [67], translation efficiency [68], etc. For instance, YTHDF1/2/3 are dominantly found in the cytoplasm, and the binding of YTHDF1 and YHTDF3 to each other can promote the translation efficiency of m6A-modified mRNAs to facilitate protein synthesis and eventually mediate the mRNA degradation due to YTHDF2 [11]. Meanwhile, YTHDF2 mediates the degradation of target gene mRNA to modulate the stability of mRNA, thereby regulating the level of gene expression in somatic cells. Notably, it can directly recruit and bind to the SH domain of the scaffold subunit CNOT1 of the CCR4-NOT (the carbon catabolite repression 4-negative on TATA-less complex) deadenylase complex and subsequently transfer to the RNA containing the m6A modification to degrade that RNA, or it can use the heat-responsive protein HRSP12 (heat-responsive protein 12) as a bridge to shear the interior of the RNA by RNaseP/MRP (ribonuclease P and RNase MRP) nucleases [55,70].
In 2019, Gao et al. [71] revealed that a segment of the disordered region upstream of the YTH domain in the YTHDF protein, i.e., the low complexity domain (LCD), is capable of m6A-dependent phase separation, which may be related to the stress phenomenon [71]. However, recently Zaccara et al. [72] proposed that YTHDF1/2/3 are not involved in the regulation of translation, and their protein structures are nearly identical, suggesting a non-specific recognition of m6A modifications for RNA degradation in a redundant manner. In addition, an analysis conducted by another laboratory using the QTLs(quantitative trait loci) significantly showed that m6A has a complex effect on translation and does not simply promote or repress it [73].
Interestingly, YTHDC1, belonging to the YTH family, is a protein localized to the nucleus. It can interact with the precursor mRNA splicing factors serine/arginine-rich splicing factor 3 (SRSF3) and SRSF10 [74] and then binds to the m6A site of mRNA to regulate the selective splicing of mRNA in the nucleus. Furthermore, the mRNA exit factor NXF1 binds closely to SRSF3 and promotes the exit of m6A-modified mRNAs from the nucleus [74,75]. On the other hand, Li et al. [76] found that YTHDC2 is present in the cytoplasm, and it not only promotes the translation of m6A-modified mRNA but also accelerates the degradation of m6A-modified mRNA. In conclusion, YTHDC2 is firmly believed to regulate the translation efficiency and stability of RNA.
Apart from the above, there are other factors that are also involved in the mechanism of m6A modification, and more still need to be investigated in the future. Among them, one class of binding proteins belongs to the family of heterogeneous nuclear ribonucleoproteins (HNRNPs), including HNRNPC/G and HNRNPA2/B1. In particular, HNRNPA2/B1 possesses two tandem RNA recognition motifs (RRM) and a low complexity region (LC) at the C-terminus [77]. As an intranuclear m6A reader, HNRNPA2/B1 recognizes the RGm6AC motif (the m6A core motif RGAC) and regulates the selective shearing of RNA in a METTL3-dependent manner. Moreover, it is also able to mediate and facilitate the processing of precursor miRNAs; when knockdown of HNRNPA2/B1 occurs, the variable shearing of cells will be abnormal, ultimately leading to a significant reduction of mature miRNAs [78]. Similarly, the RGG motif (Arg-Gly-Gly) in the LC region of HNRNPG can directly bind to the phosphorylated carboxy-terminal domain (CTD) of RNA polymerase II and recognize m6A modifications near the nascent RNA splice site, finally interacting with the nascent RNA to regulate the selective splicing of transcripts [79]. Overall, m6A modifications can reshape RNA structure and subsequently regulate RNA–protein interactions around or near it, which is referred to as the “m6A-switch” [80].
In recent years, it has been shown that insulin-like growth factor 2 messenger RNA (mRNA)-binding proteins (IGF2BPs) are identified as an additional class of readers. As the conserved family of single-stranded RNA-binding proteins (RBPs), it structurally contains six RNA-binding domains, including two RRM domains and four K-homology (KH) domains [81]. It has been reported experimentally that the KH3-4 domains of IGF2BPs proteins are essential for the recognition of m6A-modified mRNAs in vitro. In addition, IGF2BPs have the function of stabilizing mRNA, which may be achieved by their ability to recruit some RNA stabilizing proteins such as ELAV-like RNA-binding protein 1(ELAVL1), matrin3(MATR3) to function [82]. Furthermore, some recent studies have also explored other binding proteins, e.g., fragile X mental retardation protein (FMRP) [83], proline-rich coiled-coil 2A (PRRC2A) [84], eukaryotic initiation factor 3 (eIF3) [85], etc., which have a role in regulating RNA stability and RNA exit from the nucleus. However, the specific mechanisms need to be further studied.
2.3. m6A Modifications in Virus-Host Interaction
It has recently been revealed that abnormalities in m6A modification play an essential role in the development of human diseases, especially in diseases caused by pathogenic viruses. For instance, some viruses that pose a serious threat to human health, including human immunodeficiency virus type 1 (HIV-1), Epstein–Barr virus (EBV), hepatitis B virus (HBV), hepatitis C virus (HCV), human t-cell leukemia virus (HTLV), and human herpes virus 8 (HHV 8), act as a major cause of disease [86,87,88,89]. Upon their infection of human host cells, their RNAs are all subjected to varying degrees of m6A modifications. Thus, m6A modifications are involved in modulating the life cycle of viruses by affecting the process of their replication. Mechanistically, m6A modification in host cells affects the replication of pathogenic viruses and the expression of related important genes; simultaneously, the immune response of the host to viruses is also greatly influenced by m6A methylation. Importantly, some viral infections, such as HIV-1, EBV, HTLV, etc., can contribute to the occurrence of some hematologic malignancies (e.g., lymphoma, leukemia, etc.) [89].
3. m6A Methylation Modification and Normal Hematopoietic Regulation
To date, further studies on the function of m6A-related enzymes confirmed that abnormal activity of these genes led to abnormal expression of thousands of genes, firmly suggesting an important role of m6A in RNA metabolism. Specifically, m6A may affect lipid metabolism [90], sperm development [91], tumorigenesis, stem cell-directed differentiation [92], cellular reprogramming [93], biological clock rhythms, cell division, memory, and neurodevelopment [94], as well as several other life processes. Therefore, given the critical role of m6A in the regulation of epigenetic events, these classes of enzymes have been identified to be involved in a variety of cellular life activities, particularly in the normal regulation of hematopoiesis in the hematological system (Figure 3).
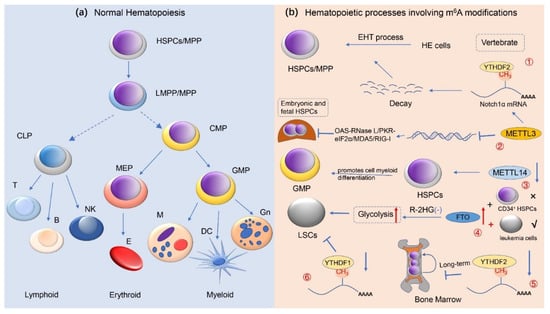
Figure 3.
m6A methylation modifications and normal hematopoietic regulation. This image depicts the role of RNA m6A methylation in normal human hematopoiesis. (a) The development and differentiation process with respect to human normal hematopoietic stem/progenitor cells (HSPCs) is presented here, i.e., from HSPCs to terminally differentiated erythroid, lymphoid, and myeloid cells; (b) It has revealed that m6A modification modulates the normal hematopoietic system. ① METTL3-mediated m6A modifications regulate HSPC fate specification via suppressing Notch signaling during early definitive hematopoiesis. ② During the expansion of HSPC in the fetal liver, METTL3 deletion promotes the formation of dsRNA, activates the OAS RNase L and PKR-eIF2a pathways, and upregulates MDA5/RIG-I, resulting in hematopoietic failure. ③ METTL14 is highly expressed during the development of normal CD34+ HSPCs cells, and silencing METTL14 facilitates terminal myeloid differentiation of HSPCs cells. ④ In R-2HG-sensitive leukemic cells but not in normal CD34+ HSPCs, overexpression of FTO reverses the effect of R-2HG-induced glycolysis inhibition, leading to leukemogenesis in vivo. ⑤ Deficiency of YTHDF2 causes the failure of hematopoietic stem cells during serial transplantation and prolonged activation of pro-inflammatory pathways, ultimately contributing to progressive bone marrow bias. ⑥ YTHDC1 deletion impedes the proliferation and survival of LSCs in vivo, supporting the oncogenic role of YTHDC1 in leukemias (e.g., AML). Abbreviations: HSPCs hematopoietic stem/progenitor cells; MPP multipotent progenitors; LMPP lymphoid-primed multipotential progenitors; CMP common myeloid progenitor; CLP common lymphoid progenitor; GMPs granulocyte/monocyte progenitor; MEPs megakaryocytes/erythroid progenitor; Gn granulocyte; DC dendritic cell; M monocyte; E erythrocyte; NK natural killer cell; EHT endothelial-to-hematopoietic transition; LSCs leukemia stem cells; HE hemogenic endothelial; R-2HG R-2-hydroxyglutarate.
3.1. m6A Methylation Modification Regulates Hematopoietic Stem/Progenitor Cell Differentiation
Indeed, the hematopoietic system guarantees a lifelong source of blood cells, which are often derived from rarely occurring multipotent hematopoietic stem/progenitor cells (HSPCs) populations with long-term self-renewal capacity and differentiation potential [95]. The mature blood cells that differentiate from HSPCs are of two main types: myeloid and lymphoid. Within these two categories, the former include granulocytes, erythroid cells, monocytes, and megakaryocytes, while the latter contains T lymphocytes, B lymphocytes, and natural killer cells (NK cells). The hematopoietic process of the body normally begins in early embryonic development and is strictly regulated during this time. Genetic alterations such as genetic mutations and chromosomal translocations during the hematopoietic process can lead to the development of hematologic tumors if the differentiation of HSPCs is blocked or their proliferation is abnormal [96].
Recent studies have demonstrated that RNA m6A methylation is closely related to normal hematopoiesis and related processes in vertebrates [97]. Vertebrate HSPCs are generated from hemogenic endothelial (HE) cells through the endothelial-to-hematopoietic transition (EHT) process [98]. In 2017, Zhang et al. [99] found in zebrafish blood and vascular tissues that m6A could regulate the differentiation process of HSPCs by affecting the balance of HE cells gene expression during EHT through YTHDF2-mediated degradation of notch receptor 1α (Notch1α) mRNA. Furthermore, emerging evidence also indicated that deletion of METTL3 in vascular endothelial cells significantly represses the EHT process by reducing the level of m6A methylation modification of Notch1αmRNA, thereby hindering the generation of HSPCs. Hence, m6A modulates the production of HSPCs during the hematopoietic process identified early in zebrafish embryogenesis. Additionally, a similar phenotype was observed in mouse models with knockdown of METTL3 [45,100]. All of the above findings indicate that m6A methylation modifications are tightly associated with the homeostatic regulation of gene expression in HE cells.
More recently, another study [101] showed that the role of METTL3 is remarkable for the development of mammalian hematopoiesis. In the Vav-Cre+-Mettl3fl/fl (vcMettl3−/−) mouse model, deficiency of METTL3 and m6A resulted in hematopoietic failure accompanied by the proliferation of Lin−Sca-1+c-Kit+ (LSK) HSPCs in the fetal liver that is defective in hematopoiesis. Based on further analysis, deletion of m6A can lead to significantly upregulated transcription of the interferon-stimulated genes (ISGs) and 2′,5′-oligoadenylate synthetase (Oas) genes, which induces a poor double-stranded RNA (dsRNA)-mediated innate immune response. Conversely, by utilizing m6A modification, i.e., METTL3 supplementation, the OAS-RNase L and PKR-eIF2α signaling pathways, and the upregulation of the dsRNA sensor MDA5 and RIG-I in HSPCs could be suppressed, thereby preventing the formation of endogenous dsRNA and the deleterious innate immune response, ultimately avoiding hematopoietic failure and perinatal lethality. Similar to METTL3, METTL14 [102] is a key component of the m6A methyltransferase complex and is significantly increased in expression in HSPCs. However, silencing METTL14 significantly promotes cell myeloid differentiation during the development of normal CD34+ HSPCs cells. Taken together, this result highlights the critical role of METTL14 and m6A modifications in normal hematopoiesis.
Of course, the m6A eraser (e.g., FTO) is dispensable for normal HSPCs. At present, it has been reported that overexpression of FTO [103] in R-2HG-sensitive leukemic cells but not normal CD34+ HSPCs attenuates R-2-hydroxyglutarate-(R-2HG)-induced glycolysis inhibition, thereby promoting leukemogenesis in vivo. In addition, as an m6A demethylase, knockdown of ALKBH5 has been proven not to affect normal hematopoiesis and the function of HSPCs in the steady-state of the organism [65,66]. In contrast, compared to normal HSPCs or other types of stem cells, Shen et al. [65] revealed that ALKBH5 deficiency significantly inhibits tumor proliferation in many types of tumors, such as breast cancer, brain tumors, and acute myeloid leukemia (AML). In fact, the role of the m6A mRNA reader YTHDF2 in hematopoiesis is highly complex. Furthermore, accumulating evidence [104,105,106] has shown that YTHDF2 deficiency facilitates the massive expansion of HSPCs in human cord blood and mice and demonstrates for the first time the function of YTHDF2 in the maintenance of adult hematopoietic stem cells (HSCs), thus providing therapeutic potential for future clinical applications. Interestingly, based on the analysis of a recent study on young mice, Mapperley et al. [107] also demonstrated that YTHDF2 deficiency causes the failure of HSCs during serial transplantation and the prolonged activation of pro-inflammatory pathways which in turn eventually leads to progressive hematopoietic loss. Thus, this study emphasizes the importance of YTHDF2 in the long-term maintenance of HSCs expansion. Apart from YTHDC2, another study [108] identified that YTHDC1 is also essential for maintaining the normal hematopoiesis and functional development of HSPCs in vivo. In an experiment on three mouse models, i.e., Ythdc1fl/+ (WT), Ythdc1fl/+Mx1Cre (Ythdc1 HET), and Ythdc1fl/flMx1Cre (Ythdc1 KO), whole blood cells as well as mature cells (e.g., myeloid, B cells, and T cells) in the serum of Ythdc1 KO mice were markedly reduced, and all mice died within three weeks. These results indicate that YTHDC1 is required for HSPC survival and that YTHDC1 deficiency leads to rapid hematopoietic failure. Further studies revealed that YTHDC1 KO but not YTHDC1 haploinsufficiency has a significant negative effect on the maintenance of HSPCs. However, when YTHDC1 is overexpressed, it hinders the differentiation of HSPCs and increases the proliferation/self-renewal of HSPCs, which supports the oncogenic role of YTHDC1 in HSPCs. However, the molecular mechanisms of other m6A readers with regard to the regulation of normal hematopoiesis are still unclear.
To conclude, the above studies unveil that RNA m6A methylation is essential in modulating the differentiation of HSPCs.
3.2. m6A Methylation Modification Regulates the Differentiation of Bone Marrow Mesenchymal Stem Cells
Bone marrow mesenchymal stem cells (BMSCs), which are characterized by their self-renewal and multidirectional differentiation potential, are a different type of stem cells from HSPCs that exist in the bone marrow and can differentiate into a variety of bone marrow stromal cells, such as osteoblasts, chondrocytes, adipocytes, etc., under different induction conditions [109]. Currently, emerging studies have indicated that RNA m6A also plays a crucial role in regulating the differentiation of BMSCs. For example, Yao et al. [110] found that METTL3 regulates the expression of Janus kinase 1 (JAK1) in BMSCs in an m6A methylation-dependent manner, while YTHDF2 affects the expression level of JAK1 by regulating the stability of JAK1 gene mRNA. Meanwhile, in this study, it has been proposed that reducing the m6A level of JAK1 gene mRNA in BMSCs can be achieved by regulating the expression of Janus kinase 1/signal transducer and activator of transcription 5/the promoter of CCAAT/enhancer binding proteinβ (JAK1/STAT5/C/EBPβ) signaling pathway to promote the differentiation of BMSCs to adipocytes. In another study, Cen et al. [111] revealed that TRAF4 could negatively regulate adipogenesis in mesenchymal stem cells (MSCs). Recently, it has been reported that TRAF4 binds to PKM2 and activates β-catenin signaling by activating the kinase activity of PKM2, which then inhibits adipogenesis in vitro and in vivo. Additionally, the expression of TRAF4 is reduced during adipogenesis, regulated by ALKBH5-mediated RNA m6A demethylation.
Specifically, BMSCs belong to the multipotential stromal cells in the hematopoietic microenvironment, which are involved in regulating the homeostasis of the bone marrow hematopoietic microenvironment. Through the synthesis and secretion of various hematopoietic factors, they can contribute to the self-renewal, proliferation, and differentiation of HSPCs, thus maintaining the balance of the hematopoietic process. In addition, osteoblasts not only are capable of maintaining the activity of HSPCs but also participate in maintaining the normal hematopoietic function of the body. In the METTL3 knockout mouse model constructed by Wu et al. [112], mouse skeletons showed a pathological phenotype associated with osteoporosis, suggesting that METTL3-mediated m6A methylation modifications play a critical role in regulating the differentiation of BMSCs. Mechanistically, the above study also observed that parathyroid hormone/parathyroid hormone receptor-1 (PTH/PTH1R) is a downstream signaling pathway of m6A, and the deletion of METTL3 inhibits PTH1R expression only at the translational level, which in turn indicates that m6A can affect osteogenic and lipogenic differentiation of BMSCs by positively regulating the PTH/PTH1R signaling pathway.
3.3. m6A Methylation Modification Regulates Cellular Reprogramming
Cellular reprogramming, on the one hand, refers to the process by which terminally differentiated cells are reversed under specific conditions and then restored to a totipotent state, ultimately either forming an embryonic stem cell line or developing further into a new individual. On the other hand, under embryonic stem cell (ESC) culture conditions, induced pluripotent stem cells (iPSCs) are defined as: mouse embryonic fibroblast (MEF) cells that are transferred with a total of four transcription factors [e.g., octamer-binding transcription factor 4 (Oct4), SYR-box 2 (Sox2), v-myc avian myelocytomatosis viral oncogene homolog (c-Myc), and Kruppel-like factor 4 (Klf4)] were subsequently induced into cells with ESC characteristics [113]. Actually, Chen et al. [57] showed that in MEF cells that express four pluripotency factors Oct4, Sox2, c-Myc, and Klf4, overexpression of METTL3 significantly increases m6A levels; moreover, the expression of OCT4, SOX2, and NANOG homologous frame protein pluripotency factors was also increased in METTL3 overexpressing MEF cells, indicating that m6A may modulate the expression levels of pluripotency factors in somatic cells, thereby promoting the reprogramming of somatic cells into pluripotent stem cells.
Simply put, c-MYC is a major regulator of self-renewal and differentiation in normal hematopoietic and leukemic cells. In particular, another recent finding [114] has confirmed that METTL3 can identically enhance the expression of c-MYC protein in leukemic cells by means of increasing the level of m6A modification of c-Myc gene mRNA, thereby inhibiting cell differentiation and promoting the self-renewal of leukemic cells, and exerting oncogenic effects in hematological tumors. Conversely, the deletion of METTL3 could increase the level of phosphorylated AKT, which contributes to promoting differentiation and apoptosis in METTL3-deficient cells. Overall, these results provide a research rationale for METTL3 exploration regarding the regulation of HSPCs function or the treatment of myeloid leukemia [114].
4. m6A Methylation Modifications and Hematological Malignancies
Hematologic malignancies, a heterogeneous group of malignancies, occur as a result of malignant transformation of HSPCs, which lose the ability to further differentiate and mature, and thus are blocked at different nodes [115]. They can be divided into two types: myeloid and lymphocytic. Furthermore, the specific disease classification consists of the following four major areas: various types of leukemia, multiple myeloma, malignant lymphoma, and myelodysplastic syndromes. Among them, multiple myeloma accounts for ~10% of the overall incidence in hematologic malignancies [116]. However, the incidence of MM has steadily increased in recent years.
Recent studies have shown that RNA m6A methylation is closely correlated with various malignant diseases and their related processes. Here, we concentrate only on RNA m6A methylation in human cancers related to the hematological system. At present, a large number of studies have confirmed the considerable place of aberrant RNA m6A expression in the development, progression, and recurrence of different types of hematologic malignancies (Table 1).

Table 1.
The functional roles of RNA m6A methylation modification in various types of hematologic malignancies.
4.1. The Role of m6A Methylation Modifications in Acute Leukemia
4.1.1. RNA m6A Methylation and Acute Lymphoblastic Leukemia
Acute lymphoblastic leukemia (ALL) is a group of hematologic neoplasms characterized by the malignant proliferation of primitive naïve lymphocytes. It has been shown that ALL has a peak incidence in childhood (1 to 9 years) and constitutes >70% of childhood leukemias. In particular, BCR-ABL fusion gene-positive ALL (BCR-ABL+ ALL) accounts for 20–30% of ALL in adults and approximately 3–5% of ALL in children, with the poor effect of conventional chemotherapy, highly susceptible to relapse, and extremely poor prognosis [139].
A recent study found a high association between the expression level of m6A catalytic enzymes genes and disease burden in ALL. Mechanistically, using real-time quantitative polymerase chain reaction (qPCR) experiments prior to disease induction therapy, Wang et al. [117] detected increased gene expression levels of m6A-modified methylases (e.g., METTL3, METTL14, WTAP) and demethylases (e.g., FTO and ALKBH5) in ETV6/RUNX1-positive ALL patients, but decreased significantly after induction therapy. Thus, the present study suggests that dysregulation of m6A may also be involved in ALL. However, the complete mechanism of whether m6A modification levels affect the development of ALL still deserves to be further investigated. Similarly, in 2021, another recent study [119] has demonstrated that ubiquitin-specific proteases (USPs) are highly correlated with the development of t-cell acute lymphoblastic leukemia (T-ALL) and are chronically resistant to chemotherapeutic drugs. Importantly, it has been shown that ALKBH5 plays an oncogenic role in the development of human cancers, which in turn would increase the stability of USPs mRNA. In an in vitro experiment, Gong et al. [119] reported that high expression of ubiquitin-specific protease 1 (USP1) was associated with poor prognosis in glucocorticoid (GC)-resistant T-ALL patients. On the contrary, the knockdown of USP1 improved CEM-C1 cells’ response to dexamethasone (Dex) sensitivity, thus contributing to promoting apoptosis and exerting oncogenic effects in ALL. Simultaneously, it was also indicated that drug resistance in T-ALL patients could be mediated by the interaction and deubiquitination between USP1 and Aurora B. Mechanistically, further studies confirmed that ALKBH5 enhanced USP1 expression via modulating USP1 RNA m6A demethylation levels and elevating mRNA stability; downregulation of ALKBH5 could decrease USP1 and Aurora B levels to promote the sensitivity of CEM-C1 cells to Dex and inhibited the proliferation of tumor cells. However, when USP1 was overexpressed in CEM-C1 cells, the effect of ALKBH5 could be reversed. Furthermore, in the in vivo experiments in mice, using tail vein injection of sh-USP1, the growth of tumors in mice was apparently inhibited, thereby resulting in a significantly prolonged survival period, which is of reference value for clinical researches on tumor therapies.
Similarly, m6A modifications are essential in the pathogenesis of pediatric ALL (P-ALL). Using real-time qPCR (RT-qPCR), Sun et al. [118] detected that the expression levels of METTL3 and METTL14 are decreased in ETV6/RUNX1-positive P-ALL, indicating a possible role in the pathogenesis and progression of E/R-positive P-ALL, and thus laying the groundwork for specific treatments targeting patients with possible E/R-positive relapses. In two five-center case-control studies, m6A modifications were equally closely associated with increased tumor risk in P-ALL [140,141]. Using single nucleotide polymorphism (SNP) analysis, they found that METTL3 and METTL14 gene polymorphisms can elevate the risk of developing P-ALL, suggesting that their genetic polymorphisms may be potential biomarkers for P-ALL susceptibility and selection of oncologic chemotherapeutic drugs.
Taken together, the above results show that abnormalities of m6A modification-related enzymes represented by ALKBH5 may be a potential condition for the development, recurrence and drug resistance of ALL, which provides a novel research direction for its clinical diagnosis and treatment.
4.1.2. RNA m6A Methylation and Acute Myeloid Leukemia
Acute myeloid leukemia (AML) belongs to a myeloid neoplasm with a highly heterogeneous malignant clonal proliferation of HSPCs and is the most common type of adult acute leukemia [142]. Importantly, leukemic cells are characterized by enhanced self-renewal, uncontrolled proliferation, impaired differentiation, and suppressed apoptosis, and accumulation in the bone marrow leading to a decrease in normal hematopoietic cells [143]. In spite of advances in intensive chemotherapy and allogeneic stem cell transplantation(allo-SCT), the prognosis of the majority of AML patients remains poor, with a 5-year survival rate of only approximately 24%, due to refractoriness, relapse, therapy resistance, and therapy-related mortality [144].
At the molecular level, the development of AML follows a “multiple-hit” pattern [145]. In particular, dysregulation of m6A-related components induces oncogene expression and thus promotes the development of malignant tumors [146]. In addition, m6A exerts an important effect in AML progression, as it enhances the self-renewal capacity of AML cells. A recent study presented by Sheng’s group [108] found that YTHDC1, a nuclear m6A reader, is highly expressed in AML and that it contributes to the maintenance of AML cell proliferation and progression; knockdown of the YTHDC1 gene significantly blocked the proliferation of primary AML cells via modulating MCM complex-mediated DNA replication, as well as the self-renewal of LSCs in vivo in mice, thus achieving control of leukemogenesis. Collectively, this finding highlights the unique oncogenic mechanism of the m6A modifier YTHDC1 in AML. Consistently, another evidence [120] demonstrated that YTHDC1 is proven to be an essential m6A reader in myeloid leukemia by applying genome-wide CRISPR screening technology. Under the mediation of mRNA m6A, the production of nuclear YTHDC1-m6A condensates (nYACs) is markedly increased in AML cells compared to normal HSPCs, thereby maintaining the cell survival and undifferentiated state, which is essential for AML development. Additionally, it has been reported that nYACs can contribute to maintaining the stability of mRNA m6A and avoid degradation by PAXT complex and exosome-related RNAs. Hence, the result shows that m6A can maintain mRNA stability and play a vital role in the pathogenesis of AML.
In 2017, Vu et al. [114] found that METTL3 is highly expressed in AML cells and can promote AML cell proliferation, inhibit cell differentiation, and ultimately play an oncogenic role in AML through mediating m6A methylation modification; downregulation of METTL3 gene can induce the differentiation and apoptosis of AML cells by increasing the phosphorylation level of phosphate protein kinase B (PKB/AKT), but not apoptosis in normal hematopoietic cells. Meanwhile, the study also found that m6A modification could exert oncogenic effects via elevating the translation level of related mRNAs [e.g., c-MYC, B-cell lymphoma/leukemia-2 (BCL2), phosphatase and tensin homolog deleted on chromosome ten (PTEN) gene, etc.] in AML cells, thus further suggesting that METTL3 can act as an oncogenic factor in myeloid malignancies, which is an important reference value for clinical treatment. Similarly, the molecular mechanism between METTL3 and adipogenesis in mesenchymal stem cells of AML patients (AML-MSCs) has been currently identified [147]. Based on the analysis of m6A epigenetic changes in MSCs identified by RIP-qPCR and MeRIP-qPCR, deletion of METTL3 in AML-MSCs can facilitate increased production of AKT protein through controlling m6A modifications of AKT1-mRNA, leading to excessive MSC adipogenesis and consequently making AML cells resistant to chemotherapy. METTL14 is another important component of the m6A methyltransferase complex. Weng et al. [102] showed that METTL14 is also highly expressed in AML cells and can play a critical role in the pathogenesis of both normal myelopoiesis and AML via mediating m6A methylation modifications that block the differentiation of normal myeloid cells and promote malignant hematopoiesis. Overall, abnormalities in m6A-related factors, represented by METTL3 and METTL14, may be potential factors in the development of hematologic malignancies (e.g., AML), which provide a novel research direction for their clinical diagnosis and treatment.
Apart from the aforementioned studies in m6A, the role of other m6A modifiers [122] in the genesis of AML has been recently well-recognized. For instance, Feng et al. [122] recently provided evidence showing that YBX1, the RNA binding proteins (RBPs), is responsible for promoting the survival of myeloid leukemia cells in an m6A-dependent manner; conversely, its deletion dramatically induces cell death while stunting the proliferation of human and mouse AML cells in vitro and in vivo. Mechanistically, through interacting with IGF2BPs, YBX1 stabilizes m6A-tagged RNA. Furthermore, deletion of YBX1 facilitates mRNA decay of MYC and BCL2 in an m6A-dependent manner, resulting in defective survival of AML cells. Thus, the findings reveal that YBX1 is a key factor in leukemia survival, which may provide a research basis for determining whether YBX1 is a therapeutic target for AML. Moreover, other m6A modifiers [e.g., writers (WTAP [124,148], RBM15 [149,150]), erasers (FTO [103,128,151], ALKBH5 [65,66]), readers (YTHDF2 [105], IGF2BP1 [152]), etc.] are also crucial in influencing the occurrence and development of AML in the evidence of related studies.
4.2. The Role of m6A Methylation Modifications in Chronic Leukemia
RNA m6A Methylation and Chronic Myeloid Leukemia
Chronic myeloid leukemia (CML) is a malignant myeloproliferative neoplasm originating from multipotent hematopoietic stem cells. In essence, the t (9; 22) (q34; q11.2) translocation is a characteristic chromosomal alteration in CML, and at the molecular level, the translocation results from the formation of a BCR-ABL fusion gene [153,154]. Currently, the annual incidence of CML is (0.7–1.8) per 100,000 worldwide, which is the third-highest among all types of leukemia [155,156]. In particular, the annual incidence rate in China is approximately (3.6 to 5.5) per 1 million. Depending on the disease, CML is clinically classified into three phases: chronic phase (CP), accelerated phase (AP), and blastic phase (BP or blast crisis (BC)), and patients are generally diagnosed in the chronic phase [157,158].
At present, it has been reported that METTL3, an RNA m6A modifier, is also involved in the mechanism of CML development [130]. Specifically, Ianniello et al. [130] identified that the m6A methyltransferase complex METTL3/METTL14 is highly expressed in CML patients and is essential to maintain the proliferation of primary CML cells and CML cell lines that are sensitive or resistant to tyrosine kinase inhibitors (TKIs) (e.g., imatinib). Notably, it has been reported that aberrant translation is thought to be one of the mechanisms that mediate BCR/ABL transformation and maintain the leukemic phenotype of CML cells [159,160]. Here, the study proved that METTL3 promotes the proliferation of tumor cells by indirectly affecting MYC levels or directly methylating specific mRNAs to modulate ribosome levels and translation, resulting in tumorigenesis. Conversely, METTL3 deficiency drastically damages the translation efficiency of mRNAs for metabolism-related genes in organisms. Therefore, the findings suggest that METTL3 is a novel oncogene in the pathogenesis of CML and a potential therapeutic target for TKIs-resistant CML. Another recent finding [131] also revealed that via modulating the miR-766-5p/CDKN1A axis, METTL3 could modify long non-coding RNAs (lncRNAs) nuclear-enriched transcript 1 (NEAT1), thus controlling the progressive process of CML disorder and playing an oncogenic role in CML. NEAT1 is currently known to have low expression levels in CML cell lines or PBMCs of CML patients [131]. Mechanistically, the dysfunction of m6A methyltransferase METTL3 causes the abnormal expression of NEAT1 in CML, and the overexpression of NEAT1 greatly promotes the death of CML cells. Moreover, using methylated RNA immunoprecipitation (MeRIP) assay and SRAMP, we could identify the modification sites of m6A. Overall, this study explains the role of m6A in CML and further provides a rationale for targeting therapy aimed at CML. Consistently, Lai et al. [132] also observed that dysregulation of METTL3 contributes to chemoresistance of CML cells. Based on experimental analysis of mouse models and two cell lines, KCL22 and K562, the investigators confirmed that LINC00470 is able to importantly suppress the expression of PTEN mRNA through METTL3, showing a negative regulatory relationship between LINC00470 and PTEN. Conversely, the expression level of PTEN is increased after silencing METTL3 in cancer cells. Therefore, we can conclude that METTL3 is a detrimental factor in affecting the chemotherapeutic effect of CML. In this regard, we can address the corresponding molecular targets for anticancer therapy.
4.3. The Role of m6A Methylation Modifications in Lymphoma
4.3.1. RNA m6A Methylation and Diffuse Large B-Cell Lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most prevalent subtype of non-Hodgkin’s lymphoma (NHL), accounting for approximately 30–35% of NHL. DLBCL is a heterogeneous tumor that can develop at any age, with a peak age of 50–70 years. Along with the aging of the population, the incidence of lymphoma in the elderly is increasing year by year, and >50% of DLBCL patients are >60 years old [161].
Importantly, m6A modifiers also play a crucial role in the progression of DLBCL. At present, it is generally recognized that PIWI-interacting RNAs (piRNAs) exert oncogenic effects as epigenetic effectors in human cancers [135]. In a xenograft DLBCL model, depletion of piRNA-30473 can suppress the proliferation of tumor cells. Han et al. [135] further discovered that piRNA-30473 could cause the upregulation of WTAP expression and enhance the levels of DLBCL m6A, which in turn results in the development of DLBCL disease. In addition, the upregulation of WTAP facilitates the expression of its critical target gene, hexokinase 2 (HK2), and therefore contribute to the progression of DLBCL, as verified by the experimental method m6A sequencing. To sum up, the study emphasizes that the RNA m6A methylation factor, i.e., WTAP, possesses an important function in DLBCL and will support the exploration of novel therapeutic approaches for DLBCL. As an m6A writer, METTL3 is another potential target for the treatment of DLBCL [134]. By regulating m6A modifications in pigment epithelium-derived factor (PEDF), METTL3 is functionally closely associated with the development of DLBCL. Indeed, METTL3 is significantly overexpressed in DLBCL tissues and cell lines, ultimately promoting the proliferation of tumor cells. However, overexpression of PEDF reverses the inhibitory effect of DLBCL cell proliferation due to METTL3 deficiency. Hence, the above findings suggest that METTL3 is essential in boosting the progression of DLBCL. Strikingly, a recent study of Xie’s group has clearly demonstrated a correlation between m6A modifications and the prognosis of DLBCL [133]. Based on the expression of m6A modifiers, the researchers distinguish different subtypes of DLBCL at the molecular level using a consensus clustering approach. Thus, by affecting m6A regulators correlated with survival, they observed that different DLBCL patients will possess adverse prognostic characteristics and outcomes of disease progression. Further, based on an analysis of prognostic correlations, it was determined that in DLBCL patients, high-risk m6A modifications signify more rapidly progressive tumors, thereby indicating poorer patients’ survival. From this point of view, we firmly believe that m6A-related modifiers can be well-suited indicators to determine the prognostic status of DLBCL. However, further elucidation of the risk assessment mechanism underlying m6A modifiers for tumor disease is still required.
4.3.2. RNA m6A Methylation and Other Types of Lymphoma
Apart from DLBCL mentioned above, there are currently relatively few mechanistic studies involving the relationship between m6A and other less frequent types of lymphoma [e.g., natural killer (NK)/T-cell lymphoma, B-cell lymphoma, T-cell lymphoblastic lymphoma (T-LBL), etc.]. For example, T-LBL belongs to a group of highly aggressive hematologic malignancies that originate from primitive T lymphocytes. To our knowledge, the pathogenesis of T-LBL was first confirmed in 2021 by An et al. [162] to involve modulation based on m6A modifications. In line with its role in other tumors, the m6A methyltransferase METTL14 is also essential in influencing the development of T-LBL. Numerous evidence shows that microRNAs (miRNAs) play a vital regulatory role in human cancers. Using high-throughput sequencing technology, they further reveal that miR-211 is a miRNA with down-regulated expression in T-LBL, and silencing miR-211 predicts poor disease outcome; however, overexpression of miR-211 remarkably reduces the viability and inhibits the proliferation of T-LBL cells. Mechanistically, METTL14 enables methylation modification of primary miR-211 (pri-miR-211), which unexpectedly exerts anti-cancer effects. Similarly, in xenograft tumor models, miR-211 significantly suppresses the growth of tumors in vivo. To conclude, these data definitively suggest that targeting miR-211, a target gene, may be a promising therapeutic approach for patients with T-LBL. Another study [163] has revealed that the occurrence of NK/T-cell lymphoma (NKTCL) is also importantly correlated with RNA m6A methylation. Compared to normal NK cells in the organism, the expression level of WTAP is markedly elevated in human NKTCL cell lines (e.g., YTS and SNK-6 cells). By contrast, WTAP deficiency notably inhibits the proliferation of YTS and SNK-6 cells and promotes the death of tumor cells. Specifically, Ma et al. [163] recently found that WTAP increases the m6A level of dual-specific phosphatase 6 (DUSP6) mRNA in an m6A-dependent manner, thereby enhancing its expression and ultimately causing the oncogenic effects of NKTCL. Simultaneously, WTAP also heightens the chemoresistance to the drug cisplatin. These results highlight that the treatment of NKTCL may possibly be achieved via modulating WTAP.
In addition, it has been reported that B-cell lymphomas are similarly involved in m6A methylation modifications. Wu et al. [164] showed that, either in vitro or in vivo experiments, suppression of ALKBH5 or overexpression of selected MYC-repressed genes (MRG) SPI1 or PHF12 could effectively impede the growth of tumor protein MYC-mediated B-cell lymphoma. Moreover, in this study, emerging evidence shows that MYC also modulates the translation of MRG mRNA through a particular m6A reader, YTHDF3. Notably, given that ALKBH5 and YTHDF3 are major representatives of m6A binding proteins, the findings reflect that MYC promotes the development of tumors via altering the m6A modification of target genes. Consistently, m6A reader YTHDF1 is currently identified to inhibit EBV replication in patients and play a vital anti-cancer role, thus providing a new target for future treatment of EBV-infected B-cell lymphoma [165]. To our knowledge, mantle cell lymphoma (MCL) is a highly aggressive non-Hodgkin B-cell lymphoma with a median age of diagnosis of 60 years. By analyzing the gene expression omnibus database, Zhang et al. [166] found that approximately half of the m6A regulators can predict the survival of patients with MCL, especially the m6A.index, and can classify them. Concretely, MCL patients with low m6A.index have lower survival rates and lower mRNA levels in the total transcriptome. In essence, it is probably due to the link of m6A regulators with cell division and RNA metabolic pathways. In summary, the study suggests that imbalanced expression levels of m6A regulators may be used to predict poor survival in patients with MCL, thus providing an idea for treatment.
4.4. The Role of m6A Methylation Modifications in Multiple Myeloma
Multiple myeloma (MM) is a malignant hematologic disease with abnormal proliferation of clonal plasma cells, which can destroy the bones, kidneys, and hematopoietic function of the body. Among them, renal injury is a common complication of MM and one of its major causes of death [167]. In a current study, a computational method, xPore, based directly on the analysis of RNA sequencing (RNA-seq), in the absence of matched control samples, showed that only one m6A locus manifests a remarkable difference in the rate of modification in six cell lines and MM patient samples [168]. This result demonstrates that xPore can identify differential m6A modifications and expression from a high-throughput experiment for the data in question.
Indeed, FTO is also a promising target for the treatment of MM [137]. Functionally, isocitrate dehydrogenase 2 (IDH2) regulates mRNA m6A modification by activating FTO; on the other hand, it can down-regulate the m6A level of WNT7B mRNA and increase the expression of WNT7B, thus activating the Wnt signaling pathway, which ultimately facilitates the growth of tumor cells in MM in vitro and shortens the overall survival of patients. It is apparent that m6A and its related factors (e.g., FTO) are involved in the progression of MM and provide a potential therapeutic target. Furthermore, ALKBH5 is similarly confirmed to be overexpressed in MM and predicts a worse prognosis for MM patients [136]. Functionally, the downregulation of ALKBH5 promotes tumor cell death and suppresses the growth of MM cells in vitro, exerting a tumorigenic effect. Thus, the results suggest that ALKBH5 is essential in maintaining the progression of MM tumors and provide a novel direction for scientists to investigate. Interestingly, HNRNPA2B1, a specific m6A reader, is also found to be essential in the pathogenesis of MM. In 2021, Jiang et al. [138] concluded that HNRNPA2B1 is highly expressed in MM patients and mediates m6A methylation modification of ILF3 mRNA, thereby contributing to the proliferation of MM cells in vitro and in vivo, which results in MM patients presenting a poor prognosis; conversely, genetic depletion of HNRNPA2B1 inhibits the growth of tumor cells and exerts an anti-cancer effect. Taken together, the above findings reflect that m6A modification may be of great importance for the occurrence of MM and provide a possible research direction for clinical researchers.
4.5. The Role of m6A Methylation Modifications in Myelodysplastic Syndromes
Myelodysplastic syndrome (MDS) is defined as a myeloid clonal disorder of hematopoietic stem cell origin with symptoms including ineffective hematopoiesis, intractable anemia and a high risk of AML. Current studies generally agree that the occurrence and development of MDS is a cumulative damage of multiple genetic events and that the combination of genomic abnormalities and altered epigenetic modifications can cause phenotypic heterogeneity of MDS [169]. Among these, altered expression levels of relevant genes may affect key oncogenes or tumor suppressor genes in MDS myelopoietic cells, which allow abnormal hematopoietic cells to gain a clonal proliferative advantage. Previous studies [170] have pointed out that genes encoding RNA spliceosomes may be involved in the development and progression of MDS as early driver genes. Importantly, small alterations in the spliceosome can affect the specificity of splicing and trigger changes in the peptide sequences of transcribed proteins, which subsequently affect the normal physiological functions of hematopoietic cells. However, the exact mechanism of MDS induced by aberrant RNA splicing is hitherto unknown.
Actually, YTHDC1 can mediate the mRNA splicing process by recruiting precursor mRNA splicing factors [74], which can contribute to elucidating the role of splicing abnormalities in the abnormal hematopoietic function of MDS. In addition, the reduced level of m6A modification induced by downregulation of the METTL3 gene can modify the alternative splicing (AS) pattern of precursor mRNA, which is mainly enriched in the p53 signaling pathway and apoptosis pathway [24,26]. Therefore, the above results further illustrate that m6A methylation modification is important for the exploration of the abnormal splicing mechanism for MDS. The m6A-based regulation of bone marrow hematopoiesis plays an important role in maintaining the normal hematopoietic process of bone marrow. Bone marrow is the main site of hematopoiesis in the body, and abnormal regulation of bone marrow hematopoiesis can potentially lead to the development of a series of diseases. Inherited bone marrow failure syndromes (IBMFS) are a heterogeneous group of genetic disorders characterized by common clinical features of bone marrow hematopoietic failure, congenital malformations, and high risk of progression to malignancy [171], mainly including Fanconi anemia (FA), dyskeratosis congenita (DC), Diamond–Blackfan anemia (DBA), and Shwachman–Diamond syndrome (SDS). Among them, FA is due to defective DNA repair mechanisms, while DC is caused by telomerase defects, both of which have a tendency to acquire genetic alterations and develop into MDS. Specifically, the hematologic lesions of IBMFS manifest as impaired bone marrow hematopoiesis and decreased whole blood cells or mono-lineage cells, implying that IBMFS lesions are derived from the myeloid pluripotent stem cell level. m6A and its related factors could be involved in the development and progression of IBMFS via dramatically affecting the development of bone marrow hematopoiesis. Nevertheless, the exact mechanism of action remains to be elucidated through further studies.
5. Discussion
It is widely recognized that m6A modification is a widespread form of methylation modification in eukaryotic RNA. Under the joint reversible regulation of m6A methyltransferases, m6A demethylases and m6A binding proteins, m6A methylation modification covers various stages of RNA life course such as mRNA, lncRNA, and miRNA. Through such epigenetic m6A methylation modifications, various RNAs (e.g., mRNA, lncRNA, and miRNA) are modulated to perform their corresponding functions, which in turn alter the expression of tumor cell-related genes and thus facilitate the development of hematological tumors. Notably, studies related to the effects of m6A and its related factors on hematologic malignancies (e.g., AML, DLBCL, MM, etc.) are currently a hot topic in the research of RNA epigenetics. However, the researches on the species and functions of m6A-related proteins are still in the exploratory stage. The existing studies demonstrate that: (1) m6A methylation modification is a double-edged sword, either over-modification or under-modification may result in the development of tumor; (2) the same m6A methylation-related enzymes may possess different functions in different action pathways; (3) m6A methylation modification is not only limited to mRNA but also exists in lncRNA, miRNA and other RNAs, which provides a wide scope for our research; (4) m6A methylation-related enzymes can also act as the regulatory targets of lncRNAs and miRNAs, which affect the m6A methylation levels of other RNAs; (5) In addition to modulating m6A levels, m6A methyltransferases probably also possess other independent functions. For instance, METTL3, besides having methyltransferase activity, can enhance the translation of cancer genes and affect the progression of tumors independently of their catalytic subunits. These findings also extend the research horizon of m6A for us.
Explicitly, remarkable progress has been made in exploring the role of m6A methylation in hematological malignancies. However, many opportunities and challenges exist: (1) The role of m6A in hematological tumors is still controversial, and these functional features are attributed to the fluctuating distribution of m6A in different regions of mRNA and subcellular readers. Given the extensive interactions between metabolic networks, it is particularly important to maintain the balance of the internal environment between metabolic processes, which is one of the factors to be considered in the future; (2) whether m6A and its regulators can be used as potential biomarkers for diagnosis and prognosis of hematologic tumors, and the specificity and sensitivity of these biomarkers remain to be explored; (3) some studies indicate that m6A regulators and related pathways can be considered as therapeutic targets, but specific applications in large samples of clinical practice are deficient and their side effects are largely unknown; (4) Whether there is a combined or subtractive effect between m6A methylation modifications and other epigenetic modifications needs to be further explored; (5) Most of the existing hematological tumor studies have focused on methyltransferases and demethylases, but there are fewer studies on readers, which is one of our future directions to explore.
6. Conclusions
Collectively, based on the above findings, m6A methylation modification can remarkably affect the self-renewal, proliferation, and differentiation processes of pluripotent stem cells in hematologic malignancies, but their exact biological functions in normal hematopoietic and malignant hematologic diseases are not clear. Simultaneously, the molecular mechanism of m6A methylation modification that regulates the hematopoietic system and induces hematological tumors needs to be further elucidated. This is significant for our in-depth understanding of the pathogenic mechanism in hematological malignancies and the exploration of new targeted interventions. Therefore, we are convinced that with continuous research on the functional changes after m6A modification of RNA, a reference basis for clinicians to diagnose hematologic malignancies early and search for therapeutic targets will be provided.
Author Contributions
Y.Z. and H.P. contributed to the concept and design of the study. Literature collection and collation were completed by Y.Z. The manuscript was written by Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (82070175) and the Scientific program of the Health Commission of Hunan Province (20201179).
Acknowledgments
The authors thank Wenni Dai for her experience and Juan Liu for her timely help in completing the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Prakash, G.; Kaur, A.; Malhotra, P.; Khadwal, A.; Sharma, P.; Suri, V.; Varma, N.; Varma, S. Current Role of Genetics in Hematologic Malignancies. Indian J. Hematol. Blood Transfus. 2016, 32, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Mehrpouri, M.; Pourbagheri-Sigaroodi, A.; Bashash, D. The contributory roles of histone deacetylases (HDACs) in hematopoiesis regulation and possibilities for pharmacologic interventions in hematologic malignancies. Int. Immunopharmacol. 2021, 100, 108114. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark. Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenet. 2019, 11, 81. [Google Scholar] [CrossRef]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate detection of m(6)A RNA modifications in native RNA sequences. Nat. Commun. 2019, 10, 4079. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Pan, T. N6-methyladenosine–encoded epitranscriptomics. Nat. Struct. Mol. Biol. 2016, 23, 98–102. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m(6)A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Huang, H.; Chen, J. RNA N (6)-Methyladenosine Modification in Normal and Malignant Hematopoiesis. Adv. Exp. Med. Biol. 2019, 1143, 75–93. [Google Scholar] [CrossRef]
- Fong, C.Y.; Morison, J.; Dawson, M.A. Epigenetics in the hematologic malignancies. Haematologica 2014, 99, 1772–1783. [Google Scholar] [CrossRef] [Green Version]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Haruehanroengra, P.; Zheng, Y.Y.; Zhou, Y.; Huang, Y.; Sheng, J. RNA modifications and cancer. RNA Biol. 2020, 17, 1560–1575. [Google Scholar] [CrossRef]
- Wei, C.M.; Gershowitz, A.; Moss, B. Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell 1975, 4, 379–386. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [Green Version]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Zhang, W.; Qian, Y.; Jia, G. The detection and functions of RNA modification m(6)A based on m(6)A writers and erasers. J. Biol. Chem. 2021, 297, 100973. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Dzhashiashvili, Y.; Shah, A.; Kunjamma, R.B.; Weng, Y.L.; Elbaz, B.; Fei, Q.; Jones, J.S.; Li, Y.I.; Zhuang, X.; et al. m(6)A mRNA Methylation Is Essential for Oligodendrocyte Maturation and CNS Myelination. Neuron 2020, 105, 293–309.e5. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Bodi, Z.; Bottley, A.; Archer, N.; May, S.T.; Fray, R.G. Yeast m6A Methylated mRNAs Are Enriched on Translating Ribosomes during Meiosis, and under Rapamycin Treatment. PLoS ONE 2015, 10, e0132090. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Narayan, P.; Ludwiczak, R.L.; Goodwin, E.C.; Rottman, F.M. Context effects on N6-adenosine methylation sites in prolactin mRNA. Nucleic Acids Res. 1994, 22, 419–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csepany, T.; Lin, A.; Baldick, C.J., Jr.; Beemon, K. Sequence specificity of mRNA N6-adenosine methyltransferase. J. Biol. Chem. 1990, 265, 20117–20122. [Google Scholar] [CrossRef]
- Narayan, P.; Rottman, F.M. An in vitro system for accurate methylation of internal adenosine residues in messenger RNA. Science 1988, 242, 1159–1162. [Google Scholar] [CrossRef]
- Huang, J.; Yin, P. Structural Insights into N(6)-methyladenosine (m(6)A) Modification in the Transcriptome. Genom. Proteom. Bioinform. 2018, 16, 85–98. [Google Scholar] [CrossRef]
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Hüttelmaier, S.; Liu, T. The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Ma, P.; Liu, Y.; Li, W.; Shu, Y. Multiple functions of m(6)A RNA methylation in cancer. J. Hematol. Oncol. 2018, 11, 48. [Google Scholar] [CrossRef]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedi, R.K.; Huang, D.; Eberle, S.A.; Wiedmer, L.; Śledź, P.; Caflisch, A. Small-Molecule Inhibitors of METTL3, the Major Human Epitranscriptomic Writer. ChemMedChem 2020, 15, 744–748. [Google Scholar] [CrossRef]
- Schöller, E.; Weichmann, F.; Treiber, T.; Ringle, S.; Treiber, N.; Flatley, A.; Feederle, R.; Bruckmann, A.; Meister, G. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. RNA 2018, 24, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; He, L.; Liu, Z.; Ren, X.; Qi, L.; Wan, L.; Wang, W.; Tu, C.; Li, Z. Multifaceted Functions and Novel Insight Into the Regulatory Role of RNA N(6)-Methyladenosine Modification in Musculoskeletal Disorders. Front. Cell Dev. Biol. 2020, 8, 870. [Google Scholar] [CrossRef]
- Huang, J.; Dong, X.; Gong, Z.; Qin, L.Y.; Yang, S.; Zhu, Y.L.; Wang, X.; Zhang, D.; Zou, T.; Yin, P.; et al. Solution structure of the RNA recognition domain of METTL3-METTL14 N(6)-methyladenosine methyltransferase. Protein Cell 2019, 10, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhuo, L.; Wang, J.; Zhang, Q.; Li, Q.; Li, G.; Yan, L.; Jin, T.; Pan, T.; Sui, X.; et al. METTL3 plays multiple functions in biological processes. Am. J. Cancer Res. 2020, 10, 1631–1646. [Google Scholar]
- Zheng, W.; Dong, X.; Zhao, Y.; Wang, S.; Jiang, H.; Zhang, M.; Zheng, X.; Gu, M. Multiple Functions and Mechanisms Underlying the Role of METTL3 in Human Cancers. Front. Oncol. 2019, 9, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835.e14. [Google Scholar] [CrossRef] [Green Version]
- Satterwhite, E.R.; Mansfield, K.D. RNA methyltransferase METTL16: Targets and function. Wiley Interdiscip. Rev. RNA 2021, e1681. [Google Scholar] [CrossRef]
- Ignatova, V.V.; Stolz, P.; Kaiser, S.; Gustafsson, T.H.; Lastres, P.R.; Sanz-Moreno, A.; Cho, Y.L.; Amarie, O.V.; Aguilar-Pimentel, A.; Klein-Rodewald, T.; et al. The rRNA m(6)A methyltransferase METTL5 is involved in pluripotency and developmental programs. Genes Dev. 2020, 34, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Lobo, J.; Guimarães-Teixeira, C.; Barros-Silva, D.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Oing, C.; Honecker, F.; et al. The component of the m(6)A writer complex VIRMA is implicated in aggressive tumor phenotype, DNA damage response and cisplatin resistance in germ cell tumors. J. Exp. Clin. Cancer Res. 2021, 40, 268. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, Y.; Yao, Y.; Xie, H.; Lu, G.; Du, C.; Cheng, J.; Zhou, J. VIRMA contributes to non-small cell lung cancer progression via N(6)-methyladenosine-dependent DAPK3 post-transcriptional modification. Cancer Lett. 2021, 522, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qi, X.; Liu, L.; Ma, S.; Liu, J.; Wu, J. Epigenetic Regulation of m(6)A Modifications in Human Cancer. Mol. Ther. Nucleic Acids 2020, 19, 405–412. [Google Scholar] [CrossRef]
- Gong, P.J.; Shao, Y.C.; Yang, Y.; Song, W.J.; He, X.; Zeng, Y.F.; Huang, S.R.; Wei, L.; Zhang, J.W. Analysis of N6-Methyladenosine Methyltransferase Reveals METTL14 and ZC3H13 as Tumor Suppressor Genes in Breast Cancer. Front. Oncol. 2020, 10, 578963. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m(6)A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Choe, J.; Park, O.H.; Kim, Y.K. Molecular Mechanisms Driving mRNA Degradation by m(6)A Modification. Trends Genet. 2020, 36, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Hao, Y.J.; Zhang, Y.; Li, M.M.; Wang, M.; Han, W.; Wu, Y.; Lv, Y.; Hao, J.; Wang, L.; et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 2015, 16, 289–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Weng, H.; Zhou, K.; Wu, T.; Zhao, B.S.; Sun, M.; Chen, Z.; Deng, X.; Xiao, G.; Auer, F.; et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 2019, 567, 414–419. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Aas, A.; Isakson, P.; Bindesbøll, C.; Alemu, E.A.; Klungland, A.; Simonsen, A. Nucleocytoplasmic Shuttling of FTO Does Not Affect Starvation-Induced Autophagy. PLoS ONE 2017, 12, e0168182. [Google Scholar] [CrossRef]
- Wu, B.; Li, L.; Huang, Y.; Ma, J.; Min, J. Readers, writers and erasers of N(6)-methylated adenosine modification. Curr. Opin. Struct. Biol. 2017, 47, 67–76. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, L.H.; Wang, Y.; Xiao, Y.; Liu, J.; Zhang, W.; Yan, N.; Amu, G.; Tang, X.; Zhang, L.; et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc. Natl. Acad. Sci. USA 2019, 116, 2919–2924. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, J.; Gu, Q.; Ma, Y.; Yang, Y.; Zhu, J.; Zhang, Q. The biological function of m6A demethylase ALKBH5 and its role in human disease. Cancer Cell Int. 2020, 20, 347. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Sheng, Y.; Zhu, A.C.; Robinson, S.; Jiang, X.; Dong, L.; Chen, H.; Su, R.; Yin, Z.; Li, W.; et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 2020, 27, 64–80.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Wang, P.; Han, G.; Zhang, T.; Chang, J.; Yin, R.; Shan, Y.; Wen, J.; Xie, X.; et al. Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell 2020, 27, 81–97.e8. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Sun, H.; Xu, C. YTH Domain: A Family of N(6)-methyladenosine (m(6)A) Readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016, 7, 12626. [Google Scholar] [CrossRef]
- Gao, Y.; Pei, G.; Li, D.; Li, R.; Shao, Y.; Zhang, Q.C.; Li, P. Multivalent m(6)A motifs promote phase separation of YTHDF proteins. Cell Res. 2019, 29, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Jaffrey, S.R. A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell 2020, 181, 1582–1595.e18. [Google Scholar] [CrossRef]
- Zhang, Z.; Luo, K.; Zou, Z.; Qiu, M.; Tian, J.; Sieh, L.; Shi, H.; Zou, Y.; Wang, G.; Morrison, J.; et al. Genetic analyses support the contribution of mRNA N(6)-methyladenosine (m(6)A) modification to human disease heritability. Nat. Genet. 2020, 52, 939–949. [Google Scholar] [CrossRef]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Roundtree, I.A.; Luo, G.Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 2017, 6, e31311. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, J.N.; Wang, E.H.; Gong, C.J.; Lan, K.F.; Ding, X. The m6A reader protein YTHDC2 is a potential biomarker and associated with immune infiltration in head and neck squamous cell carcinoma. PeerJ 2020, 8, e10385. [Google Scholar] [CrossRef]
- Wu, B.; Su, S.; Patil, D.P.; Liu, H.; Gan, J.; Jaffrey, S.R.; Ma, J. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat. Commun. 2018, 9, 420. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.I.; Shi, H.; Lyu, R.; Wylder, A.C.; Matuszek, Ż.; Pan, J.N.; He, C.; Parisien, M.; Pan, T. Regulation of Co-transcriptional Pre-mRNA Splicing by m(6)A through the Low-Complexity Protein hnRNPG. Mol. Cell 2019, 76, 70–81.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.L.; Wächter, K.; Mühleck, B.; Pazaitis, N.; Köhn, M.; Lederer, M.; Hüttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of cancer progression? Cell Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Kang, Y.; Wang, M.; Li, Y.; Xu, T.; Yang, W.; Song, H.; Wu, H.; Shu, Q.; Jin, P. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet. 2018, 27, 3936–3950. [Google Scholar] [CrossRef]
- Wu, R.; Li, A.; Sun, B.; Sun, J.G.; Zhang, J.; Zhang, T.; Chen, Y.; Xiao, Y.; Gao, Y.; Zhang, Q.; et al. A novel m(6)A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019, 29, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Orouji, E.; Peitsch, W.K.; Orouji, A.; Houben, R.; Utikal, J. Oncogenic Role of an Epigenetic Reader of m(6)A RNA Modification: YTHDF1 in Merkel Cell Carcinoma. Cancers 2020, 12, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichinchi, G.; Gao, S.; Saletore, Y.; Gonzalez, G.M.; Bansal, V.; Wang, Y.; Mason, C.E.; Rana, T.M. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 2016, 1, 16011. [Google Scholar] [CrossRef]
- Imam, H.; Khan, M.; Gokhale, N.S.; McIntyre, A.B.R.; Kim, G.W.; Jang, J.Y.; Kim, S.J.; Mason, C.E.; Horner, S.M.; Siddiqui, A. N6-methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc. Natl. Acad. Sci. USA 2018, 115, 8829–8834. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, N.S.; McIntyre, A.B.R.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Abreu, D.; Bordoni, A.; Zucca, E. Epidemiology of hematological malignancies. Ann. Oncol. 2007, 18 (Suppl. S1), i3–i8. [Google Scholar] [CrossRef]
- Peng, Z.; Gong, Y.; Wang, X.; He, W.; Wu, L.; Zhang, L.; Xiong, L.; Huang, Y.; Su, L.; Shi, P.; et al. METTL3-m(6)A-Rubicon axis inhibits autophagy in nonalcoholic fatty liver disease. Mol. Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Sun, H.; Guo, Y.; Zhou, Y.; Gu, X.; Jin, J.; Chen, X.; Wang, F.; Ma, H.; Guo, X.; et al. m(6) A reader protein YTHDF2 regulates spermatogenesis by timely clearance of phase-specific transcripts. Cell Prolif. 2021, e13164. [Google Scholar] [CrossRef]
- Yan, G.; Yuan, Y.; He, M.; Gong, R.; Lei, H.; Zhou, H.; Wang, W.; Du, W.; Ma, T.; Liu, S.; et al. m(6)A Methylation of Precursor-miR-320/RUNX2 Controls Osteogenic Potential of Bone Marrow-Derived Mesenchymal Stem Cells. Mol. Ther. Nucleic Acids 2020, 19, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, M.; He, J.; Wu, K.; Lin, S.; Jin, L.; Chen, Y.; Liu, H.; Shi, J.; Wang, X.; et al. The RNA m(6)A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 2021, 591, 322–326. [Google Scholar] [CrossRef]
- Mathoux, J.; Henshall, D.C.; Brennan, G.P. Regulatory Mechanisms of the RNA Modification m(6)A and Significance in Brain Function in Health and Disease. Front. Cell Neurosci. 2021, 15, 671932. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, S.; Xia, J.; Liu, F. Hematopoietic Hierarchy—An Updated Roadmap. Trends Cell Biol. 2018, 28, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.M.; Foroni, L. Core binding factor genes and human leukemia. Haematologica 2002, 87, 1307–1323. [Google Scholar]
- Kissa, K.; Herbomel, P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 2010, 464, 112–115. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, Y.; Sun, B.; Wang, L.; Yang, Y.; Ma, D.; Lv, J.; Heng, J.; Ding, Y.; Xue, Y.; et al. m(6)A modulates haematopoietic stem and progenitor cell specification. Nature 2017, 549, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhang, Y.; Gao, S.; Zhang, C.; Chen, Y.; Li, W.; Yang, Y.G.; Zhou, Q.; Liu, F. Endothelial-specific m(6)A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res. 2018, 28, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Vasic, R.; Song, Y.; Teng, R.; Liu, C.; Gbyli, R.; Biancon, G.; Nelakanti, R.; Lobben, K.; Kudo, E.; et al. m(6)A Modification Prevents Formation of Endogenous Double-Stranded RNAs and Deleterious Innate Immune Responses during Hematopoietic Development. Immunity 2020, 52, 1007–1021.e8. [Google Scholar] [CrossRef]
- Weng, H.; Huang, H.; Wu, H.; Qin, X.; Zhao, B.S.; Dong, L.; Shi, H.; Skibbe, J.; Shen, C.; Hu, C.; et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 2018, 22, 191–205.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e9. [Google Scholar] [CrossRef]
- Li, Z.; Qian, P.; Shao, W.; Shi, H.; He, X.C.; Gogol, M.; Yu, Z.; Wang, Y.; Qi, M.; Zhu, Y.; et al. Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018, 28, 904–917. [Google Scholar] [CrossRef]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 137–148.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zuo, H.; Liu, J.; Wen, F.; Gao, Y.; Zhu, X.; Liu, B.; Xiao, F.; Wang, W.; Huang, G.; et al. Loss of YTHDF2-mediated m(6)A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res. 2018, 28, 1035–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapperley, C.; van de Lagemaat, L.N.; Lawson, H.; Tavosanis, A.; Paris, J.; Campos, J.; Wotherspoon, D.; Durko, J.; Sarapuu, A.; Choe, J.; et al. The mRNA m6A reader YTHDF2 suppresses proinflammatory pathways and sustains hematopoietic stem cell function. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Wei, J.; Yu, F.; Xu, H.; Yu, C.; Wu, Q.; Liu, Y.; Li, L.; Cui, X.-L.; Gu, X.; et al. A Critical Role of Nuclear m6A Reader YTHDC1 in Leukemogenesis by Regulating MCM Complex-Mediated DNA Replication. Blood 2021, 138, 2838–2852. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Yao, Y.; Bi, Z.; Wu, R.; Zhao, Y.; Liu, Y.; Liu, Q.; Wang, Y.; Wang, X. METTL3 inhibits BMSC adipogenic differentiation by targeting the JAK1/STAT5/C/EBPβ pathway via an m(6)A-YTHDF2-dependent manner. FASEB J. 2019, 33, 7529–7544. [Google Scholar] [CrossRef]
- Cen, S.; Li, J.; Cai, Z.; Pan, Y.; Sun, Z.; Li, Z.; Ye, G.; Zheng, G.; Li, M.; Liu, W.; et al. TRAF4 acts as a fate checkpoint to regulate the adipogenic differentiation of MSCs by activating PKM2. EBioMedicine 2020, 54, 102722. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xie, L.; Wang, M.; Xiong, Q.; Guo, Y.; Liang, Y.; Li, J.; Sheng, R.; Deng, P.; Wang, Y.; et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat. Commun. 2018, 9, 4772. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Choorapoikayil, S.; Kers, R.; Herbomel, P.; Kissa, K.; den Hertog, J. Pivotal role of Pten in the balance between proliferation and differentiation of hematopoietic stem cells in zebrafish. Blood 2014, 123, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zeng, H.M.; Xue, Y.J.; Lu, A.D.; Jia, Y.P.; Zuo, Y.X.; Zhang, L.P. The gene expression level of m6A catalytic enzymes is increased in ETV6/RUNX1-positive acute lymphoblastic leukemia. Int. J. Lab. Hematol. 2021, 43, e89–e91. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Chang, L.; Liu, C.; Chen, X.; Zhu, X. The study of METTL3 and METTL14 expressions in childhood ETV6/RUNX1-positive acute lymphoblastic leukemia. Mol. Genet. Genom. Med. 2019, 7, e00933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, H.T.; Liu, L.; Cui, L.N.; Ma, H.Y.; Shen, L.Y. ALKBH5-mediated m6A-demethylation of USP1 regulated T-cell acute lymphoblastic leukemia cell glucocorticoid resistance by Aurora B. Mol. Carcinog. 2021, 60, 644–657. [Google Scholar] [CrossRef]
- Cheng, Y.M.; Xie, W.; Pickering, B.F.; Chu, K.L.; Savino, A.M.; Yang, X.J.; Luo, H.Z.; Nguyen, D.T.; Mo, S.L.; Barin, E.; et al. N-6-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 2021, 39, 958–972.e8. [Google Scholar] [CrossRef]
- Chen, Z.; Shao, Y.L.; Wang, L.L.; Lin, J.; Zhang, J.B.; Ding, Y.; Gao, B.B.; Liu, D.H.; Gao, X.N. YTHDF2 is a potential target of AML1/ETO-HIF1α loop-mediated cell proliferation in t(8;21) AML. Oncogene 2021, 40, 3786–3798. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.D.; Xie, X.Q.; Han, G.Q.; Zhang, T.T.; Li, Y.S.; Li, Y.C.; Yin, R.; Wang, Q.F.; Zhang, T.; Wang, P.P.; et al. YBX1 is required for maintaining myeloid leukemia cell survival by regulating BCL2 stability in an m(6)A-dependent manner. Blood 2021, 138, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Naren, D.; Yan, T.; Gong, Y.; Huang, J.; Zhang, D.; Sang, L.; Zheng, X.; Li, Y. High Wilms’ tumor 1 associating protein expression predicts poor prognosis in acute myeloid leukemia and regulates m(6)A methylation of MYC mRNA. J. Cancer Res. Clin. Oncol. 2021, 147, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yu, M.; Li, C.; Wang, Y.; Li, X.; Ulrich, B.; Su, R.; Dong, L.; Weng, H.; Huang, H.; et al. miR-550-1 functions as a tumor suppressor in acute myeloid leukemia via the hippo signaling pathway. Int. J. Biol. Sci. 2020, 16, 2853–2867. [Google Scholar] [CrossRef]
- Sorci, M.; Ianniello, Z.; Cruciani, S.; Larivera, S.; Ginistrelli, L.C.; Capuano, E.; Marchioni, M.; Fazi, F.; Fatica, A. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. 2018, 9, 796. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Du, Y.; Hou, Y.; Zhao, M.; Li, J.; Du, Y.; Zhang, L.; Chen, C.; Yang, H.; Yan, F.; et al. Saikosaponin D exhibits anti-leukemic activity by targeting FTO/m(6)A signaling. Theranostics 2021, 11, 5831–5846. [Google Scholar] [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e10. [Google Scholar] [CrossRef] [Green Version]
- Ianniello, Z.; Sorci, M.; Ceci Ginistrelli, L.; Iaiza, A.; Marchioni, M.; Tito, C.; Capuano, E.; Masciarelli, S.; Ottone, T.; Attrotto, C.; et al. New insight into the catalytic -dependent and -independent roles of METTL3 in sustaining aberrant translation in chronic myeloid leukemia. Cell Death Dis. 2021, 12, 870. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.Y.; Zhao, C.; Zhong, F.M.; Qin, T.Y.; Wen, F.; Li, M.Y.; Liu, J.; Huang, B.; Wang, X.Z. m(6)A Modification of lncRNA NEAT1 Regulates Chronic Myelocytic Leukemia Progression via miR-766-5p/CDKN1A Axis. Front. Oncol. 2021, 11, 679634. [Google Scholar] [CrossRef]
- Lai, X.; Wei, J.; Gu, X.Z.; Yao, X.M.; Zhang, D.S.; Li, F.; Sun, Y.Y. Dysregulation of LINC00470 and METTL3 promotes chemoresistance and suppresses autophagy of chronic myelocytic leukaemia cells. J. Cell. Mol. Med. 2021, 25, 4248–4259. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Li, M.; Hong, H.; Xu, Q.; He, Z.; Peng, Z. Expression of N(6)-methyladenosine (m(6)A) regulators correlates with immune microenvironment characteristics and predicts prognosis in diffuse large cell lymphoma (DLBCL). Bioengineered 2021, 12, 6115–6133. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Fu, Y.; Wang, Y.; Wang, J. The m6A Methyltransferase METTL3 Is Functionally Implicated in DLBCL Development by Regulating m6A Modification in PEDF. Front. Genet. 2020, 11, 955. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Fan, G.; Song, S.; Jiang, Y.; Qian, C.; Zhang, W.; Su, Q.; Xue, X.; Zhuang, W.; Li, B. piRNA-30473 contributes to tumorigenesis and poor prognosis by regulating m6A RNA methylation in DLBCL. Blood 2021, 137, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Hou, Y.; Chen, Q.; Chen, J.; Li, Y.; Zhang, E.; Gu, H.; Xu, R.; Liu, Y.; Cao, W.; et al. RNA demethylase ALKBH5 promotes tumorigenesis in multiple myeloma via TRAF1-mediated activation of NF-κB and MAPK signaling pathways. Oncogene 2021. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Fan, G.; Li, Q.; Su, Q.; Zhang, X.; Xue, X.; Wang, Z.; Qian, C.; Jin, Z.; Li, B.; et al. IDH2 contributes to tumorigenesis and poor prognosis by regulating m6A RNA methylation in multiple myeloma. Oncogene 2021, 40, 5393–5402. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Tang, X.; Tang, C.; Hua, Z.; Ke, M.; Wang, C.; Zhao, J.; Gao, S.; Jurczyszyn, A.; Janz, S.; et al. HNRNPA2B1 promotes multiple myeloma progression by increasing AKT3 expression via m6A-dependent stabilization of ILF3 mRNA. J. Hematol. Oncol. 2021, 14, 54. [Google Scholar] [CrossRef]
- Chissoe, S.L.; Bodenteich, A.; Wang, Y.F.; Wang, Y.P.; Burian, D.; Clifton, S.W.; Crabtree, J.; Freeman, A.; Iyer, K.; Jian, L.; et al. Sequence and analysis of the human ABL gene, the BCR gene, and regions involved in the Philadelphia chromosomal translocation. Genomics 1995, 27, 67–82. [Google Scholar] [CrossRef]
- Liu, X.; Huang, L.; Huang, K.; Yang, L.; Yang, X.; Luo, A.; Cai, M.; Wu, X.; Liu, X.; Yan, Y.; et al. Novel Associations Between METTL3 Gene Polymorphisms and Pediatric Acute Lymphoblastic Leukemia: A Five-Center Case-Control Study. Front. Oncol. 2021, 11, 635251. [Google Scholar] [CrossRef]
- Luo, A.; Yang, L.; Li, M.; Cai, M.; Huang, A.; Liu, X.; Yang, X.; Yan, Y.; Wang, X.; Wu, X.; et al. Genetic Variants in METTL14 are Associated with the Risk of Acute Lymphoblastic Leukemia in Southern Chinese Children: A Five-Center Case-Control Study. Cancer Manag. Res. 2021, 13, 9189–9200. [Google Scholar] [CrossRef]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.H.; Dai, Y.F.; Huang, W.H.; Zhong, Q.F.; Zhu, P.; Zhang, W.J.; Wu, Z.H.; Lin, Q.; Zhu, H.Y.; Cui, L.Z.; et al. Prognostic Value of MicroRNA-20b in Acute Myeloid Leukemia. Front. Oncol. 2021, 10, 3467. [Google Scholar] [CrossRef]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.M.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Gilliland, D.G. Genetic’s of myeloid leukemias. Annu. Rev. Genom. Hum. Genet. 2002, 3, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Eckert, M.A.; Harada, B.T.; Liu, S.M.; Lu, Z.K.; Yu, K.K.; Tienda, S.M.; Chryplewicz, A.; Zhu, A.C.; Yang, Y.; et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018, 20, 1074–1083. [Google Scholar] [CrossRef]
- Pan, Z.P.; Wang, B.; Hou, D.Y.; You, R.L.; Wang, X.T.; Xie, W.H.; Huang, H.F. METTL3 mediates bone marrow mesenchymal stem cell adipogenesis to promote chemoresistance in acute myeloid leukaemia. FEBS Open Bio 2021, 11, 1659–1672. [Google Scholar] [CrossRef]
- Bansal, H.; Yihua, Q.; Iyer, S.P.; Ganapathy, S.; Proia, D.A.; Penalva, L.O.; Uren, P.J.; Suresh, U.; Carew, J.S.; Karnad, A.B.; et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 2014, 28, 1171–1174. [Google Scholar] [CrossRef] [Green Version]
- Raffel, G.D.; Mercher, T.; Shigematsu, H.; Williams, I.R.; Cullen, D.E.; Akashi, K.; Bernard, O.A.; Gilliland, D.G. Ott1(Rbm15) has pleiotropic roles in hematopoietic development. Proc. Natl. Acad. Sci. USA 2007, 104, 6001–6006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Renda, M.J.; Wang, L.; Cheng, E.C.; Niu, C.; Morris, S.W.; Chi, A.S.; Krause, D.S. Rbm15 modulates Notch-induced transcriptional activation and affects myeloid differentiation. Mol. Cell Biol. 2007, 27, 3056–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 2018, 172, 90–105.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elcheva, I.A.; Wood, T.; Chiarolanzio, K.; Chim, B.; Wong, M.; Singh, V.; Gowda, C.P.; Lu, Q.; Hafner, M.; Dovat, S.; et al. RNA-binding protein IGF2BP1 maintains leukemia stem cell properties by regulating HOXB4, MYB, and ALDH1A1. Leukemia 2020, 34, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef]
- Höglund, M.; Sandin, F.; Simonsson, B. Epidemiology of chronic myeloid leukaemia: An update. Ann. Hematol. 2015, 94 (Suppl. S2), S241–S247. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [Green Version]
- Trela, E.; Glowacki, S.; Błasiak, J. Therapy of chronic myeloid leukemia: Twilight of the imatinib era? ISRN Oncol. 2014, 2014, 596483. [Google Scholar] [CrossRef] [Green Version]
- Perrotti, D.; Turturro, F.; Neviani, P. BCR/ABL, mRNA translation and apoptosis. Cell Death Differ. 2005, 12, 534–540. [Google Scholar] [CrossRef]
- Prabhu, S.; Saadat, D.; Zhang, M.; Halbur, L.; Fruehauf, J.P.; Ong, S.T. A novel mechanism for Bcr-Abl action: Bcr-Abl-mediated induction of the eIF4F translation initiation complex and mRNA translation. Oncogene 2007, 26, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.Q.; Zeng, H.M.; Zheng, R.S.; Zhang, S.W.; He, J. Cancer incidence and mortality in china, 2007. Chin. J. Cancer Res. 2012, 24, 1–8. [Google Scholar] [CrossRef] [Green Version]
- An, L.; Li, X.; Yang, J. MicroRNA-211 attenuates cell proliferation in T-cell lymphoblastic lymphoma through targeting TCF12. Leuk. Res. 2021, 110, 106653. [Google Scholar] [CrossRef]
- Ma, H.; Shen, L.; Yang, H.; Gong, H.; Du, X.; Li, J. m6A methyltransferase Wilms’ tumor 1-associated protein facilitates cell proliferation and cisplatin resistance in NK/T cell lymphoma by regulating dual-specificity phosphatases 6 expression via m6A RNA methylation. IUBMB Life 2021, 73, 108–117. [Google Scholar] [CrossRef]
- Wu, G.; Suo, C.; Yang, Y.; Shen, S.; Sun, L.; Li, S.T.; Zhou, Y.; Yang, D.; Wang, Y.; Cai, Y.; et al. MYC promotes cancer progression by modulating m(6) A modifications to suppress target gene translation. EMBO Rep. 2021, 22, e51519. [Google Scholar] [CrossRef]
- Xia, T.L.; Li, X.; Wang, X.; Zhu, Y.J.; Zhang, H.; Cheng, W.; Chen, M.L.; Ye, Y.; Li, Y.; Zhang, A.; et al. N(6)-methyladenosine-binding protein YTHDF1 suppresses EBV replication and promotes EBV RNA decay. EMBO Rep. 2021, 22, e50128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; He, X.; Hu, J.; Yang, P.; Liu, C.; Wang, J.; An, R.; Zhen, J.; Pang, M.; Hu, K.; et al. Dysregulation of N(6)-methyladenosine regulators predicts poor patient survival in mantle cell lymphoma. Oncol. Lett. 2019, 18, 3682–3690. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Ntanasis-Stathopoulos, I. Multiple Myeloma: Clinical Updates From the American Society of Hematology Annual Meeting 2018. Clin. Lymphoma Myeloma Leuk. 2019, 19, e324–e336. [Google Scholar] [CrossRef] [PubMed]
- Pratanwanich, P.N.; Yao, F.; Chen, Y.; Koh, C.W.Q.; Wan, Y.K.; Hendra, C.; Poon, P.; Goh, Y.T.; Yap, P.M.L.; Chooi, J.Y.; et al. Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore. Nat. Biotechnol. 2021, 39, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Adès, L.; Itzykson, R.; Fenaux, P. Myelodysplastic syndromes. Lancet 2014, 383, 2239–2252. [Google Scholar] [CrossRef]
- Fei, D.L.; Zhen, T.; Durham, B.; Ferrarone, J.; Zhang, T.; Garrett, L.; Yoshimi, A.; Abdel-Wahab, O.; Bradley, R.K.; Liu, P.; et al. Impaired hematopoiesis and leukemia development in mice with a conditional knock-in allele of a mutant splicing factor gene U2af1. Proc. Natl. Acad. Sci. USA 2018, 115, E10437–E10446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, H.; Nakanishi, K.; Kojima, S. Inherited bone marrow failure syndromes in 2012. Int. J. Hematol. 2013, 97, 20–29. [Google Scholar] [CrossRef] [PubMed]
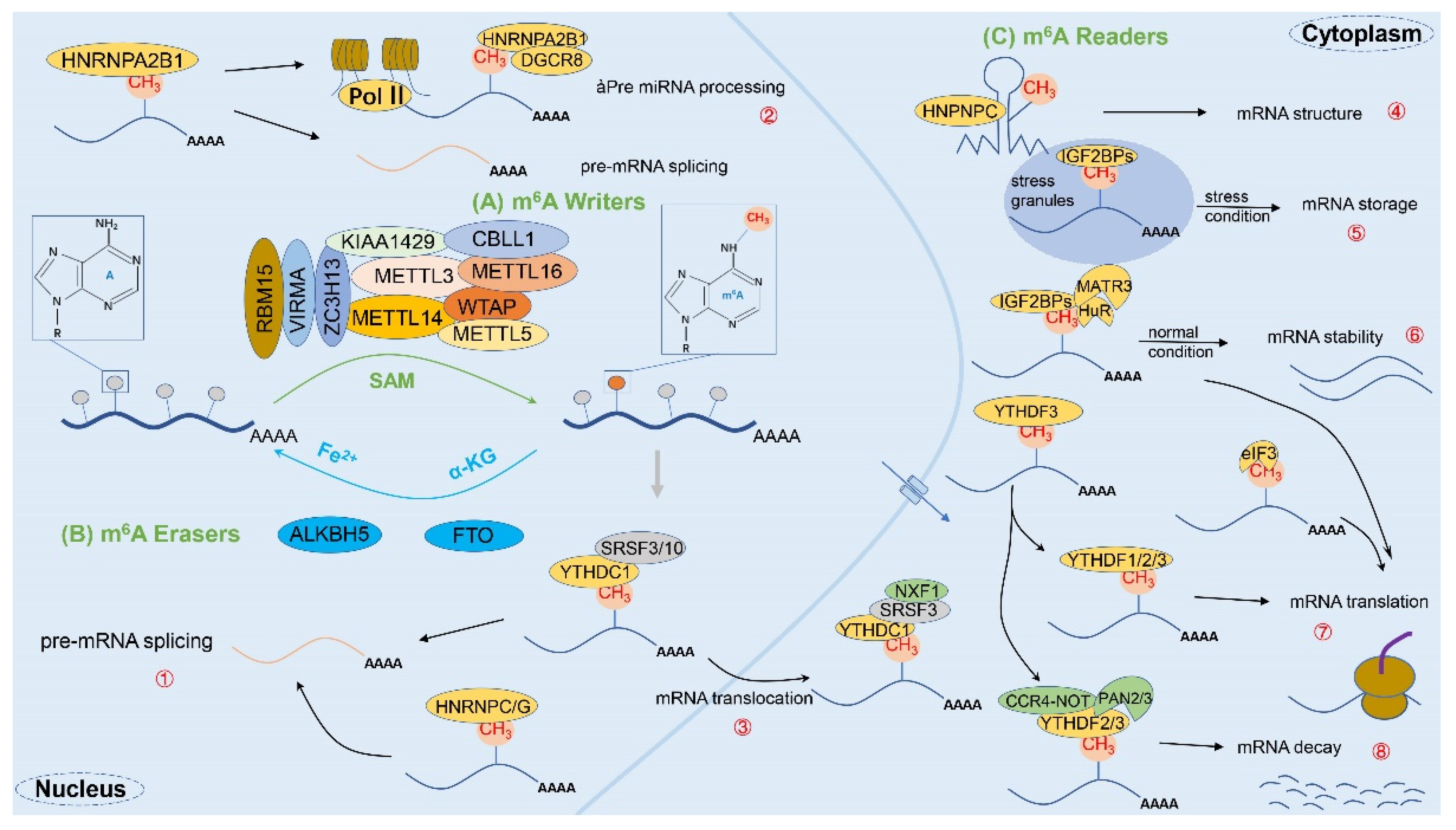
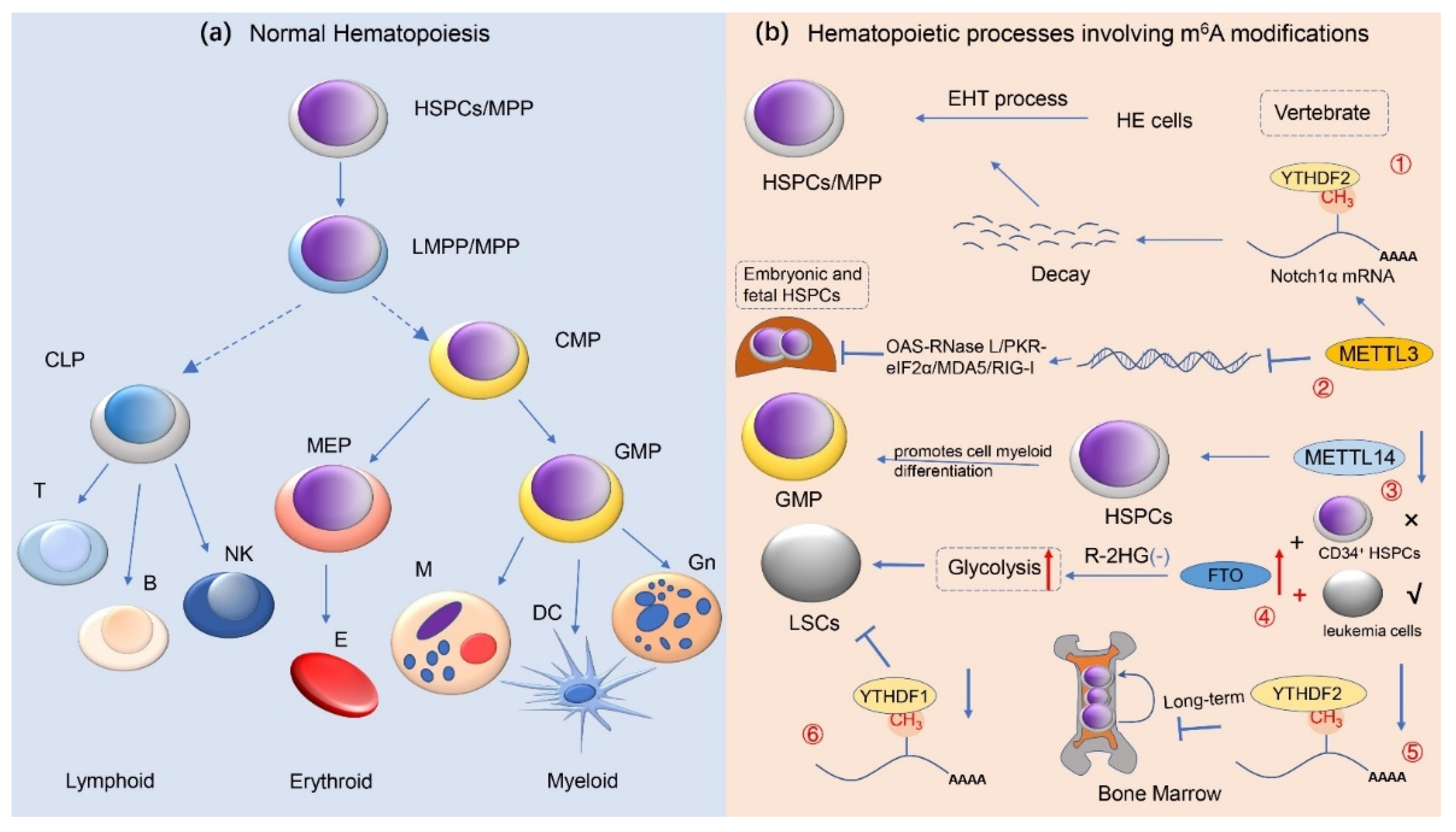
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).