
The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity



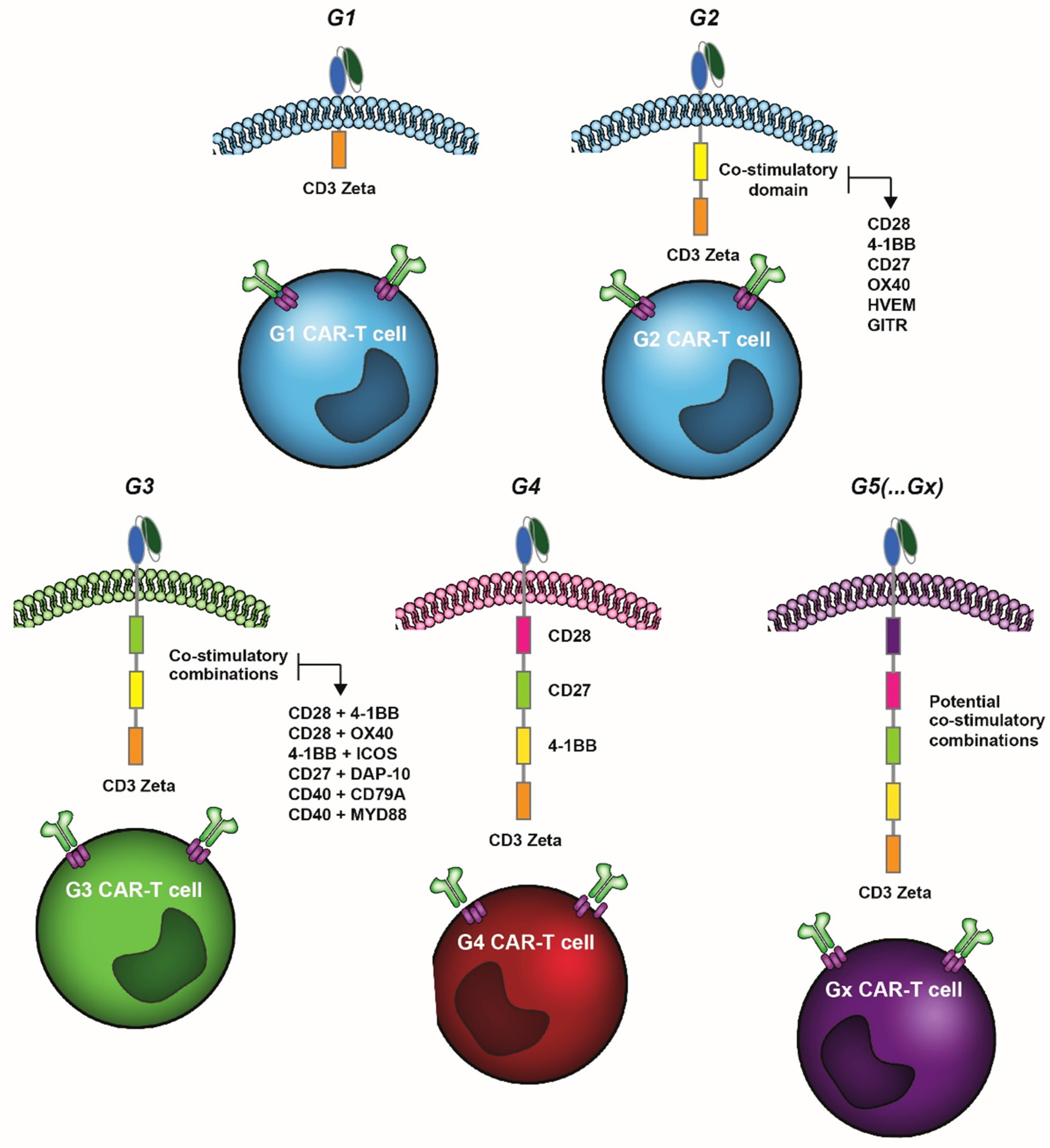
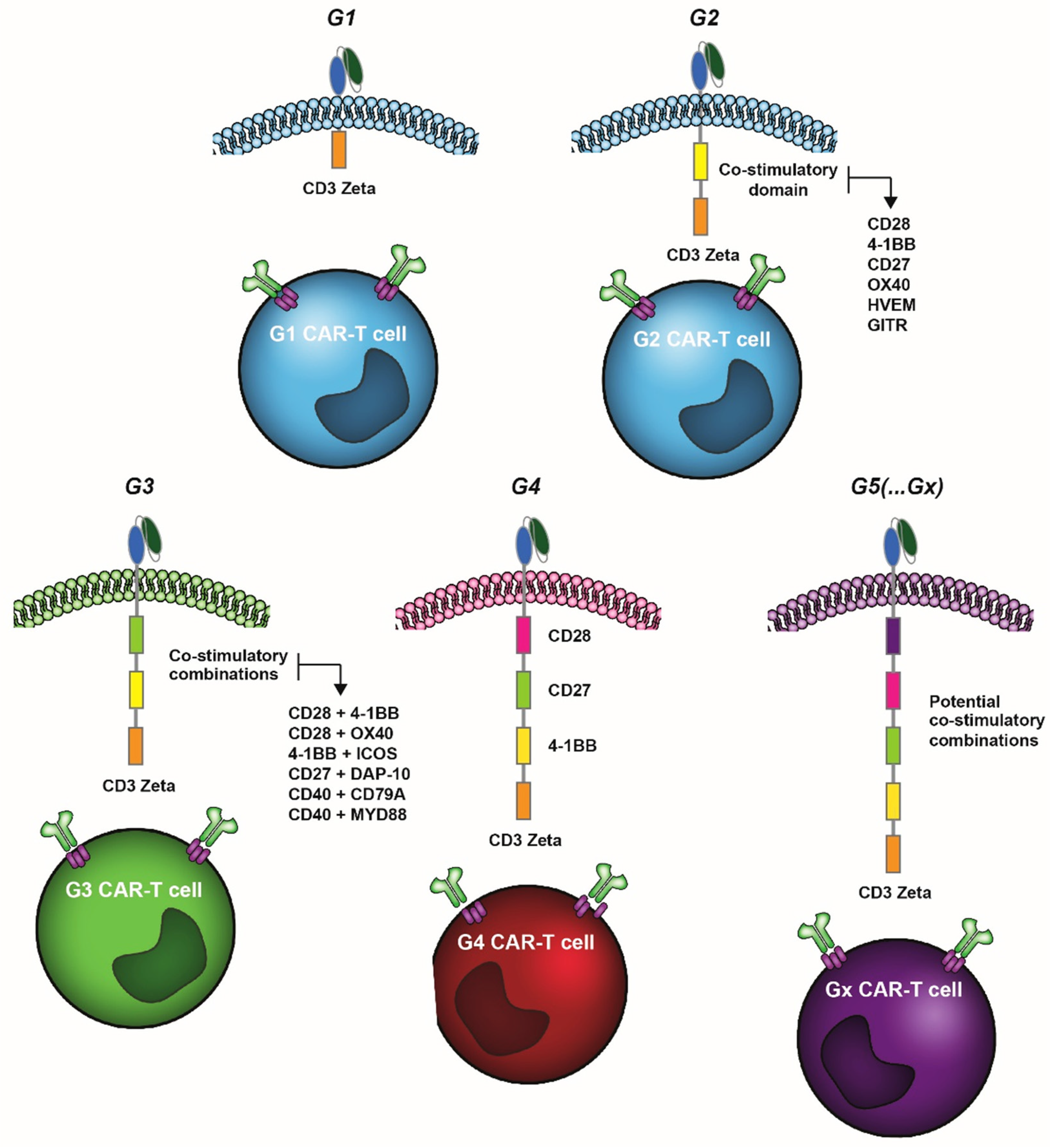{kind=link}
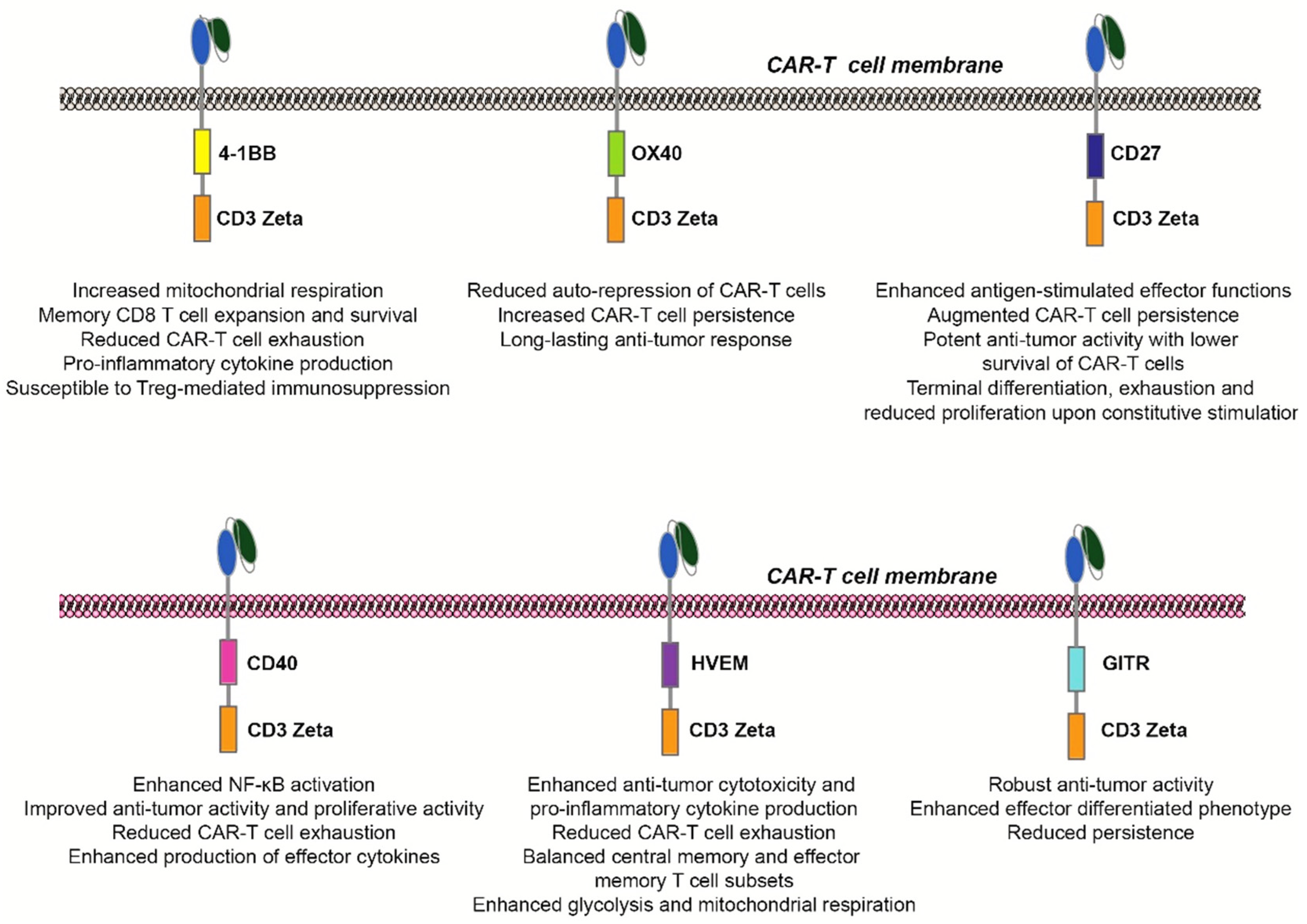
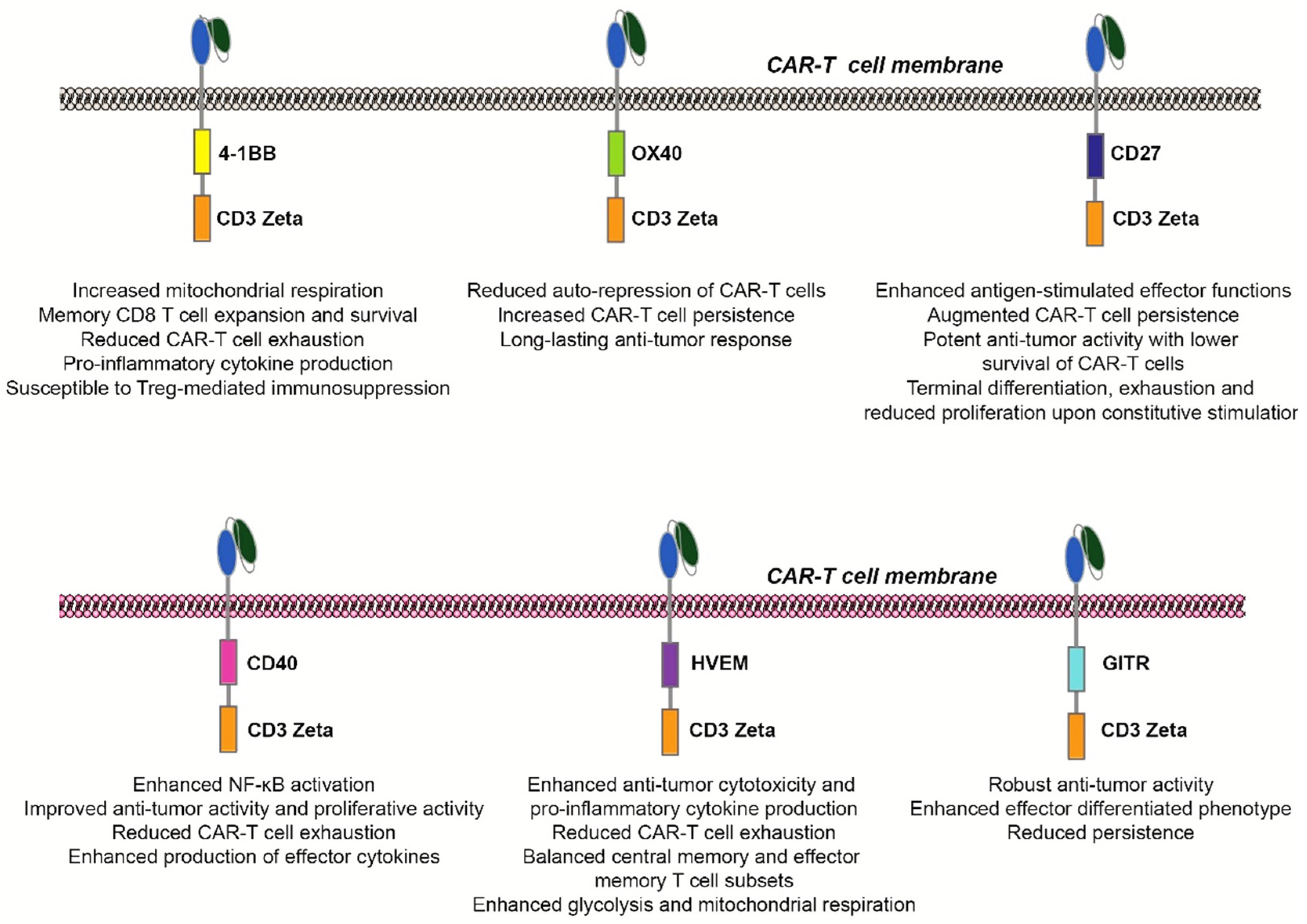{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. 4-1BB as Co-Stimulatory Domain in CAR-T Cells
3. OX40 as Co-Stimulatory Domain in CAR-T Cells
4. CD27 as Co-Stimulatory Domain in CAR-T Cells
5. CD40 as Co-Stimulatory Domain in CAR-T Cells
6. HVEM and GITR as Co-Stimulatory Domains in CAR-T Cells
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Moritz, D.; Wels, W.; Mattern, J.; Groner, B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4318–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.C.; Popplewell, L.P.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008, 112, 2261–2271. [Google Scholar] [CrossRef] [Green Version]
- Tokarew, N.; Ogonek, J.; Endres, S.; Von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2018, 120, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Zhao, J.; Song, Y. Engineering switchable and programmable universal CARs for CAR T therapy. J. Hematol. Oncol. 2019, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.; Xiong, T.; Wu, D.; Hu, P.; Alabanza, L.; Brittany, S.; Mahmud, H.; Anthony-Gonda, K.; Krueger, W.; Zhu, Z.; et al. Trispecific CD19-CD20-CD22–targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci. Transl. Med. 2021, 13, eabc6401. [Google Scholar] [CrossRef] [PubMed]
- Kyung, T.; Gordon, K.S.; Perez, C.R.; Holec, P.V.; Ramos, A.; Zhang, A.Q.; Liu, Y.; Koch, C.; Starchenko, A.; Joughin, B.; et al. CARPOOL: A library-based platform to rapidly identify next generation chimeric antigen receptors. bioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.07.09.450900v1 (accessed on 29 November 2021).
- Tang, X.Y.; Sun, Y.; Zhang, A.; Hu, G.L.; Cao, W.; Wang, D.H.; Zhang, B.; Chen, H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: A non-randomised, open-label phase I trial protocol. BMJ Open 2016, 6, e013904. [Google Scholar] [CrossRef] [Green Version]
- Bôle-Richard, E.; Fredon, M.; Biichlé, S.; Anna, F.; Certoux, J.; Renosi, F.; Tsé, F.; Molimard, C.; Valmary-Degano, S.; Jenvrin, A.; et al. CD28/4-1BB CD123 CAR T cells in blastic plasmacytoid dendritic cell neoplasm. Leukemia 2020, 34, 3228–3241. [Google Scholar] [CrossRef] [PubMed]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O' Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philipson, B.I.; O'Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-kappaB signaling. Sci. Signal. 2020, 13, eaay8248. [Google Scholar] [CrossRef] [PubMed]
- Kintz, H.; Nylen, E.; Barber, A. Inclusion of Dap10 or 4-1BB costimulation domains in the chPD1 receptor enhances anti-tumor efficacy of T cells in murine models of lymphoma and melanoma. Cell. Immunol. 2020, 351, 104069. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, F.; Cao, J.; Wang, X.; Cheng, H.; Qi, K.; Wang, G.; Xu, K.; Zheng, J.; Fu, Y.-X.; et al. A chimeric antigen receptor with antigen-independent OX40 signaling mediates potent antitumor activity. Sci. Transl. Med. 2021, 13, aba7308. [Google Scholar] [CrossRef]
- Kim, D.-H.; Chang, W.-S.; Lee, Y.-S.; Lee, K.-A.; Kim, Y.-K.; Kwon, B.S.; Kang, C.-Y. 4-1BB engagement costimulates NKT cell activation and exacerbates NKT cell ligand-induced airway hyperresponsiveness and inflammation. J. Immunol. 2008, 180, 2062–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, T.; Bukczynski, J.; Watts, T.H. 4-1BB Ligand-Mediated Costimulation of Human T Cells Induces CD4 and CD8 T Cell Expansion, Cytokine Production, and the Development of Cytolytic Effector Function. J. Immunol. 2002, 168, 4897–4906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futagawa, T.; Akiba, H.; Kodama, T.; Takeda, K.; Hosoda, Y.; Yagita, H.; Okumura, K. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int. Immunol. 2002, 14, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, R.G.; Din, W.S.; Davis-Smith, T.; Anderson, D.M.; Gimpel, S.D.; Sato, T.A.; Maliszewski, C.R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; et al. Molecular cloning of a ligand for the inducible T cell gene 4-1BB: A member of an emerging family of cytokines with homology to tumor necrosis factor. Eur. J. Immunol. 1993, 23, 2631–2641. [Google Scholar] [CrossRef]
- Zapata, J.M.; Perez-Chacon, G.; Carr-Baena, P.; Martinez-Forero, I.; Azpilikueta, A.; Otano, I.; Melero, I. CD137 (4-1BB) Signalosome: Complexity Is a Matter of TRAFs. Front. Immunol. 2018, 9, 2618. [Google Scholar] [CrossRef]
- Croft, M. The TNF family in T cell differentiation and function—Unanswered questions and future directions. Semin. Immunol. 2014, 26, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellström, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar] [CrossRef]
- Houot, R.; Goldstein, M.J.; Kohrt, H.E.; Myklebust, J.H.; Alizadeh, A.A.; Lin, J.T.; Irish, J.M.; Torchia, J.A.; Kolstad, A.; Chen, L.; et al. Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by Treg depletion. Blood 2009, 114, 3431–3438. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.M.; Trivedi, S.; Concha-Benavente, F.; Gibson, S.P.; Reeder, C.; Ferrone, S.; Ferris, R.L. CD137 Stimulation Enhances Cetuximab-Induced Natural Killer: Dendritic Cell Priming of Anti-tumor T-Cell Immunity in Patients with Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lee, L.-F.; Fisher, T.S.; Jessen, B.; Elliott, M.; Evering, W.; Logronio, K.; Tu, G.H.; Tsaparikos, K.; Li, X.; et al. Combination of 4-1BB Agonist and PD-1 Antagonist Promotes Antitumor Effector/Memory CD8 T Cells in a Poorly Immunogenic Tumor Model. Cancer Immunol. Res. 2015, 3, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 Blockade and 4-1BB Activation Enhances Tumor Rejection by Increasing T-Cell Infiltration, Proliferation, and Cytokine Production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Gato-Cañas, M.; Melero, I.; Delgoffe, G.M. Metabolic Consequences of T-cell Costimulation in Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 1564–1569. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.-A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther.-Oncolytics 2020, 18, 272–281. [Google Scholar] [CrossRef]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol. Ther.-Oncolytics 2019, 15, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Wherry, E. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Dai, Q.; Han, P.; Qi, X.; Li, F.; Li, M.; Fan, L.; Zhang, H.; Zhang, X.; Yang, X. 4-1BB Signaling Boosts the Anti-Tumor Activity of CD28-Incorporated 2nd Generation Chimeric Antigen Receptor-Modified T Cells. Front. Immunol. 2020, 11, 539654. [Google Scholar] [CrossRef]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Recep-tor-Redirected T Cells. Cancer Cell 2020, 37, 216–225.e6. [Google Scholar] [CrossRef]
- Kegler, A.; Koristka, S.; Bergmann, R.; Berndt, N.; Arndt, C.; Feldmann, A.; Hoffmann, A.; Bornhäuser, M.; Schmitz, M.; Bachmann, M.P. T cells engrafted with a UniCAR 28/z outperform UniCAR BB/z-transduced T cells in the face of regulatory T cell-mediated immunosuppression. OncoImmunology 2019, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.S.; Rooney, C.M.; Orange, J.S. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vec-tor-Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Pulsipher, M.A.; Boyer, M.W.; Grupp, S.A.; Davies, M.S.M.; Phillips, C.L.; Verneris, M.R.; August, M.K.J.; Schlis, K.; Driscoll, T.A.; et al. Efficacy and Safety of CTL019 in the First US Phase II Multicenter Trial in Pediatric Relapsed/Refractory Acute Lymphoblastic Leukemia: Results of an Interim Analysis. Blood 2016, 128, 2801. [Google Scholar] [CrossRef]
- Zhang, J.; Li, J.; Ma, Q.; Yang, H.; Signorovitch, J.; Wu, E. A Review of Two Regulatory Approved Anti-CD19 CAR T-Cell Therapies in Diffuse Large B-Cell Lym-phoma: Why Are Indirect Treatment Comparisons Not Feasible? Adv. Ther. 2020, 37, 3040–3058. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; LiaPalomba, M.; Gordon, L.I.; LunningDO, M.A.; Wang, M.L.; Arnason, J.E.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Pivotal Safety and Efficacy Results from Transcend NHL 001, a Multicenter Phase 1 Study of Li-socabtagene Maraleucel (liso-cel) in Relapsed/Refractory (R/R) Large B Cell Lymphomas. Blood 2019, 134 (Suppl. 1), 241. [Google Scholar] [CrossRef]
- Abramson, J.S.; Gordon, L.I.; Palomba, M.L.; Lunning, M.A.; Arnason, J.E.; Forero-Torres, A.; Wang, M.; Maloney, D.G.; Sehgal, A.; Andreadis, C.; et al. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of li-socabtagene maraleucel (JCAR017) in R/R aggressive NHL. J. Clin. Oncol. 2018, 36 (Suppl. 15), 7505. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Delforge, M.; Baz, R.C.; Cavo, M.; Callander, N.S.; Ghobadi, A.; Rodriguez-Otero, P.; Mateos, M.-V.; Massaro, M.; Ding, L.; Patel, P.; et al. KarMMa-3: A Phase 3 Study of Idecabtagene Vicleucel (ide-cel, bb2121), a BCMA-Directed CAR T Cell Therapy Vs Standard Regimens in Relapsed and Refractory Multiple Myeloma. Blood 2020, 136, 24–25. [Google Scholar] [CrossRef]
- Jagannath, S.; Lin, Y.; Goldschmidt, H.; Reece, D.; Nooka, A.; Senin, A.; Rodriguez-Otero, P.; Powles, R.; Matsue, K.; Shah, N.; et al. KarMMa-RW: Comparison of idecabtagene vicleucel with real-world outcomes in relapsed and re-fractory multiple myeloma. Blood Cancer J. 2021, 11, 116. [Google Scholar] [CrossRef]
- Karlsson, H.; Svensson, E.; Gigg, C.; Jarvius, M.; Olsson-Strömberg, U.; Savoldo, B.; Dotti, G.; Loskog, A. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS ONE 2015, 10, e0144787. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, A.; Shahinian, A.; Bladt, F.; Yoshinaga, S.K.; Jordana, M.; Wakeham, A.; Boucher, L.-M.; Bouchard, D.; Chan, V.S.F.; Duncan, G.S.; et al. ICOS is essential for effective T-helper-cell responses. Nature 2001, 409, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, S.P.; Kim, S.; Motz, G.T.; Alatzoglou, D.; Melita, I.; Irving, M.; Powell, D.J., Jr.; Coukos, G. T Cells Bearing a Chimeric Antigen Receptor against Prostate-Specific Membrane Antigen Mediate Vascular Disruption and Result in Tumor Regression. Cancer Immunol. Res. 2015, 3, 68–84. [Google Scholar] [CrossRef] [Green Version]
- Cappell, K.M.; Kochenderfer, J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costim-ulatory domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Control of Immunity by the TNFR-Related Molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [Green Version]
- Gramaglia, I.; Weinberg, A.D.; Lemon, M.; Croft, M. Ox-40 ligand: A potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998, 161, 6510–6517. [Google Scholar]
- Dawicki, W.; Bertram, E.M.; Sharpe, A.H.; Watts, T.H. 4-1BB and OX40 Act Independently to Facilitate Robust CD8 and CD4 Recall Responses. J. Immunol. 2004, 173, 5944–5951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serghides, L.; Bukczynski, J.; Wen, T.; Wang, C.; Routy, J.; Boulassel, M.; Sekaly, R.; Ostrowski, M.; Bernard, N.F.; Watts, T.H.; et al. Evaluation of OX40 ligand as a costimulator of human antiviral memory CD8 T cell responses: Comparison with B7.1 and 4-1BBL. J. Immunol. 2005, 175, 6368–6377. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; So, T.; Croft, M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and sur-vival. J. Immunol. 2008, 180, 7240–7248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, P.R.; Song, J.; Gramaglia, I.; Killeen, N.; Croft, M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001, 15, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Weatherill, A.R.; Maxwell, J.R.; Takahashi, C.; Weinberg, A.D.; Vella, A.T. OX40 Ligation Enhances Cell Cycle Turnover of Ag-Activated CD4 T Cells in Vivo. Cell. Immunol. 2001, 209, 63–75. [Google Scholar] [CrossRef]
- Xiao, X.; Gong, W.; Demirci, G.; Liu, W.; Spoerl, S.; Chu, X.; Bishop, D.K.; Turka, L.A.; Li, X.C. New Insights on OX40 in the Control of T Cell Immunity and Immune Tolerance In Vivo. J. Immunol. 2011, 188, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, J.; Xiao, Y.; Rossen, J.W.A.; Van Der Sluijs, K.F.; Sugamura, K.; Ishii, N.; Borst, J. During Viral Infection of the Respiratory Tract, CD27, 4-1BB, and OX40 Collectively Determine Formation of CD8+ Memory T Cells and Their Capacity for Secondary Expansion. J. Immunol. 2005, 175, 1665–1676. [Google Scholar] [CrossRef] [Green Version]
- Arch, R.H.; Thompson, C.B. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor re-ceptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol. Cell Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-W.; Park, Y.; Song, A.; Cheroutre, H.; Kwon, B.S.; Croft, M. Functional dichotomy between OX40 and 4-1BB in modulating effector CD8 T cell responses. J. Immunol. 2006, 177, 4464–4472. [Google Scholar] [CrossRef] [Green Version]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+T cells. OncoImmunology 2012, 1, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Terai, M.; Tamura, Y.; Alexeev, V.; Mastrangelo, M.J.; Selvan, S.R. Interleukin 10 in the tumor microenvironment: A target for anticancer immunotherapy. Immunol. Res. 2011, 51, 170–182. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.A.; Abken, H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int. J. Cancer 2011, 129, 2935–2944. [Google Scholar] [CrossRef]
- Hombach, A.A.; Chmielewski, M.; Rappl, G.; Abken, H. Adoptive immunotherapy with redirected T cells produces CCR7- cells that are trapped in the periphery and benefit from combined CD28-OX40 costimulation. Hum. Gene Ther. 2013, 24, 259–269. [Google Scholar] [CrossRef]
- De Angelis, B.; Guercio, M.; Orlando, D.; Di Cecca, S.; Sinibaldi, M.; Boffa, I.; Caruso, S.; Abbaszadeh, Z.; Camera, A.; Biancamaria, C.; et al. A New Promising Third Generation CAR-CD30 T-Cell Therapy for CD30+ Lymphoma. Blood 2019, 134, 2069. [Google Scholar] [CrossRef]
- Guercio, M.; Orlando, D.; Di Cecca, S.; Sinibaldi, M.; Boffa, I.; Caruso, S.; Abbaszadeh, Z.; Camera, A.; Cembrola, B.; Bovetti, K.; et al. CD28.OX40 co-stimulatory combination is associated with long in vivo persistence and high activity of CAR.CD30 T-cells. Haematologica 2020, 106, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Zhang, Y.; Yu, L.; Wu, D. Cytotoxic effect of CLL-1 CAR-T cell immunotherapy with PD-1 silencing on relapsed/refractory acute myeloid leukemia. Mol. Med. Rep. 2021, 23, 208. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.; Hendriks, J.; Xiao, Y. CD27 and CD70 in T cell and B cell activation. Curr. Opin. Immunol. 2005, 17, 275–281. [Google Scholar] [CrossRef]
- Roberts, D.J.; Franklin, N.A.; Kingeter, L.M.; Yagita, H.; Tutt, A.L.; Glennie, M.J.; Bullock, T.N. Control of Established Melanoma by CD27 Stimulation Is Associated With Enhanced Effector Function and Persistence, and Reduced PD-1 Expression of Tumor Infiltrating CD8+ T Cells. J. Immunother. 2010, 33, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, V.; Sundarapandiyan, K.; Zhao, B.; Bylesjo, M.; Marsh, H.C.; Keler, T. Characterization of the human T cell response to in vitro CD27 costimulation with varlilumab. J. Immunother. Cancer 2015, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J., Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Han, Y.; Xie, W.; Song, D.-G.; Powell, D.J., Jr. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J. Hematol. Oncol. 2018, 11, 92. [Google Scholar] [CrossRef]
- Duong, C.P.M.; Westwood, J.A.; Yong, C.S.M.; Murphy, A.; Devaud, C.; John, L.B.; Darcy, P.K.; Kershaw, M.H. Engineering T Cell Function Using Chimeric Antigen Receptors Identified Using a DNA Library Approach. PLoS ONE 2013, 8, e63037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.; Cao, T.M.; Baker, J.A.; Verneris, M.R.; Soares, L.; Negrin, R.S. Silencing human NKG2D, DAP10, and DAP12 reduces cytotoxicity of activated CD8+ T cells and NK cells. J. Immunol. 2005, 175, 7819–7828. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Cheng, L.; Jiang, Z.; Wei, X.; Li, B.; Wu, Q.; Wang, S.; Lin, S.; Long, Y.; Zhang, X.; et al. DNAX-activating protein 10 co-stimulation enhances the anti-tumor efficacy of chimeric antigen receptor T cells. OncoImmunology 2018, 8, e1509173. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ma, X.; Liu, Y.; Zhao, W.; Yu, L.; Qin, M.; Zhu, G.; Wang, K.; Shi, X.; Zhang, Z. Chimeric Antigen Receptor 4SCAR-GD2-Modified T Cells Targeting High-Risk and Recurrent Neuroblastoma: A Phase II Multi-Center Trial in China. Blood 2017, 130 (Suppl. 1), 3335. [Google Scholar]
- Zhang, H.; Wang, P.; Li, Z.; He, Y.; Gan, W.; Jiang, H. Anti-CLL1 Chimeric Antigen Receptor T-Cell Therapy in Children with Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 3549–3555. [Google Scholar] [CrossRef]
- Jiao, C.; Zvonkov, E.; Lai, X.; Zhang, R.; Liu, Y.; Qin, Y.; Savchenko, V.; Gabeeva, N.; Chung, T.-H.; Sheng, L.; et al. 4SCAR2.0: A multi-CAR-T therapy regimen for the treatment of relapsed/refractory B cell lymphomas. Blood Cancer J. 2021, 11, 59. [Google Scholar] [CrossRef]
- Shaffer, D.R.; Savoldo, B.; Yi, Z.; Chow, K.K.H.; Kakarla, S.; Spencer, D.M.; Dotti, G.; Wu, M.-F.; Liu, H.; Kenney, S.; et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood 2011, 117, 4304–4314. [Google Scholar] [CrossRef] [Green Version]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Gottschalk, S.; Rooney, C.M. CD70-Specific CAR T Cells Have Potent Activity Against Acute Myeloid Leukemia (AML) without HSC Toxicity. Blood 2019, 134, 1932. [Google Scholar] [CrossRef]
- Wasiuk, A.; Testa, J.; Weidlick, J.; Sisson, C.; Vitale, L.; Widger, J.; Crocker, A.; Thomas, L.J.; Goldstein, J.; Marsh, H.C.; et al. CD27-Mediated Regulatory T Cell Depletion and Effector T Cell Costimulation Both Contribute to Antitumor Efficacy. J. Immunol. 2017, 199, 4110–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Brito, A.; Alizadeh, D.; Starr, R.; Aguilar, B.; Badie, B.; Forman, S.J.; Brown, C.E. Abstract 2321: Dual-function of CD27-CD70 costimulatory signal in CAR T cell therapy. Cancer Res. 2019, 79 (Suppl. 13), 2321. [Google Scholar]
- Mach, F.; Schönbeck, U.; Sukhova, G.K.; Bourcier, T.; Bonnefoy, J.Y.; Pober, J.S.; Libby, P. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and mac-rophages: Implications for CD40-CD40 ligand signaling in atherosclerosis. Proc. Natl. Acad. Sci. USA 1997, 94, 1931–1936. [Google Scholar] [CrossRef] [Green Version]
- Vanichakarn, P.; Blair, P.; Wu, C.; Freedman, J.E.; Chakrabarti, S. Neutrophil CD40 enhances platelet-mediated inflammation. Thromb. Res. 2008, 122, 346–358. [Google Scholar] [CrossRef]
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998, 393, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Carbone, E.; Ruggiero, G.; Terrazzano, G.; Palomba, C.; Manzo, C.; Fontana, S.; Spits, H.; Kärre, K.; Zappacosta, S. A New Mechanism of NK Cell Cytotoxicity Activation: The CD40–CD40 Ligand Interaction. J. Exp. Med. 1997, 185, 2053–2060. [Google Scholar] [CrossRef]
- Bennett, S.R.M.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.A.P.; Heath, W. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480. [Google Scholar] [CrossRef]
- Schoenberger, S.P.; Toes, R.; Van Der Voort, E.I.H.; Offringa, R.; Melief, C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Munroe, M.; Bishop, G.A. A Costimulatory Function for T Cell CD40. J. Immunol. 2007, 178, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Ara, A.; Ahmed, K.A.; Xiang, J. Multiple effects of CD40–CD40L axis in immunity against infection and cancer. ImmunoTargets Ther. 2018, 7, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.; Cheng, X.; Truong, B.; Sun, L.; Yang, X.; Wang, H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol. Ther. 2021, 219, 107709. [Google Scholar] [CrossRef]
- McWhirter, S.M.; Pullen, S.S.; Holton, J.M.; Crute, J.J.; Kehry, M.R.; Alber, T. Crystallographic analysis of CD40 recognition and signaling by human TRAF2. Proc. Natl. Acad. Sci. USA 1999, 96, 8408–8413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostager, B.S.; Catlett, I.; Bishop, G. Recruitment of CD40 and Tumor Necrosis Factor Receptor-associated Factors 2 and 3 to Membrane Microdomains during CD40 Signaling. J. Biol. Chem. 2000, 275, 15392–15398. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488.e6. [Google Scholar] [CrossRef] [Green Version]
- Levin-Piaeda, O.; Levin, N.; Pozner, S.; Danieli, A.; Weinstein-Marom, H.; Gross, G. The Intracellular Domain of CD40 is a Potent Costimulatory Element in Chimeric Antigen Receptors. J. Immunother. 2021, 44, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Bishop, G.A.; Warren, W.D.; Berton, M.T. Signaling via major histocompatibility complex class II molecules and antigen receptors enhances the B cell response to gp39/CD40 ligand. Eur. J. Immunol. 1995, 25, 1230–1238. [Google Scholar] [CrossRef]
- Julamanee, J.; Terakura, S.; Umemura, K.; Adachi, Y.; Miyao, K.; Okuno, S.; Takagi, E.; Sakai, T.; Koyama, D.; Goto, T.; et al. Composite CD79A/CD40 co-stimulatory endodomain enhances CD19CAR-T cell proliferation and survival. Mol. Ther. 2021, 29, 2677–2690. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.E.; Mahendravada, A.; Shinners, N.P.; Chang, W.; Crisostomo, J.; Lu, A.; Khalil, M.; Morschl, E.; Shaw, J.L.; Saha, S.; et al. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Mole-cule-Dependent Inducible MyD88/CD40. Mol. Ther. 2017, 25, 2176–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata, M.; Gerken, C.; Nguyen, P.; Krenciute, G.; Spencer, D.M.; Gottschalk, S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017, 7, 1306–1319. [Google Scholar] [CrossRef] [Green Version]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef]
- Zhou, X.; Dotti, G.; Krance, R.A.; Martinez, C.A.; Naik, S.; Kamble, R.T.; Durett, A.G.; Dakhova, O.; Savoldo, B.; Di Stasi, A.; et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015, 125, 4103–4113. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.; Ballard, B.; Yi, X.; Malankar, A.; Collinson-Pautz, M.R.; Becerra, C.R.; Woodard, J.P.; Foster, A.E. Tumor infiltration and cytokine biomarkers of prostate stem cell antigen (PSCA)-directed GOCAR-T cells in patients with advanced pancreatic tumors. J. Clin. Oncol. 2020, 38, 734. [Google Scholar] [CrossRef]
- Wang, X.; Jasinski, D.L.; Medina, J.L.; Spencer, D.M.; Foster, A.E.; Bayle, J.H. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020, 4, 1950–1964. [Google Scholar] [CrossRef] [PubMed]
- Collinson-Pautz, M.R.; Chang, W.; Lu, A.; Khalil, M.; Crisostomo, J.W.; Lin, P.; Mahendravada, A.; Shinners, N.P.; Brandt, M.E.; Zhang, M.; et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia 2019, 33, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.M.; Nelson, C.A.; Sedy, J. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat. Rev. Immunol. 2006, 6, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Shui, J.-W.; Kronenberg, M. HVEM is a TNF Receptor with Multiple Regulatory Roles in the Mucosal Immune System. Immune Netw. 2014, 14, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Freeman, G.J. The CD160, BTLA, LIGHT/HVEM pathway: A bidirectional switch regulating T-cell activation. Immunol. Rev. 2009, 229, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Stiles, K.M.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Herpes Simplex Virus Glycoprotein D Interferes with Binding of Herpesvirus Entry Mediator to Its Ligands through Downregulation and Direct Competition. J. Virol. 2010, 84, 11646–11660. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.; Hutchinson, T.; Murphy, K.M.; Ware, C.F.; Croft, M.; Salek-Ardakani, S. CD8 T Cell Memory to a Viral Pathogen Requires Trans. Cosignaling between HVEM and BTLA. PLoS ONE 2013, 8, e77991. [Google Scholar] [CrossRef]
- Park, J.-J.; Anand, S.; Zhao, Y.; Matsumura, Y.; Sakoda, Y.; Kuramasu, A.; Strome, S.E.; Chen, L.; Tamada, K. Expression of anti-HVEM single-chain antibody on tumor cells induces tumor-specific immunity with long-term memory. Cancer Immunol. Immunother. 2011, 61, 203–214. [Google Scholar] [CrossRef]
- Nunoya, J.-I.; Masuda, M.; Ye, C.; Su, L. Chimeric Antigen Receptor T Cell Bearing Herpes Virus Entry Mediator Co-stimulatory Signal Domain Exhibits High Functional Potency. Mol. Ther.-Oncolytics 2019, 14, 27–37. [Google Scholar] [CrossRef]
- Fisicaro, P.; Barili, V.; Montanini, B.; Acerbi, G.; Ferracin, M.; Guerrieri, F.; Salerno, D.; Boni, C.; Massari, M.; Cavallo, M.C.; et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017, 23, 327–336. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8 + T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Nocentini, G.; Riccardi, C. GITR: A Modulator of Immune Response and Inflammation. Adv. Exp. Med. Biol. 2009, 647, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Chakraborty, N. GITR expression onT-cell receptor-stimulated human CD8+T cell in a JNK-dependent pathway. Indian J. Hum. Genet. 2009, 15, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrier, Y.; Whitters, M.J.; Miyashiro, J.S.; Labranche, T.P.; Ramon, H.E.; Benoit, S.E.; Ryan, M.S.; Keegan, S.P.; Guay, H.; Douhan, J.; et al. Enhanced GITR/GITRL interactions augment IL-27 expression and induce IL-10-producing Tr-1 like cells. Eur. J. Immunol. 2012, 42, 1393–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronchetti, S.; Ricci, E.; Petrillo, M.G.; Cari, L.; Migliorati, G.; Nocentini, G.; Riccardi, C. Glucocorticoid-Induced Tumour Necrosis Factor Receptor-Related Protein: A Key Marker of Functional Regulatory T Cells. J. Immunol. Res. 2015, 2015, 171520. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.-H.; Kim, I.-K.; Shin, K.-S.; Jeon, I.; Song, B.; Lee, J.-M.; Bae, E.-A.; Seo, H.; Kang, T.-S.; Kim, B.-S.; et al. GITR agonism triggers antitumor immune responses through IL21-expressing follicular helper T cells. Cancer Immunol. Res. 2020, 8, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Golubovskaya, V.M.; Berahovich, R.; Xu, Q.; Zhou, H.; Xu, S.; Guan, J.; Harto, H.; Li, L.; Wu, L. GITR domain inside CAR co-stimulates activity of CAR-T cells against cancer. Front. Biosci. 2018, 23, 2245–2254. [Google Scholar] [CrossRef]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.E.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D., Jr. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Jang, I.K.; Lee, Z.H.; Kim, Y.J.; Kim, S.H.; Kwon, B.S. Human 4-1BB (CD137) signals are mediated by TRAF2 and activate nuclear factor-kappa B. Biochem. Biophys. Res. Commun. 1998, 242, 613–620. [Google Scholar] [CrossRef]
- Saoulli, K.; Lee, S.Y.; Cannons, J.L.; Yeh, W.C.; Santana, A.; Goldstein, M.D.; Bangia, N.; Debenedette, M.A.; Mak, T.W.; Choi, Y.; et al. CD28-independent, TRAF2-dependent Costimulation of Resting T Cells by 4-1BB Ligand. J. Exp. Med. 1998, 187, 1849–1862. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, A.; Labiano, S.; Garasa, S.; Etxeberria, I.; Santamaría, E.; Rouzaut, A.; Enamorado, M.; Azpilikueta, A.; Inoges, S.; Bolanos-Mateo, E.; et al. Mitochondrial Morphological and Functional Reprogramming Following CD137 (4-1BB) Costimulation. Cancer Immunol. Res. 2018, 6, 798–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Snyder, K.M.; Suhoski, M.M.; Maus, M.V.; Kapoor, V.; June, C.H.; Mackall, C.L. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J. Immunol. 2007, 179, 4910–4918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murad, J.P.; Tilakawardane, D.; Park, A.K.; Lopez, L.S.; Young, C.A.; Gibson, J.; Yamaguchi, Y.; Lee, H.J.; Kennewick, K.T.; Gittins, B.J.; et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol. Ther. 2021, 29, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. eBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Vlaming, M.; van Meerten, T.; Bremer, E. The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers 2022, 14, 299. https://doi.org/10.3390/cancers14020299
He Y, Vlaming M, van Meerten T, Bremer E. The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers. 2022; 14(2):299. https://doi.org/10.3390/cancers14020299
Chicago/Turabian StyleHe, Yuan, Martijn Vlaming, Tom van Meerten, and Edwin Bremer. 2022. "The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity" Cancers 14, no. 2: 299. https://doi.org/10.3390/cancers14020299
APA StyleHe, Y., Vlaming, M., van Meerten, T., & Bremer, E. (2022). The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers, 14(2), 299. https://doi.org/10.3390/cancers14020299