
Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications

 , ,
, , 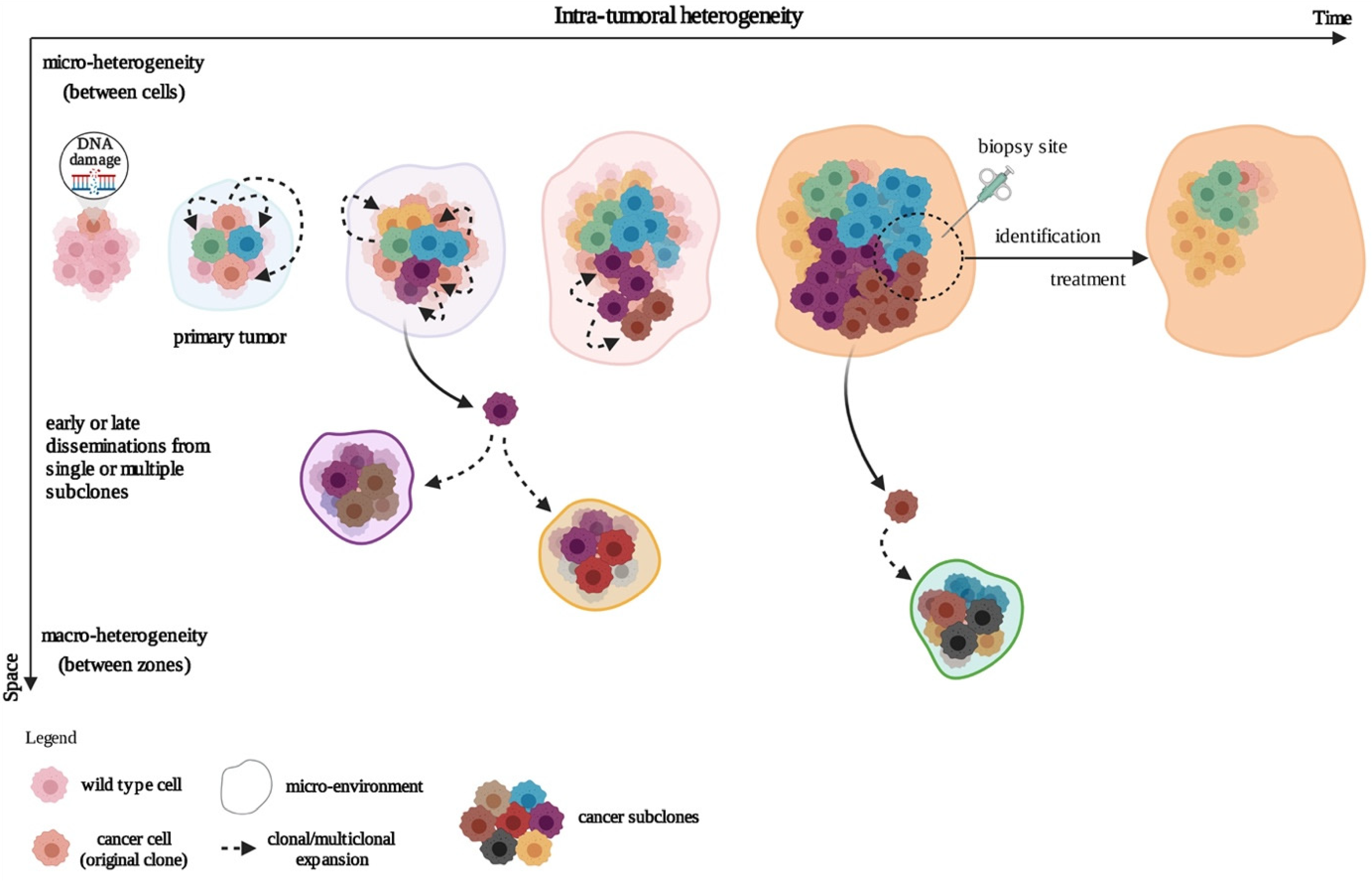{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Types and Mechanisms of Heterogeneity
2.1. Genetic and Epigenetic Heterogeneity
2.2. Transcriptional Heterogeneity
2.3. Tumor Ecosystem: Microenvironment and Metabolism
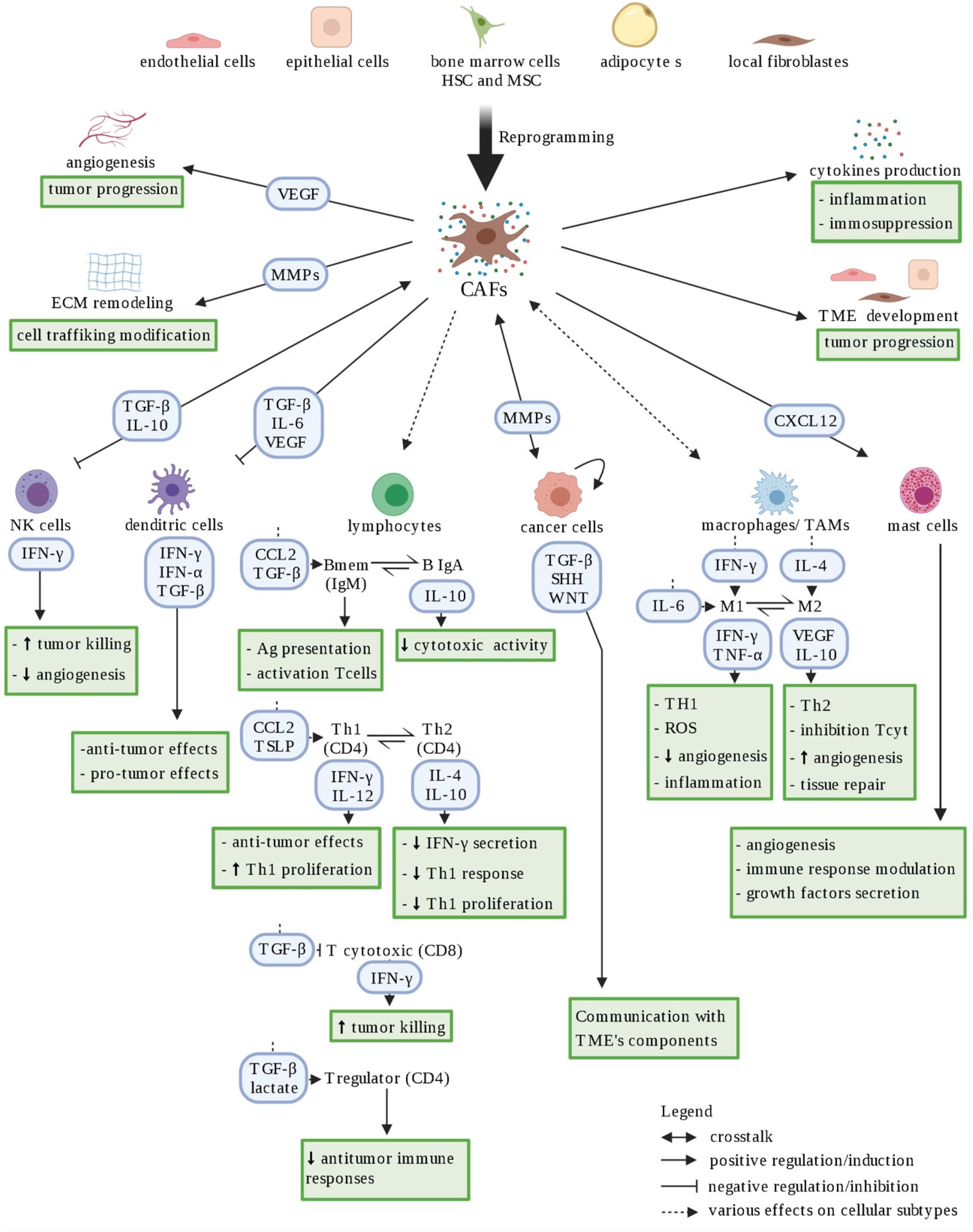
2.3.1. CAFs Support Cancer Cell Development
2.3.2. Cancer Cells: Immune System “Hijackers”
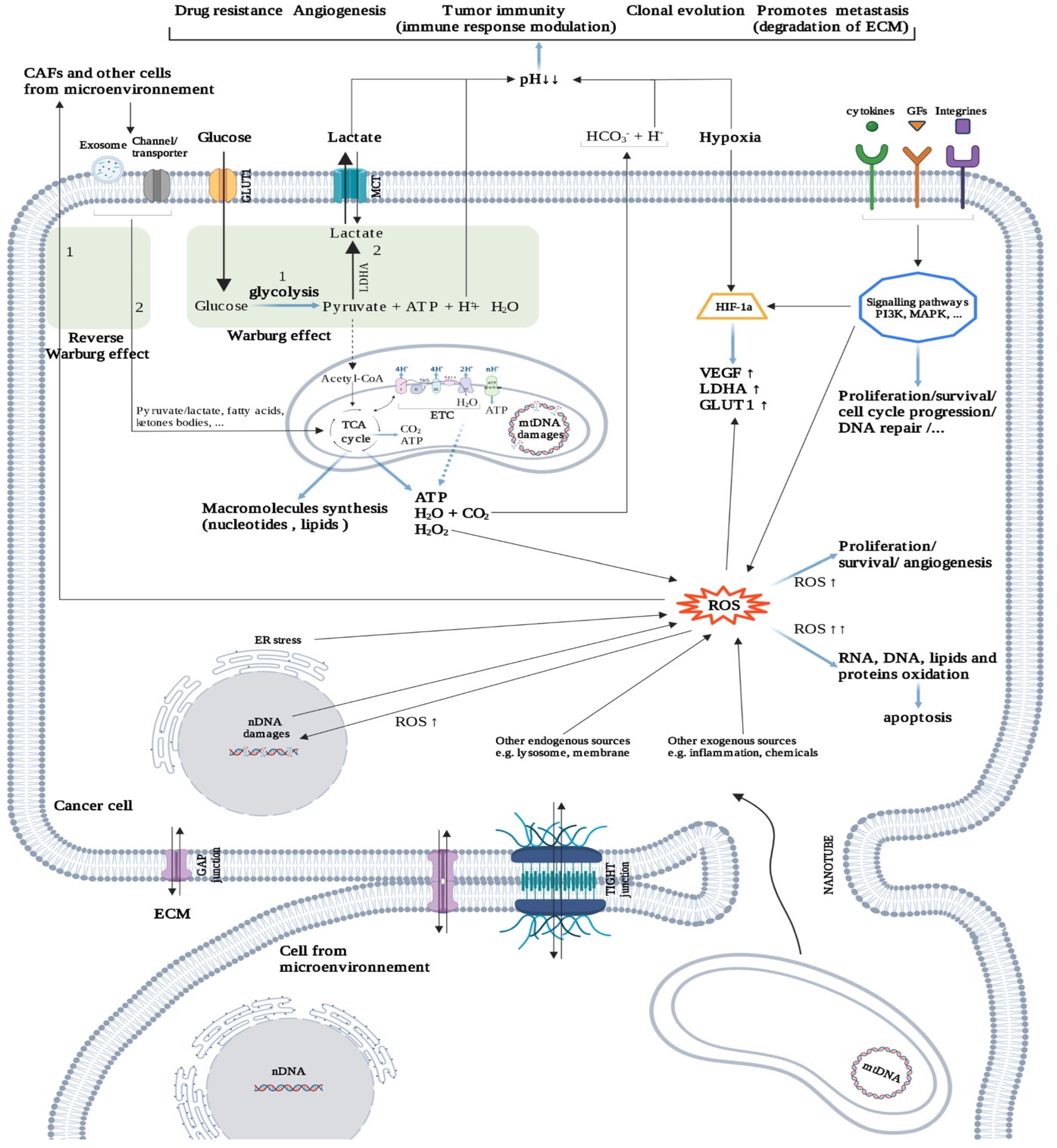
2.3.3. Cancer Cells: “Metabolic Parasites”
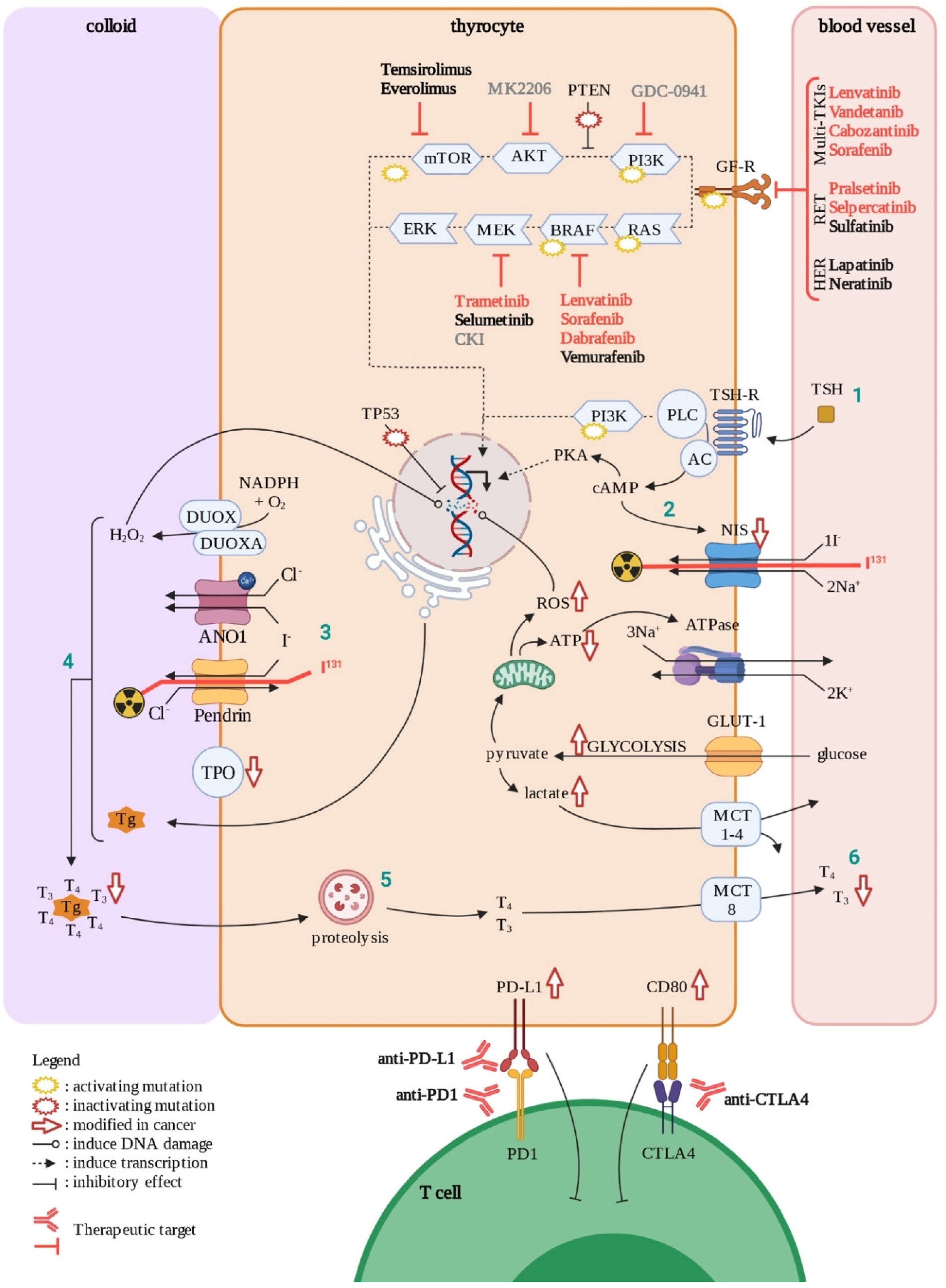
3. Thyroid Carcinoma as an Example: Various Patterns of Heterogeneity
3.1. Papillary Thyroid Carcinomas
3.2. Follicular Thyroid Carcinomas
3.3. Anaplastic Thyroid Carcinomas
3.4. Diagnosing Thyroid Cancer
3.5. Current and Future Therapies for Thyroid Cancer
4. Conceptual, Diagnostic, and Therapeutic Implications of the Dynamic Heterogeneity of Cancer
4.1. Conceptual Point of View
4.2. Diagnosis and Cancer Heterogeneity
4.3. Therapeutic Approaches vs. Cancer Heterogeneity
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.; Firlej, V.; Allory, Y.; Roméo, P.-H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef]
- Haffner, M.C.; Zwart, W.; Roudier, M.P.; True, L.D.; Nelson, W.G.; Epstein, J.I.; De Marzo, A.M.; Nelson, P.S.; Yegnasubramanian, S. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2021, 18, 79–92. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1616288 (accessed on 22 June 2021).
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef]
- O’Connor, J.P.B. Cancer heterogeneity and imaging. Semin. Cell Dev. Biol. 2017, 64, 48–57. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Cancer Evolution Constrained by the Immune Microenvironment. Cell 2017, 170, 825–827. [Google Scholar] [CrossRef]
- Suvà, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Baez-Ortega, A.; Gori, K.; Strakova, A.; Allen, J.L.; Allum, K.M.; Bansse-Issa, L.; Bhutia, T.N.; Bisson, J.L.; Briceño, C.; Domracheva, A.C.; et al. Somatic evolution and global expansion of an ancient transmissible cancer lineage. Science 2019, 365, eaau9923. [Google Scholar] [CrossRef] [PubMed]
- Stammnitz, M.R.; Coorens, T.H.H.; Gori, K.C.; Hayes, D.; Fu, B.; Wang, J.; Martin-Herranz, D.E.; Alexandrov, L.B.; Baez-Ortega, A.; Barthorpe, S.; et al. The Origins and Vulnerabilities of Two Transmissible Cancers in Tasmanian Devils. Cancer Cell 2018, 33, 607–619.e15. [Google Scholar] [CrossRef] [PubMed]
- Bullman, S.; Zou, T.; Meyerson, M. Mechanistic Insights into Transmissible Cancers of Mammals. Cancer Cell 2018, 33, 543–544. [Google Scholar] [CrossRef] [PubMed]
- Pellacani, D.; Tan, S.; Lefort, S.; Eaves, C.J. Transcriptional regulation of normal human mammary cell heterogeneity and its perturbation in breast cancer. EMBO J. 2019, 38, e100330. [Google Scholar] [CrossRef]
- Chmielik, E.; Rusinek, D.; Oczko-Wojciechowska, M.; Jarzab, M.; Krajewska, J.; Czarniecka, A.; Jarzab, B. Heterogeneity of Thyroid Cancer. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2018, 85, 117–129. [Google Scholar] [CrossRef]
- González-Silva, L.; Quevedo, L.; Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Available online: https://www.cell.com/trends/cancer/fulltext/S2405-8033(19)30258-4 (accessed on 22 June 2021).
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark. Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Cheng, S. Epigenetic Modifications: Novel Therapeutic Approach for Thyroid Cancer. Endocrinol. Metab. 2017, 32, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Bateman, N.W.; Conrads, T.P. Recent advances and opportunities in proteomic analyses of tumour heterogeneity. J. Pathol. 2018, 244, 628–637. [Google Scholar] [CrossRef]
- Frezza, C. Metabolism and cancer: The future is now. Br. J. Cancer 2020, 122, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.-S.; Zhang, T.-T.; Xue, D.-X.; Wu, W.-L.; Wang, Y.-L.; Wang, Y.; Ji, Q.-H.; Zhu, Y.-X.; Qu, N.; Shi, R.-L. Metabolic reprogramming and its clinical application in thyroid cancer. Oncol. Lett. 2019, 18, 1579–1584. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Yuan, Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb. Perspect. Med. 2016, 6, a026583. [Google Scholar] [CrossRef]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018, 18, 19–32. [Google Scholar] [CrossRef]
- Gottlieb, B.; Trifiro, M.; Batist, G. Why Tumor Genetic Heterogeneity May Require Rethinking Cancer Genesis and Treatment. Trends Cancer 2021, 7, 400–409. [Google Scholar] [CrossRef]
- Gawin, M.; Kurczyk, A.; Stobiecka, E.; Frątczak, K.; Polańska, J.; Pietrowska, M.; Widłak, P. Molecular Heterogeneity of Papillary Thyroid Cancer: Comparison of Primary Tumors and Synchronous Metastases in Regional Lymph Nodes by Mass Spectrometry Imaging. Endocr. Pathol. 2019, 30, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Jing, Y.; Cai, M.-C.; Ma, P.; Zhang, Y.; Xu, C.; Zhang, M.; Di, W.; Zhuang, G. Clonality, Heterogeneity, and Evolution of Synchronous Bilateral Ovarian Cancer. Cancer Res. 2017, 77, 6551–6561. [Google Scholar] [CrossRef]
- Ciccarelli, F.D. Mutations differ in normal and cancer cells of the oesophagus. Nature 2019, 565, 301–303. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.C.; Cunningham, J.J.; Bui, M.M.; Gillies, R.J.; Brown, J.S.; Gatenby, R.A. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection Forces. Cancer Res. 2016, 76, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.C.; Torres, M.; Real, F.X. Somatic mosaicism: On the road to cancer. Nat. Rev. Cancer 2016, 16, 43–55. [Google Scholar] [CrossRef]
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta 2017, 1867, 151–161. [Google Scholar] [CrossRef]
- Galofré, C.; Gönül Geyik, Ö.; Asensio, E.; Wangsa, D.; Hirsch, D.; Parra, C.; Saez, J.; Mollà, M.; Yüce, Z.; Castells, A.; et al. Tetraploidy-Associated Genetic Heterogeneity Confers Chemo-Radiotherapy Resistance to Colorectal Cancer Cells. Cancers 2020, 12, E1118. [Google Scholar] [CrossRef]
- Herrera, E.L.; Azzam, S.Z.; Berger, M.C.; Diaz-Martinez, L.A. Genomic heterogeneity meets cellular energetics: Crosstalk between the mitochondria and the cell cycle. J. Cancer Metastasis Treat. 2018, 4, 42. [Google Scholar] [CrossRef][Green Version]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef]
- Kumar, S.; Warrell, J.; Li, S.; McGillivray, P.D.; Meyerson, W.; Salichos, L.; Harmanci, A.; Martinez-Fundichely, A.; Chan, C.W.Y.; Nielsen, M.M.; et al. Passenger mutations in 2500 cancer genomes: Overall molecular functional impact and consequences. Cell 2020, 180, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, J.; Martincorena, I.; Koonin, E.V. Cancer-mutation network and the number and specificity of driver mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E6010–E6019. [Google Scholar] [CrossRef] [PubMed]
- Koch, L. Cancer genomics: The driving force of cancer evolution. Nat. Rev. Genet. 2017, 18, 703. [Google Scholar] [CrossRef]
- Zhao, Z.-M.; Zhao, B.; Bai, Y.; Iamarino, A.; Gaffney, S.G.; Schlessinger, J.; Lifton, R.P.; Rimm, D.L.; Townsend, J.P. Early and multiple origins of metastatic lineages within primary tumors. Proc. Natl. Acad. Sci. USA 2016, 113, 2140–2145. [Google Scholar] [CrossRef]
- Townsend, J.P. Evolution Research Could Revolutionize Cancer Therapy. Scientific American, 18 April.
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Siegel, M.B.; Kanchi, K.L.; Miller, C.A.; Ding, L.; Zhao, W.; He, X.; Parker, J.S.; Wendl, M.C.; Fulton, R.S.; et al. Tumor Evolution in Two Patients with Basal-like Breast Cancer: A Retrospective Genomics Study of Multiple Metastases. PLoS Med. 2016, 13, e1002174. [Google Scholar] [CrossRef]
- Ulintz, P.J.; Greenson, J.K.; Wu, R.; Fearson, E.R.; Hardiman, K.M. Lymph Node Metastases in Colon Cancer Are Polyclonal. Available online: https://clincancerres.aacrjournals.org/content/24/9/2214.full (accessed on 7 June 2021).
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef]
- Assenov, Y.; Brocks, D.; Gerhäuser, C. Intratumor heterogeneity in epigenetic patterns. Semin. Cancer Biol. 2018, 51, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Miyamoto, D.T.; Ting, D.T.; Toner, M.; Maheswaran, S.; Haber, D.A. Single-Cell Analysis of Circulating Tumor Cells as a Window into Tumor Heterogeneity. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 269–274. [Google Scholar] [CrossRef]
- Sarkar, S.; Sabhachandani, P.; Ravi, D.; Potdar, S.; Purvey, S.; Beheshti, A.; Evens, A.M.; Konry, T. Dynamic Analysis of Human Natural Killer Cell Response at Single-Cell Resolution in B-Cell Non-Hodgkin Lymphoma. Front. Immunol. 2017, 8, 1736. [Google Scholar] [CrossRef]
- Dominiak, A.; Chełstowska, B.; Olejarz, W.; Nowicka, G. Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers 2020, 12, 1232. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Houthuijzen, J.M.; Jonkers, J. Cancer-associated fibroblasts as key regulators of the breast cancer tumor microenvironment. Cancer Metastasis Rev. 2018, 37, 577–597. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef]
- Strickaert, A.; Corbet, C.; Spinette, S.-A.; Craciun, L.; Dom, G.; Andry, G.; Larsimont, D.; Wattiez, R.; Dumont, J.E.; Feron, O.; et al. Reprogramming of Energy Metabolism: Increased Expression and Roles of Pyruvate Carboxylase in Papillary Thyroid Cancer. Thyroid Off. J. Am. Thyroid Assoc. 2019, 29, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bouchard, G.; Yu, A.; Shafiq, M.; Jamali, M.; Shrager, J.B.; Ayers, K.; Bakr, S.; Gentles, A.J.; Diehn, M.; et al. GFPT2-Expressing Cancer-Associated Fibroblasts Mediate Metabolic Reprogramming in Human Lung Adenocarcinoma. Cancer Res. 2018, 78, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. Cancer-associated fibroblasts: Secretions, interactions, and therapy. J. Cell. Biochem. 2019, 120, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Coelho, F.; André, S.; Félix, A.; Serpa, J. Breast cancer metabolic cross-talk: Fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol. Cell. Endocrinol. 2018, 462, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Zhao, X.; Gupta, V.K.; Sharma, N.; Kesh, K.; Gnamlin, P.; Dudeja, V.; Vickers, S.M.; Banerjee, S.; Saluja, A. Inactivation of Cancer-Associated-Fibroblasts Disrupts Oncogenic Signaling in Pancreatic Cancer Cells and Promotes Its Regression. Cancer Res. 2018, 78, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and TAM functions-it takes two to tango. FEBS J. 2018, 285, 700–716. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Muenst, S.; Läubli, H.; Soysal, S.D.; Zippelius, A.; Tzankov, A.; Hoeller, S. The immune system and cancer evasion strategies: Therapeutic concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef] [PubMed]
- Ulisse, S.; Tuccilli, C.; Sorrenti, S.; Antonelli, A.; Fallahi, P.; D’Armiento, E.; Catania, A.; Tartaglia, F.; Amabile, M.I.; Giacomelli, L.; et al. PD-1 Ligand Expression in Epithelial Thyroid Cancers: Potential Clinical Implications. Int. J. Mol. Sci. 2019, 20, 1405. [Google Scholar] [CrossRef]
- Shrihari, T. Dual role of inflammatory mediators in cancer. Ecancermedicalscience 2017, 11, 721. [Google Scholar] [CrossRef]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Fallahi, P.; Galdiero, M.R.; Ruffilli, I.; Elia, G.; Ragusa, F.; Paparo, S.R.; Patrizio, A.; Mazzi, V.; Varricchi, G.; et al. Immune and Inflammatory Cells in Thyroid Cancer Microenvironment. Int. J. Mol. Sci. 2019, 20, 4413. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. CANTOS Trial Group Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet Lond. Engl. 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 13, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Loffredo, S.; Marone, G.; Modestino, L.; Fallahi, P.; Ferrari, S.M.; de Paulis, A.; Antonelli, A.; Galdiero, M.R. The Immune Landscape of Thyroid Cancer in the Context of Immune Checkpoint Inhibition. Int. J. Mol. Sci. 2019, 20, 3934. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef]
- Jung, J.G.; Le, A. Targeting Metabolic Cross Talk Between Cancer Cells and Cancer-Associated Fibroblasts. Adv. Exp. Med. Biol. 2021, 1311, 205–214. [Google Scholar] [CrossRef]
- Murray, P.J. Nonresolving macrophage-mediated inflammation in malignancy. FEBS J. 2018, 285, 641–653. [Google Scholar] [CrossRef]
- Schmidt, E.M.; Lamprecht, S.; Blaj, C.; Schaaf, C.; Krebs, S.; Blum, H.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Targeting tumor cell plasticity by combined inhibition of NOTCH and MAPK signaling in colon cancer. J. Exp. Med. 2018, 215, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mims, J.; Huang, X.; Singh, N.; Motea, E.; Planchon, S.M.; Beg, M.; Tsang, A.W.; Porosnicu, M.; Kemp, M.L.; et al. Modulators of Redox Metabolism in Head and Neck Cancer. Antioxid. Redox Signal. 2018, 29, 1660–1690. [Google Scholar] [CrossRef] [PubMed]
- Dovmark, T.H.; Hulikova, A.; Niederer, S.A.; Vaughan-Jones, R.D.; Swietach, P. Normoxic cells remotely regulate the acid-base balance of cells at the hypoxic core of connexin-coupled tumor growths. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Zilionis, R.; Da Silva Martins, J.; Bos, S.A.; Courties, G.; Rickelt, S.; Severe, N.; Baryawno, N.; Faget, J.; et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science 2017, 358, 442–446. [Google Scholar] [CrossRef]
- Ombrato, L.; Nolan, E.; Kurelac, I.; Mavousian, A.; Bridgeman, V.L.; Heinze, I.; Chakravarty, P.; Horswell, S.; Gonzalez-Gualda, E.; Matacchione, G.; et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 2019, 572, 603–608. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 76–86. [Google Scholar] [CrossRef]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef]
- Heiland, D.H.; Gaebelein, A.; Börries, M.; Wörner, J.; Pompe, N.; Franco, P.; Heynckes, S.; Bartholomae, M.; hAilín, D.Ó.; Carro, M.S.; et al. Microenvironment-Derived Regulation of HIF Signaling Drives Transcriptional Heterogeneity in Glioblastoma Multiforme. Mol. Cancer Res. MCR 2018, 16, 655–668. [Google Scholar] [CrossRef]
- Assi, M. The differential role of reactive oxygen species in early and late stages of cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R646–R653. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Neveu, M.-A.; De Preter, G.; Marchand, V.; Bol, A.; Brender, J.R.; Saito, K.; Kishimoto, S.; Porporato, P.E.; Sonveaux, P.; Grégoire, V.; et al. Multimodality Imaging Identifies Distinct Metabolic Profiles In Vitro and In Vivo. Neoplasia 2016, 18, 742–752. [Google Scholar] [CrossRef]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, Consequences, and Therapy of Tumors Acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Pinto, A.; Martherus, R.; Santiago de Jesus, J.P.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef]
- Roswall, P.; Bocci, M.; Bartoschek, M.; Li, H.; Kristiansen, G.; Jansson, S.; Lehn, S.; Sjölund, J.; Reid, S.; Larsson, C.; et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat. Med. 2018, 24, 463–473. [Google Scholar] [CrossRef] [PubMed]
- McQuerry, J.A.; Chang, J.T.; Bowtell, D.D.L.; Cohen, A.; Bild, A.H. Mechanisms and clinical implications of tumor heterogeneity and convergence on recurrent phenotypes. J. Mol. Med. 2017, 95, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Fugazzola, L.; Muzza, M.; Pogliaghi, G.; Vitale, M. Intratumoral Genetic Heterogeneity in Papillary Thyroid Cancer: Occurrence and Clinical Significance. Cancers 2020, 12, 383. [Google Scholar] [CrossRef]
- MacDonald, L.; Jenkins, J.; Purvis, G.; Lee, J.; Franco, A.T. The Thyroid Tumor Microenvironment: Potential Targets for Therapeutic Intervention and Prognostication. Horm. Cancer 2020, 11, 205–217. [Google Scholar] [CrossRef]
- Ameziane El Hassani, R.; Buffet, C.; Leboulleux, S.; Dupuy, C. Oxidative stress in thyroid carcinomas: Biological and clinical significance. Endocr. Relat. Cancer 2019, 26, R131–R143. [Google Scholar] [CrossRef]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on Fundamental Mechanisms of Thyroid Cancer. Front. Endocrinol. 2020, 11, 102. [Google Scholar] [CrossRef]
- Zaballos, M.A.; Santisteban, P. Key signaling pathways in thyroid cancer. J. Endocrinol. 2017, 235, R43–R61. [Google Scholar] [CrossRef]
- Tuccilli, C.; Baldini, E.; Sorrenti, S.; Catania, A.; Antonelli, A.; Fallahi, P.; Tartaglia, F.; Barollo, S.; Mian, C.; Palmieri, A.; et al. CTLA-4 and PD-1 Ligand Gene Expression in Epithelial Thyroid Cancers. Available online: https://www.hindawi.com/journals/ije/2018/1742951/abs/ (accessed on 12 July 2019).
- Masoodi, T.; Siraj, A.K.; Siraj, S.; Azam, S.; Qadri, Z.; Parvathareddy, S.K.; Al-Sobhi, S.S.; AlDawish, M.; Alkuraya, F.S.; Al-Kuraya, K.S. Evolution and Impact of Subclonal Mutations in Papillary Thyroid Cancer. Am. J. Hum. Genet. 2019, 105, 959–973. [Google Scholar] [CrossRef]
- Li, T.; Ma, Z.; Lu, C.; Zhou, Q.; Feng, Z.; Wu, X.; Luo, Y.; Li, D.; Cheng, X.; Liu, X. Chest wall lymph node metastasis from follicular thyroid carcinoma: A rare case report. Diagn. Pathol. 2019, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ma, Y.; Gong, Y.; Gong, R.; Li, Z.; Zhu, J. Multiple Simultaneous Rare Distant Metastases as the Initial Presentation of Papillary Thyroid Carcinoma: A Case Report. Front. Endocrinol. 2019, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Kakudo, K.; Liu, Z.; Bychkov, A.; Jung, C.K. Nuclear Features of Papillary Thyroid Carcinoma (BRAF-like Tumors), Noninvasive Follicular Thyroid Neoplasm with Papillary-Like Nuclear Features (RAS-like Tumors), and Follicular Adenoma/Follicular Thyroid Carcinoma (RAS-like Tumors); Springer: Singapore, 2019; pp. 173–179. ISBN 9789811318962. [Google Scholar]
- Tarabichi, M.; Antoniou, A.; Le Pennec, S.; Gacquer, D.; de Saint Aubain, N.; Craciun, L.; Cielen, T.; Laios, I.; Larsimont, D.; Andry, G.; et al. Distinctive Desmoplastic 3D Morphology Associated With BRAFV600E in Papillary Thyroid Cancers. J. Clin. Endocrinol. Metab. 2018, 103, 1102–1111. [Google Scholar] [CrossRef]
- Xie, Z.; Li, X.; He, Y.; Wu, S.; Wang, S.; Sun, J.; He, Y.; Lun, Y.; Zhang, J. Immune Cell Confrontation in the Papillary Thyroid Carcinoma Microenvironment. Front. Endocrinol. 2020, 11, 570604. [Google Scholar] [CrossRef] [PubMed]
- Jolly, L.A.; Novitskiy, S.; Owens, P.; Massoll, N.; Cheng, N.; Fang, W.; Moses, H.L.; Franco, A.T. Fibroblast-mediated collagen remodeling within the tumor microenvironment facilitates progression of thyroid cancers driven by BrafV600E and Pten loss. Cancer Res. 2016, 76, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Strickaert, A.; Saiselet, M.; Dom, G.; De Deken, X.; Dumont, J.E.; Feron, O.; Sonveaux, P.; Maenhaut, C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 2017, 36, 2637–2642. [Google Scholar] [CrossRef]
- Pasanen, I.; Lehtonen, S.; Sormunen, R.; Skarp, S.; Lehtilahti, E.; Pietilä, M.; Sequeiros, R.B.; Lehenkari, P.; Kuvaja, P. Breast cancer carcinoma-associated fibroblasts differ from breast fibroblasts in immunological and extracellular matrix regulating pathways. Exp. Cell Res. 2016, 344, 53–66. [Google Scholar] [CrossRef]
- Icard, P.; Lincet, H. A global view of the biochemical pathways involved in the regulation of the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1826, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Dom, G.; Frank, S.; Floor, S.; Kehagias, P.; Libert, F.; Hoang, C.; Andry, G.; Spinette, A.; Craciun, L.; de Saint Aubin, N.; et al. Thyroid follicular adenomas and carcinomas: Molecular profiling provides evidence for a continuous evolution. Oncotarget 2017, 9, 10343–10359. [Google Scholar] [CrossRef]
- Parameswaran, R.; Shulin Hu, J.; Min En, N.; Tan, W.; Yuan, N. Patterns of metastasis in follicular thyroid carcinoma and the difference between early and delayed presentation. Ann. R. Coll. Surg. Engl. 2017, 99, 151–154. [Google Scholar] [CrossRef]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef]
- Takano, T.; Amino, N. Fetal cell carcinogenesis: A new hypothesis for better understanding of thyroid carcinoma. Thyroid Off. J. Am. Thyroid Assoc. 2005, 15, 432–438. [Google Scholar] [CrossRef]
- Gianì, F.; Vella, V.; Tumino, D.; Malandrino, P.; Frasca, F. The Possible Role of Cancer Stem Cells in the Resistance to Kinase Inhibitors of Advanced Thyroid Cancer. Cancers 2020, 12, 2249. [Google Scholar] [CrossRef]
- Hébrant, A.; Dom, G.; Dewaele, M.; Andry, G.; Trésallet, C.; Leteurtre, E.; Dumont, J.E.; Maenhaut, C. mRNA Expression in Papillary and Anaplastic Thyroid Carcinoma: Molecular Anatomy of a Killing Switch. PLoS ONE 2012, 7, e37807. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Menicali, E.; Guzzetti, M.; Morelli, S.; Moretti, S.; Puxeddu, E. Immune Landscape of Thyroid Cancers: New Insights. Front. Endocrinol. 2021, 11, 7826. [Google Scholar] [CrossRef]
- Fozzatti, L.; Alamino, V.A.; Park, S.; Giusiano, L.; Volpini, X.; Zhao, L.; Stempin, C.C.; Donadio, A.C.; Cheng, S.; Pellizas, C.G. Interplay of fibroblasts with anaplastic tumor cells promotes follicular thyroid cancer progression. Sci. Rep. 2019, 9, 8028. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Luo, Y.; Zhou, M.; Huo, L.; Chen, H.; Zuo, Q.; Deng, W. Comparison of Diagnostic Accuracy of Thyroid Cancer With Ultrasound-Guided Fine-Needle Aspiration and Core-Needle Biopsy: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2020, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Nylén, C.; Mechera, R.; Maréchal-Ross, I.; Tsang, V.; Chou, A.; Gill, A.J.; Clifton-Bligh, R.J.; Robinson, B.G.; Sywak, M.S.; Sidhu, S.B.; et al. Molecular Markers Guiding Thyroid Cancer Management. Cancers 2020, 12, 2164. [Google Scholar] [CrossRef] [PubMed]
- Tarabichi, M.; Demetter, P.; Craciun, L.; Maenhaut, C.; Detours, V. Thyroid cancer under the scope of emerging technologies. Mol. Cell. Endocrinol. 2022, 541, 111491. [Google Scholar] [CrossRef] [PubMed]
- Naoum, G.E.; Morkos, M.; Kim, B.; Arafat, W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol. Cancer 2018, 17, 51. [Google Scholar] [CrossRef]
- Laha, D.; Nilubol, N.; Boufraqech, M. New Therapies for Advanced Thyroid Cancer. Front. Endocrinol. 2020, 11, 82. [Google Scholar] [CrossRef]
- San Román Gil, M.; Pozas, J.; Molina-Cerrillo, J.; Gómez, J.; Pian, H.; Pozas, M.; Carrato, A.; Grande, E.; Alonso-Gordoa, T. Current and Future Role of Tyrosine Kinases Inhibition in Thyroid Cancer: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 4951. [Google Scholar] [CrossRef]
- Sugishita, Y.; Kammori, M.; Yamada, O.; Poon, S.S.S.; Kobayashi, M.; Onoda, N.; Yamazaki, K.; Fukumori, T.; Yoshikawa, K.-I.; Onose, H.; et al. Amplification of the human epidermal growth factor receptor 2 gene in differentiated thyroid cancer correlates with telomere shortening. Int. J. Oncol. 2013, 42, 1589–1596. [Google Scholar] [CrossRef]
- Ensinger, C.; Prommegger, R.; Kendler, D.; Gabriel, M.; Spizzo, G.; Mikuz, G.; Kremser, R. Her2/neu expression in poorly-differentiated and anaplastic thyroid carcinomas. Anticancer Res. 2003, 23, 2349–2353. [Google Scholar]
- Wei, W.; Jiang, D.; Rosenkrans, Z.T.; Barnhart, T.E.; Engle, J.W.; Luo, Q.; Cai, W. HER2-targeted multimodal imaging of anaplastic thyroid cancer. Am. J. Cancer Res. 2019, 9, 2413–2427. [Google Scholar] [PubMed]
- Ruggeri, R.M.; Campennì, A.; Giuffrè, G.; Giovanella, L.; Siracusa, M.; Simone, A.; Branca, G.; Scarfì, R.; Trimarchi, F.; Ieni, A.; et al. HER2 Analysis in Sporadic Thyroid Cancer of Follicular Cell Origin. Int. J. Mol. Sci. 2016, 17, 2040. [Google Scholar] [CrossRef]
- Buffet, C.; Wassermann, J.; Hecht, F.; Leenhardt, L.; Dupuy, C.; Groussin, L.; Lussey-Lepoutre, C. Redifferentiation of radioiodine-refractory thyroid cancers. Endocr. Relat. Cancer 2020, 27, R113–R132. [Google Scholar] [CrossRef] [PubMed]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Bogdanoff, D.; Wang, Y.; Hartoularos, G.C.; Woo, J.M.; Mowery, C.T.; Nisonoff, H.M.; Lee, D.S.; Sun, Y.; Lee, J.; et al. XYZeq: Spatially resolved single-cell RNA sequencing reveals expression heterogeneity in the tumor microenvironment. Sci. Adv. 2021, 7, eabg4755. [Google Scholar] [CrossRef]
- Chervoneva, I.; Peck, A.R.; Yi, M.; Freydin, B.; Rui, H. Quantification of spatial tumor heterogeneity in immunohistochemistry staining images. Bioinformatics 2021, 37, 1452–1460. [Google Scholar] [CrossRef]
- Bonin, S.; Stanta, G. Pre-analytics and tumor heterogeneity. New Biotechnol. 2020, 55, 30–35. [Google Scholar] [CrossRef]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Saiselet, M.; Rodrigues-Vitória, J.; Tourneur, A.; Craciun, L.; Spinette, A.; Larsimont, D.; Andry, G.; Lundeberg, J.; Maenhaut, C.; Detours, V. Transcriptional output, cell-type densities, and normalization in spatial transcriptomics. J. Mol. Cell Biol. 2020, 12, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Demirci, U.; Chen, P. Emerging organoid models: Leaping forward in cancer research. J. Hematol. Oncol. 2019, 12, 142. [Google Scholar] [CrossRef]
- Porter, R.J.; Murray, G.I.; McLean, M.H. Current concepts in tumour-derived organoids. Br. J. Cancer 2020, 123, 1209–1218. [Google Scholar] [CrossRef]
- Yang, C.; Xia, B.-R.; Jin, W.-L.; Lou, G. Circulating tumor cells in precision oncology: Clinical applications in liquid biopsy and 3D organoid model. Cancer Cell Int. 2019, 19, 341. [Google Scholar] [CrossRef]
- Treglia, G.; Annunziata, S.; Muoio, B.; Salvatori, M.; Ceriani, L.; Giovanella, L. The Role of Fluorine-18-Fluorodeoxyglucose Positron Emission Tomography in Aggressive Histological Subtypes of Thyroid Cancer: An Overview. Int. J. Endocrinol. 2013, 2013, 856189. [Google Scholar] [CrossRef]
- Shen, K.; Liu, B.; Zhou, X.; Ji, Y.; Chen, L.; Wang, Q.; Xue, W. The Evolving Role of 18F-FDG PET/CT in Diagnosis and Prognosis Prediction in Progressive Prostate Cancer. Front. Oncol. 2021, 11, 2436. [Google Scholar] [CrossRef]
- Larg, M.I.; Barbus, E.; Gabora, K.; Pestean, C.; Cheptea, M.; Piciu, D. 18F-FDG PET/CT IN DIFFERENTIATED THYROID CARCINOMA. Acta Endocrinol. Buchar. 2019, 15, 203–208. [Google Scholar] [CrossRef]
- Kaushik, A.K.; DeBerardinis, R.J. Applications of metabolomics to study cancer metabolism. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 2–14. [Google Scholar] [CrossRef]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Heiden, M.G.V.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA. Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef] [PubMed]
- van Manen, L.; Dijkstra, J.; Boccara, C.; Benoit, E.; Vahrmeijer, A.L.; Gora, M.J.; Mieog, J.S.D. The clinical usefulness of optical coherence tomography during cancer interventions. J. Cancer Res. Clin. Oncol. 2018, 144, 1967–1990. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Aimono, E.; Tanishima, S.; Imai, M.; Nagatsuma, A.K.; Hayashi, H.; Yoshimura, Y.; Nakayama, K.; Kyo, S.; Nishihara, H. Intratumoral Genomic Heterogeneity May Hinder Precision Medicine Strategies in Patients with Serous Ovarian Carcinoma. Diagnostics 2020, 10, 200. [Google Scholar] [CrossRef]
- Raspollini, M.R.; Montagnani, I.; Montironi, R.; Castiglione, F.; Martignoni, G.; Cheng, L.; Lopez-Beltran, A. Intratumoural heterogeneity may hinder precision medicine strategies in patients with clear cell renal cell carcinoma. J. Clin. Pathol. 2018, 71, 467–471. [Google Scholar] [CrossRef]
- Hoffman, M.; Gillmor, A.H.; Kunz, D.J.; Johnston, M.J.; Nikolic, A.; Narta, K.; Zarrei, M.; King, J.; Ellestad, K.; Dang, N.H.; et al. Intratumoral Genetic and Functional Heterogeneity in Pediatric Glioblastoma. Cancer Res. 2019, 79, 2111–2123. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.W.; Amin, R.; Deasy, S.; Ha, N.-H.; Wakefield, L. Genetic insights into the morass of metastatic heterogeneity. Nat. Rev. Cancer 2018, 18, 211–223. [Google Scholar] [CrossRef]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef]
- Gambardella, V.; Fleitas, T.; Tarazona, N.; Cejalvo, J.M.; Gimeno-Valiente, F.; Martinez-Ciarpaglini, C.; Huerta, M.; Roselló, S.; Castillo, J.; Roda, D.; et al. Towards precision oncology for HER2 blockade in gastroesophageal adenocarcinoma. Ann. Oncol. 2019, 30, 1254–1264. [Google Scholar] [CrossRef]
- Sheng, S.; Margarida Bernardo, M.; Dzinic, S.H.; Chen, K.; Heath, E.I.; Sakr, W.A. Tackling tumor heterogeneity and phenotypic plasticity in cancer precision medicine: Our experience and a literature review. Cancer Metastasis Rev. 2018, 37, 655–663. [Google Scholar] [CrossRef]
- Baliu-Piqué, M.; Pandiella, A.; Ocana, A. Breast Cancer Heterogeneity and Response to Novel Therapeutics. Cancers 2020, 12, 3271. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Capaldo, B.D.; Vaillant, F.; Pal, B.; van Ineveld, R.; Dawson, C.A.; Chen, Y.; Nolan, E.; Fu, N.Y.; Jackling, F.C.; et al. Intraclonal Plasticity in Mammary Tumors Revealed through Large-Scale Single-Cell Resolution 3D Imaging. Cancer Cell 2019, 35, 618–632.e6. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Tourneau, C.L.; Borcoman, E.; Kamal, M. Molecular profiling in precision medicine oncology. Nat. Med. 2019, 25, 711. [Google Scholar] [CrossRef]
- Barok, M.; Puhka, M.; Vereb, G.; Szollosi, J.; Isola, J.; Joensuu, H. Cancer-derived exosomes from HER2-positive cancer cells carry trastuzumab-emtansine into cancer cells leading to growth inhibition and caspase activation. BMC Cancer 2018, 18, 504. [Google Scholar] [CrossRef]
- Wu, P.; Gao, W.; Su, M.; Nice, E.C.; Zhang, W.; Lin, J.; Xie, N. Adaptive Mechanisms of Tumor Therapy Resistance Driven by Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 641469. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, H.; Yan, Y.; Liu, Y.; Su, C.; Chen, H.; Yan, Y.; Adhikari, R.; Wu, Q.; Zhang, J. The Role of Exosomal microRNA in Cancer Drug Resistance. Front. Oncol. 2020, 10, 472. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.-C.; Prawira, A.; de Braud, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A.; et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer 2019, 19, 196. [Google Scholar] [CrossRef]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Melanoma Patients. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Cheng, R.; Pan, Y.; Zhang, H.; He, Y.; Su, C.; Ren, S.; Zhou, C. Heterogeneity of neoantigen landscape between primary lesions and their matched metastases in lung cancer. Transl. Lung Cancer Res. 2020, 9, 246–256. [Google Scholar] [CrossRef]
- Bernards, R. Finding effective cancer therapies through loss of function genetic screens. Curr. Opin. Genet. Dev. 2014, 24, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jin, H.; Gao, D.; Lieftink, C.; Evers, B.; Jin, G.; Xue, Z.; Wang, L.; Beijersbergen, R.L.; Qin, W.; et al. Phospho-ERK is a biomarker of response to a synthetic lethal drug combination of sorafenib and MEK inhibition in liver cancer. J. Hepatol. 2018, 69, 1057–1065. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquemin, V.; Antoine, M.; Dom, G.; Detours, V.; Maenhaut, C.; Dumont, J.E. Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications. Cancers 2022, 14, 280. https://doi.org/10.3390/cancers14020280
Jacquemin V, Antoine M, Dom G, Detours V, Maenhaut C, Dumont JE. Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications. Cancers. 2022; 14(2):280. https://doi.org/10.3390/cancers14020280
Chicago/Turabian StyleJacquemin, Valerie, Mathieu Antoine, Geneviève Dom, Vincent Detours, Carine Maenhaut, and Jacques E. Dumont. 2022. "Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications" Cancers 14, no. 2: 280. https://doi.org/10.3390/cancers14020280
APA StyleJacquemin, V., Antoine, M., Dom, G., Detours, V., Maenhaut, C., & Dumont, J. E. (2022). Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications. Cancers, 14(2), 280. https://doi.org/10.3390/cancers14020280