
Impact of Cancer Stem Cells and Cancer Stem Cell-Driven Drug Resiliency in Lung Tumor: Options in Sight



Abstract
Simple Summary
Abstract
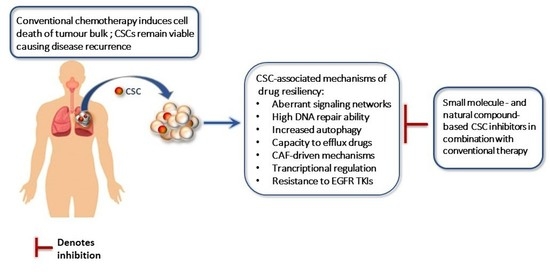
1. Introduction
2. Identification of CSCs in Lung Cancer
3. CSC-Associated Cellular and Molecular Mechanisms of Chemoresistance
3.1. Modulation through Signaling Networks
3.2. Transcriptional Control
3.3. Cancer-Associated Fibroblast (CAF)-Driven Mechanisms
3.4. High DNA Repair Ability
3.5. Resistance to EGFR TKIs
3.6. Elevated Autophagy
3.7. Drug Efflux through ABC Transporters
4. CSC Inhibitors in Lung Cancer
5. Immunological Attributes of CSCs
6. Immunotherapy against CSCs
7. Conclusions and Future Insights
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.M.; Mortensen, J.; Hansen, H.; Vilmann, P.; Larsen, S.S.; Loft, A.; Bertelsen, A.K.; Ravn, J.; Clementsen, P.F.; Høegholm, A.; et al. Multimodality approach to mediastinal staging in non-small cell lung cancer. Faults and benefits of PET-CT: A randomised trial. Thorax 2011, 66, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Rivera, S.; Loriot, Y.; Vozenin, M.-C.; Deutsch, E. Lung Cancer Stem Cell: New Insights on Experimental Models and Preclinical Data. J. Oncol. 2010, 2011, 549181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zakaria, N.; Satar, N.A.; Abu Halim, N.H.; Ngalim, S.H.; Yusoff, N.M.; Lin, J.; Yahaya, B.H. Targeting Lung Cancer Stem Cells: Research and Clinical Impacts. Front. Oncol. 2017, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef] [PubMed]
- MacDonagh, L.; Gray, S.; Breen, E.; Cuffe, S.; Finn, S.; O’Byrne, K.J.; Barr, M.P. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016, 372, 147–156. [Google Scholar] [CrossRef]
- Alamgeer, M.; Peacock, C.D.; Matsui, W.; Ganju, V.; Watkins, D.N. Cancer stem cells in lung cancer: Evidence and controversies. Respirology 2013, 18, 757–764. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef]
- Salcido, C.D.; LaRochelle, A.; Taylor, B.J.; Dunbar, C.E.; Varticovski, L. Molecular characterisation of side population cells with cancer stem cell-like characteristics in small-cell lung cancer. Br. J. Cancer 2010, 102, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The Biology of Cancer Stem Cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.J.; Steel, J.; Morris, J.C. Sphere Culture of Murine Lung Cancer Cell Lines Are Enriched with Cancer Initiating Cells. PLoS ONE 2012, 7, e49752. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1107. [Google Scholar] [PubMed]
- Vinogradov, S.; Wei, X. Cancer stem cells and drug resistance: The potential of nanomedicine. Nanomedicine 2012, 7, 597–615. [Google Scholar] [CrossRef]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Bruttel, V.S.; Wischhusen, J.; Wischhusen, J. Cancer Stem Cell Immunology: Key to Understanding Tumorigenesis and Tumor Immune Escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Roninson, I.B. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell 1991, 66, 85–94. [Google Scholar] [CrossRef]
- Levina, V.; Marrangoni, A.M.; Demarco, R.; Gorelik, E.; Lokshin, A.E. Drug-Selected Human Lung Cancer Stem Cells: Cytokine Network, Tumorigenic and Metastatic Properties. PLoS ONE 2008, 3, e3077. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef] [PubMed]
- Sarvi, S.; MacKinnon, A.C.; Avlonitis, N.; Bradley, M.; Rintoul, R.; Rassl, D.M.; Wang, W.; Forbes, S.; Gregory, C.; Sethi, T. CD133+ Cancer Stem-like Cells in Small Cell Lung Cancer Are Highly Tumorigenic and Chemoresistant but Sensitive to a Novel Neuropeptide Antagonist. Cancer Res. 2014, 74, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.K.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2014, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Y.; Tang, J.-N.; Xie, H.-X.; Du, Y.-A.; Huang, L.; Yu, P.-F.; Cheng, X.-D. 5-Fluorouracil Chemotherapy of Gastric Cancer Generates Residual Cells with Properties of Cancer Stem Cells. Int. J. Biol. Sci. 2015, 11, 284–294. [Google Scholar] [CrossRef]
- Hu, X.; Ghisolfi, L.; Keates, A.C.; Zhang, J.; Xiang, S.; Lee, D.K.; Li, C.J. Induction of cancer cell stemness by chemotherapy. Cell Cycle 2012, 11, 2691–2698. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-Induced Reprogramming of Breast Cancer Cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef]
- Bousquet, G.; El Bouchtaoui, M.; Sophie, T.; Leboeuf, C.; de Bazelaire, C.; Ratajczak, P.; Giacchetti, S.; de Roquancourt, A.; Bertheau, P.; Verneuil, L.; et al. Targeting autophagic cancer stem-cells to reverse chemoresistance in human triple negative breast cancer. Oncotarget 2017, 8, 35205–35221. [Google Scholar] [CrossRef]
- Bleau, A.-M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.; Holland, E.C. PTEN/PI3K/Akt Pathway Regulates the Side Population Phenotype and ABCG2 Activity in Glioma Tumor Stem-like Cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Mayea, Y.G.; Mir, C.; Masson, F.; Paciucci, R.; Lleonart, M. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. Resistance to Cell Death and Its Modulation in Cancer Stem Cells. Crit. Rev. Oncog. 2016, 21, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, L.N.; Chow, E.K.-H. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Frank, M.H. Antitumor Immunity and Cancer Stem Cells. Ann. N. Y. Acad. Sci. 2009, 1176, 154–169. [Google Scholar] [CrossRef]
- Müller, L.; Tunger, A.; Plesca, I.; Wehner, R.; Temme, A.; Westphal, D.; Meier, F.; Bachmann, M.; Schmitz, M. Bidirectional Crosstalk Between Cancer Stem Cells and Immune Cell Subsets. Front. Immunol. 2020, 11, 140. [Google Scholar] [CrossRef]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning Cancer Fate: Tumor Microenvironment’s Role in Cancer Stem Cell Quiescence and Reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Carney, D.N.; Bunn, P.A.; Gazdar, A.F.; Pagan, J.A.; Minna, J.D. Selective growth in serum-free hormone-supplemented medium of tumor cells obtained by biopsy from patients with small cell carcinoma of the lung. Proc. Natl. Acad. Sci. USA 1981, 78, 3185–3189. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Hsu, H.-S.; Chen, Y.-W.; Tsai, T.-H.; How, C.-K.; Wang, C.-Y.; Hung, S.-C.; Chang, Y.-L.; Tsai, M.-L.; Lee, Y.-Y.; et al. Oct-4 Expression Maintained Cancer Stem-Like Properties in Lung Cancer-Derived CD133-Positive Cells. PLoS ONE 2008, 3, e2637. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2007, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Setrerrahmane, S.; Xu, H. Enrichment and characterization of cancer stem cells from a human non-small cell lung cancer cell line. Oncol. Rep. 2015, 34, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Wang, J.; Chen, D.; Chen, Y.J. CD133 is a temporary marker of cancer stem cells in small cell lung cancer, but not in non-small cell lung cancer. Oncol. Rep. 2011, 25, 701–708. [Google Scholar] [CrossRef][Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Shi, Y. Aldehyde dehydrogenase-1 is a specific marker for stem cells in human lung adenocarcinoma. Med Oncol. 2011, 29, 633–639. [Google Scholar] [CrossRef]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde Dehydrogenase 1 Is a Tumor Stem Cell-Associated Marker in Lung Cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef]
- Ucar, D.; Cogle, C.R.; Zucali, J.R.; Ostmark, B.; Scott, E.W.; Zori, R.; Gray, B.A.; Moreb, J.S. Aldehyde dehydrogenase activity as a functional marker for lung cancer. Chem. Interact. 2009, 178, 48–55. [Google Scholar] [CrossRef]
- Li, Z.; Xiang, Y.; Xiang, L.; Xiao, Y.; Li, F.; Hao, P. ALDH Maintains the Stemness of Lung Adenoma Stem Cells by Suppressing the Notch/CDK2/CCNE Pathway. PLoS ONE 2014, 9, e92669. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Spinola, M.; Dodge, M.; Raso, M.G.; Behrens, C.; Gao, B.; Schuster, K.; Shao, C.; Larsen, J.; Sullivan, L.A.; et al. Aldehyde Dehydrogenase Activity Selects for Lung Adenocarcinoma Stem Cells Dependent on Notch Signaling. Cancer Res. 2010, 70, 9937–9948. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Z.; Wong, S.K.M.; Tin, V.P.-C.; Ho, K.-Y.; Wang, J.; Sham, M.-H.; Wong, M.P. Lung Cancer Tumorigenicity and Drug Resistance Are Maintained through ALDHhiCD44hi Tumor Initiating Cells. Oncotarget 2013, 4, 1698–1711. [Google Scholar] [CrossRef]
- Nishino, M.; Ozaki, M.; Hegab, A.E.; Hamamoto, J.; Kagawa, S.; Arai, D.; Yasuda, H.; Naoki, K.; Soejima, K.; Saya, H.; et al. Variant CD44 expression is enriching for a cell population with cancer stem cell-like characteristics in human lung adenocarcinoma. J. Cancer 2017, 8, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-P.; Tsai, M.-F.; Chang, T.-H.; Tang, W.-C.; Chen, S.-Y.; Lai, H.-H.; Lin, T.-Y.; Yang, J.C.-H.; Yang, P.-C.; Shih, J.-Y.; et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013, 328, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Satar, N.A.; Fakiruddin, K.S.; Lim, M.N.; Mok, P.L.; Zakaria, N.; Fakharuzi, N.A.; Rahman, A.Z.A.; Zakaria, Z.; Yahaya, B.; Baharuddin, P. Novel triple-positive markers identified in human non-small cell lung cancer cell line with chemotherapy-resistant and putative cancer stem cell characteristics. Oncol. Rep. 2018, 40, 669–681. [Google Scholar] [CrossRef]
- Gutova, M.; Najbauer, J.; Gevorgyan, A.; Metz, M.Z.; Weng, Y.; Shih, C.-C.; Aboody, K.S. Identification of uPAR-positive Chemoresistant Cells in Small Cell Lung Cancer. PLoS ONE 2007, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Goodell, M.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side Population in Human Lung Cancer Cell Lines and Tumors Is Enriched with Stem-like Cancer Cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, Y.; Qi, Y.; Liu, D.; Wu, K.; Wen, F.; Zhao, S. Side population cells separated from A549 lung cancer cell line possess cancer stem cell-like properties and inhibition of autophagy potentiates the cytotoxic effect of cisplatin. Oncol. Rep. 2015, 34, 929–935. [Google Scholar] [CrossRef]
- Xie, T.; Mo, L.; Li, L.; Mao, N.; Li, D.; Liu, D.; Zuo, C.; Huang, D.; Pan, Q.; Yang, L.; et al. Identification of side population cells in human lung adenocarcinoma A549 cell line and elucidation of the underlying roles in lung cancer. Oncol. Lett. 2018, 15, 4900–4906. [Google Scholar] [CrossRef]
- Sung, J.-M.; Cho, H.-J.; Yi, H.; Lee, C.-H.; Kim, H.-S.; Kim, D.-K.; El-Aty, A.M.A.; Kim, J.-S.; Landowski, C.; Hediger, M.A.; et al. Characterization of a stem cell population in lung cancer A549 cells. Biochem. Biophys. Res. Commun. 2008, 371, 163–167. [Google Scholar] [CrossRef]
- Yang, B.; Ma, Y.-F.; Liu, Y. Elevated Expression of Nrf-2 and ABCG2 Involved in Multi-drug Resistance of Lung Cancer SP Cells. Drug Res. 2014, 65, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Luo, H.; Zhou, X.; Zhu, B.; Wang, Y.; Bian, X. Identification of CD90 as a marker for lung cancer stem cells in A549 and H446 cell lines. Oncol. Rep. 2013, 30, 2733–2740. [Google Scholar] [CrossRef]
- Swart, G.W. Activated leukocyte cell adhesion molecule (CD166/ALCAM): Developmental and mechanistic aspects of cell clustering and cell migration. Eur. J. Cell Biol. 2002, 81, 313–321. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine Decarboxylase Activity Drives Non-Small Cell Lung Cancer Tumor-Initiating Cells and Tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef]
- Tachezy, M.; Zander, H.; Wolters-Eisfeld, G.; Müller, J.; Wicklein, D.; Gebauer, F.; Izbicki, J.R.; Bockhorn, M. Activated Leukocyte Cell Adhesion Molecule (CD166): An “Inert” Cancer Stem Cell Marker for Non-Small Cell Lung Cancer? Stem Cells 2014, 32, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, M.; Si, J.; Xiong, Y.; Lu, F.; Zhang, J.; Zhang, L.; Zhang, P.; Yang, Y. Blockade of Notch3 inhibits the stem-like property and is associated with ALDH1A1 and CD44 via autophagy in non-small lung cancer. Int. J. Oncol. 2016, 48, 2349–2358. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, W.; Guo, H.; Zhang, Y.; He, Y.; Lee, S.H.; Song, X.; Li, X.; Guo, Y.; Zhao, Y.; et al. NOTCH1 Signaling Regulates Self-Renewal and Platinum Chemoresistance of Cancer Stem–like Cells in Human Non–Small Cell Lung Cancer. Cancer Res. 2017, 77, 3082–3091. [Google Scholar] [CrossRef]
- Zhai, Y.; Wei, R.; Sha, S.; Wang, H.; Jiang, X.; Liu, G. Effect of NELL1 on lung cancer stem-like cell differentiation. Oncol. Rep. 2019, 41, 1817–1826. [Google Scholar] [CrossRef]
- Chen, C.-J.; Yang, C.-J.; Yang, S.-F.; Huang, M.-S.; Liu, Y.-P. The MyoD family inhibitor domain-containing protein enhances the chemoresistance of cancer stem cells in the epithelial state by increasing β-catenin activity. Oncogene 2020, 39, 2377–2390. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Huang, J.; Dong, Q. Wnt signaling regulation of stem-like properties in human lung adenocarcinoma cell lines. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Wu, S.; Tang, W.; Qian, H.; Zhou, H.; Guo, T. Reduced SLC27A2 induces cisplatin resistance in lung cancer stem cells by negatively regulating Bmi1-ABCG2 signaling. Mol. Carcinog. 2015, 55, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, G.; Zhang, H.; Zhang, F.; Zhou, B.; Ning, F.; Wang, H.-S.; Cai, S.-H.; Du, J. Acquisition of epithelial–mesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/β-catenin/Snail signaling pathway. Eur. J. Pharmacol. 2014, 723, 156–166. [Google Scholar] [CrossRef]
- Hsu, H.-S.; Lin, J.-H.; Huang, W.-C.; Hsu, T.-W.; Su, K.; Chiou, S.-H.; Tsai, Y.-T. Chemoresistance of lung cancer stemlike cells depends on activation of Hsp27. Cancer 2010, 117, 1516–1528. [Google Scholar] [CrossRef]
- Panneerselvam, J.; Mohandoss, P.; Patel, R.; Gillan, H.; Li, M.; Kumar, K.; Nguyen, D.; Weygant, N.; Qu, D.; Pitts, K.; et al. DCLK1 Regulates Tumor Stemness and Cisplatin Resistance in Non-small Cell Lung Cancer via ABCD-Member-4. Mol. Ther. -Oncolytics 2020, 18, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Lu, K.; Nayak, M.K.; Bhuniya, A.; Ghosh, T.; Kundu, S.; Ghosh, S.; Baral, R.; Dasgupta, P.S.; Basu, S. Activation of D2 Dopamine Receptors in CD133+ve Cancer Stem Cells in Non-small Cell Lung Carcinoma Inhibits Proliferation, Clonogenic Ability, and Invasiveness of These Cells. J. Biol. Chem. 2017, 292, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Voronkova, M.A.; Rojanasakul, L.W.; Kiratipaiboon, C.; Rojanasakul, Y. The SOX9-Aldehyde Dehydrogenase Axis Determines Resistance to Chemotherapy in Non-Small-Cell Lung Cancer. Mol. Cell. Biol. 2020, 40, e00307-19. [Google Scholar] [CrossRef]
- Cao, S.; Wang, Z.; Gao, X.; He, W.; Cai, Y.; Chen, H.; Xu, R. FOXC1 induces cancer stem cell-like properties through upregulation of beta-catenin in NSCLC. J. Exp. Clin. Cancer Res. 2018, 37, 220. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Pandey, A.; Sharma, S.; Chatterjee, I.; Mehrotra, R.; Sehgal, A.; Sharma, J.K. Genetic polymorphism of glutathione S-transferase P1 (GSTP1) in Delhi population and comparison with other global populations. Meta Gene 2014, 2, 134–142. [Google Scholar] [CrossRef]
- Li, J.; Ye, T.; Liu, Y.; Kong, L.; Sun, Z.; Liu, D.; Wang, J.; Xing, H.R. Transcriptional Activation of Gstp1 by MEK/ERK Signaling Confers Chemo-Resistance to Cisplatin in Lung Cancer Stem Cells. Front. Oncol. 2019, 9, 476. [Google Scholar] [CrossRef]
- Domen, A.; Quatannens, D.; Zanivan, S.; Deben, C.; Van Audenaerde, J.; Smits, E.; Wouters, A.; Lardon, F.; Roeyen, G.; Verhoeven, Y.; et al. Cancer-Associated Fibroblasts as a Common Orchestrator of Therapy Resistance in Lung and Pancreatic Cancer. Cancers 2021, 13, 987. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Chen, W.-J.; Ho, C.-C.; Chang, Y.-L.; Chen, H.-Y.; Lin, C.-A.; Ling, T.-Y.; Yu, S.-L.; Yuan, S.-S.; Chen, Y.-J.L.; Lin, C.-Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Lou, H.; Dean, M. Targeted therapy for cancer stem cells: The patched pathway and ABC transporters. Oncogene 2007, 26, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012, 19, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.K.; Wang, Z.; Fong, C.-C.; Liu, D.; Yip, T.-C.; Au, S.-K.; Zhu, G.; Yang, M. Chemoresistant lung cancer stem cells display high DNA repair capability to remove cisplatin-induced DNA damage. Br. J. Pharmacol. 2017, 174, 302–313. [Google Scholar] [CrossRef]
- Kobayashi, I.; Takahashi, F.; Nurwidya, F.; Nara, T.; Hashimoto, M.; Murakami, A.; Yagishita, S.; Tajima, K.; Hidayat, M.; Shimada, N.; et al. Oct4 plays a crucial role in the maintenance of gefitinib-resistant lung cancer stem cells. Biochem. Biophys. Res. Commun. 2016, 473, 125–132. [Google Scholar] [CrossRef]
- Silva, V.R.; Neves, S.P.; Santos, L.D.S.; Dias, R.B.; Bezerra, D.P. Challenges and Therapeutic Opportunities of Autophagy in Cancer Therapy. Cancers 2020, 12, 3461. [Google Scholar] [CrossRef]
- Smith, A.; MacLeod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Liu, G.; Tian, G. Autophagy inhibition of cancer stem cells promotes the efficacy of cisplatin against non-small cell lung carcinoma. Ther. Adv. Respir. Dis. 2019, 13, 1753466619866097. [Google Scholar] [CrossRef] [PubMed]
- Goebel, J.; Chmielewski, J.; Hrycyna, C.A. The roles of the human ATP-binding cassette transporters P-glycoprotein and ABCG2 in multidrug resistance in cancer and at endogenous sites: Future opportunities for structure-based drug design of inhibitors. Cancer Drug Resist. 2021, 4, 784–804. [Google Scholar] [CrossRef]
- O’Flaherty, J.D.; Barr, M.; Fennell, D.; Richard, D.; Reynolds, J.; O’Leary, J.; O’Byrne, K. The Cancer Stem-Cell Hypothesis: Its Emerging Role in Lung Cancer Biology and Its Relevance for Future Therapy. J. Thorac. Oncol. 2012, 7, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Bora-Singhal, N.; Mohankumar, D.; Saha, B.; Colin, C.M.; Lee, J.Y.; Martin, M.W.; Zheng, X.; Coppola, D.; Chellappan, S. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci. Rep. 2020, 10, 4722. [Google Scholar] [CrossRef] [PubMed]
- Larzabal, L.; El-Nikhely, N.; Redrado, M.; Seeger, W.; Savai, R.; Calvo, A. Differential Effects of Drugs Targeting Cancer Stem Cell (CSC) and Non-CSC Populations on Lung Primary Tumors and Metastasis. PLoS ONE 2013, 8, e79798. [Google Scholar] [CrossRef]
- Wang, Y. Effects of Salinomycin on Cancer Stem Cell in Human Lung Adenocarcinoma A549 Cells. Med. Chem. 2011, 7, 106–111. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Naujokat, C.; Steinhart, R. Salinomycin as a Drug for Targeting Human Cancer Stem Cells. J. Biomed. Biotechnol. 2012, 2012, 950658. [Google Scholar] [CrossRef] [PubMed]
- Khan, P.; Bhattacharya, A.; Sengupta, D.; Banerjee, S.; Adhikary, A.; Das, T. Aspirin enhances cisplatin sensitivity of resistant non-small cell lung carcinoma stem-like cells by targeting mTOR-Akt axis to repress migration. Sci. Rep. 2019, 9, 16913. [Google Scholar] [CrossRef]
- Zhang, X.; Du, R.; Luo, N.; Xiang, R.; Shen, W. Aspirin mediates histone methylation that inhibits inflammation-related stemness gene expression to diminish cancer stemness via COX-independent manner. Stem Cell Res. Ther. 2020, 11, 370. [Google Scholar] [CrossRef]
- Udoh, K.; Parte, S.; Carter, K.; Mack, A.; Kakar, S.S. Targeting of Lung Cancer Stem Cell Self-Renewal Pathway by a Small Molecule Verrucarin J. Stem Cell Rev. Rep. 2019, 15, 601–611. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gray, S.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. BBI608 inhibits cancer stemness and reverses cisplatin resistance in NSCLC. Cancer Lett. 2018, 428, 117–126. [Google Scholar] [CrossRef]
- Becerra, C.; Spira, A.I.; Conkling, P.R.; Richey, S.L.; Hanna, W.T.; Cote, G.M.; Heist, R.S.; Langleben, A.; Laurie, S.A.; Edenfield, W.J.; et al. A phase Ib/II study of cancer stemness inhibitor napabucasin (BB608) combined with weekly paclitaxel in advanced non-small cell lung cancer. J. Clin. Oncol. 2016, 34, 9093. [Google Scholar] [CrossRef]
- Yakisich, J.S.; Azad, N.; Kaushik, V.; O’Doherty, G.A.; Iyer, A.K.V. Nigericin decreases the viability of multidrug-resistant cancer cells and lung tumorspheres and potentiates the effects of cardiac glycosides. Tumor Biol. 2017, 39, 1010428317694310. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.R.; Raffa, S.; de Vitis, C.; Roscilli, G.; Malpicci, D.; Coluccia, P.; Di Napoli, A.; Ricci, A.; Giovagnoli, M.R.; Aurisicchio, L.; et al. Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells. Cell Death Dis. 2013, 4, e947. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Noto, A.; De Vitis, C.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of Stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Cancer Lett. 2017, 406, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Alama, A.; Gangemi, R.; Ferrini, S.; Barisione, G.; Orengo, A.M.; Truini, M.; Bello, M.G.D.; Grossi, F. CD133-Positive Cells from Non-Small Cell Lung Cancer Show Distinct Sensitivity to Cisplatin and Afatinib. Arch. Immunol. Ther. Exp. 2015, 63, 207–214. [Google Scholar] [CrossRef]
- Tian, F.; Mysliwietz, J.; Ellwart, J.; Gamarra, F.; Huber, R.M.; Bergner, A. Effects of the Hedgehog pathway inhibitor GDC-0449 on lung cancer cell lines are mediated by side populations. Clin. Exp. Med. 2011, 12, 25–30. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, Y.; Cai, H.; Lu, W.; Liu, H.; Wang, Z. Specific inhibition of Notch1 signaling suppresses properties of lung cancer stem cells. J. Cancer Res. Ther. 2019, 15, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Di Martile, M.; Desideri, M.; De Luca, T.; Gabellini, C.; Buglioni, S.; Eramo, A.; Sette, G.; Milella, M.; Rotili, D.; Mai, A.; et al. Histone acetyltransferase inhibitor CPTH6 preferentially targets lung cancer stem-like cells. Oncotarget 2016, 7, 11332–11348. [Google Scholar] [CrossRef]
- Al Fayi, M.; Alamri, A.; Rajagopalan, P. IOX-101 Reverses Drug Resistance Through Suppression of Akt/mTOR/NF-κB Signaling in Cancer Stem Cell-Like, Sphere-Forming NSCLC Cell. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2020, 28, 177–189. [Google Scholar] [CrossRef]
- Si, J.; Ma, Y.; Bi, J.W.; Xiong, Y.; Lv, C.; Li, S.; Wu, N.; Yang, Y. Shisa3 brakes resistance to EGFR-TKIs in lung adenocarcinoma by suppressing cancer stem cell properties. J. Exp. Clin. Cancer Res. 2019, 38, 481. [Google Scholar] [CrossRef] [PubMed]
- Kolev, V.N.; Wright, Q.; Vidal, C.M.; Ring, J.E.; Shapiro, I.M.; Ricono, J.; Weaver, D.T.; Padval, M.V.; Pachter, J.A.; Xu, Q. PI3K/mTOR Dual Inhibitor VS-5584 Preferentially Targets Cancer Stem Cells. Cancer Res. 2014, 75, 446–455. [Google Scholar] [CrossRef]
- Schwarz, Y.; Bondar, E.; Bar Shai, A.; Kohen, F.; Starr, A. Molecular mechanisms of inhibition of lung cancer stem cells invasion by a novel soy isoflavone VF166. Eur. Respir. J. 2016, 48, PA2838. [Google Scholar] [CrossRef]
- Fu, Z.; Cao, X.; Liu, L.; Cui, Y.; Li, X.; Quan, M.; Ren, K.; Chen, A.; Xu, C.; Qiu, Y.; et al. Genistein inhibits lung cancer cell stem-like characteristics by modulating MnSOD and FoxM1 expression. Oncol. Lett. 2020, 20, 2506–2515. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.; Vasaiya, A.; Vora, H.; Jain, N.; Rawal, R. Curcumin Targets Circulating Cancer Stem Cells by Inhibiting Self-Renewal Efficacy in Non-Small Cell Lung Carcinoma. Anti-Cancer Agents Med. Chem. 2017, 17, 859–864. [Google Scholar] [CrossRef]
- Wu, L.; Guo, L.; Liang, Y.; Liu, X.; Jiang, L.; Wang, L. Curcumin suppresses stem-like traits of lung cancer cells via inhibiting the JAK2/STAT3 signaling pathway. Oncol. Rep. 2015, 34, 3311–3317. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Yang, X.; Chen, Y.; Jiang, Y.; Wang, S.-J.; Li, Y.; Wang, X.-Q.; Meng, Y.; Zhu, M.-M.; Ma, X.; et al. Curcumin Suppresses Lung Cancer Stem Cells via Inhibiting Wnt/β-catenin and Sonic Hedgehog Pathways. Phytother. Res. 2017, 31, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Baharuddin, P.; Satar, N.; Fakiruddin, K.S.; Zakaria, N.; Lim, M.N.; Yusoff, N.M.; Zakaria, Z.; Yahaya, B.H. Curcumin improves the efficacy of cisplatin by targeting cancer stem-like cells through p21 and cyclin D1-mediated tumour cell inhibition in non-small cell lung cancer cell lines. Oncol. Rep. 2015, 35, 13–25. [Google Scholar] [CrossRef]
- Bhummaphan, N.; Chanvorachote, P. Gigantol Suppresses Cancer Stem Cell-Like Phenotypes in Lung Cancer Cells. Evid.-Based Complement. Altern. Med. 2015, 2015, 836564. [Google Scholar] [CrossRef] [PubMed]
- Bhummaphan, N.; Pongrakhananon, V.; Sritularak, B.; Chanvorachote, P. Cancer Stem Cell–Suppressing Activity of Chrysotoxine, a Bibenzyl from Dendrobium pulchellum. J. Pharmacol. Exp. Ther. 2017, 364, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cao, X.; Liu, Z.; Guo, H.; Ren, K.; Quan, M.; Zhou, Y.; Xiang, H.; Cao, J. Casticin suppresses self-renewal and invasion of lung cancer stem-like cells from A549 cells through down-regulation of pAkt. Acta Biochim. Biophys. Sin. 2013, 46, 15–21. [Google Scholar] [CrossRef]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin M Attenuates Cancer Stem Cell-like Phenotypes in H460 Lung Cancer Cells. Anticancer Res. 2017, 37, 615–622. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Segura-Carretero, A.; Joven, J.; Martín-Castillo, B.; Barrajón-Catalán, E.; Micol, V.; Barrera, J.B.; Menendez, J.M. Stem cell-like ALDHbrightcellular states in EGFR-mutant non-small cell lung cancer: A novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibinin. Cell Cycle 2013, 12, 3390–3404. [Google Scholar] [CrossRef] [PubMed]
- Srinual, S.; Chanvorachote, P.; Pongrakhananon, V. Suppression of cancer stem-like phenotypes in NCI-H460 lung cancer cells by vanillin through an Akt-dependent pathway. Int. J. Oncol. 2017, 50, 1341–1351. [Google Scholar] [CrossRef]
- Kwon, T.; Chandimali, N.; Huynh, D.L.; Zhang, J.J.; Kim, N.; Bak, Y.; Yoon, D.-Y.; Yu, D.-Y.; Lee, J.C.; Gera, M.; et al. BRM270 inhibits cancer stem cell maintenance via microRNA regulation in chemoresistant A549 lung adenocarcinoma cells. Cell Death Dis. 2018, 9, 244. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Wang, X.; Du, Q.; Gong, H.; Yang, Y.; Wang, T.; Wu, N.; Liu, X.; Li, W.; Zhao, C.; et al. Chetomin, a Hsp90/HIF1α pathway inhibitor, effectively targets lung cancer stem cells and non-stem cells. Cancer Biol. Ther. 2020, 21, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Bhatia, A.; Kumar, Y. Cancer stem cells and tumor immunoediting: Putting two and two together. Expert Rev. Clin. Immunol. 2016, 12, 605–607. [Google Scholar] [CrossRef][Green Version]
- Vahidian, F.; Duijf, P.; Safarzadeh, E.; Derakhshani, A.; Baghbanzadeh, A.; Baradaran, B. Interactions between cancer stem cells, immune system and some environmental components: Friends or foes? Immunol. Lett. 2019, 208, 19–29. [Google Scholar] [CrossRef]
- Grau, J.J.; Mesía, R.; De La Iglesia-Vicente, M.; Williams, E.S.; Taberna, M.; Caballero, M.; Larque, A.-B.; De La Oliva, J.; Cordón-Cardo, C.; Domingo-Domenech, J. Enrichment of Cells with Cancer Stem Cell-Like Markers in Relapses of Chemoresistant Patients with Locally Advanced Head and Neck Squamous Cell Carcinoma. Oncology 2016, 90, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Chikamatsu, K.; Takahashi, G.; Sakakura, K.; Ferrone, S.; Masuyama, K. Immunoregulatory properties of CD44+ cancer stem-like cells in squamous cell carcinoma of the head and neck. Head Neck 2011, 33, 208–215. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Expression and function of immune ligand-receptor pairs in NK cells and cancer stem cells: Therapeutic implications. Cell. Oncol. 2018, 41, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24– stem cell–like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ma, S.; Cao, K.; Zhou, S.; Zhao, A.; Li, M.; Qian, F.; Zhu, C. Therapeutic approaches targeting cancer stem cells. J. Cancer Res. Ther. 2018, 14, 1469–1475. [Google Scholar] [CrossRef]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.; Turaga, S.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461.e4. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Quesnel, B. Tumor dormancy and immunoescape. APMIS 2008, 116, 685–694. [Google Scholar] [CrossRef]
- Maccalli, C.; Rasul, K.I.; Elawad, M.; Ferrone, S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin. Cancer Biol. 2018, 53, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; De Stanchina, E.; Massagué, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Sistigu, A.; Manic, G.; Rudqvist, N.-P.; Trajanoski, Z.; Galluzzi, L. Mutational and Antigenic Landscape in Tumor Progression and Cancer Immunotherapy. Trends Cell Biol. 2019, 29, 396–416. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Rasool, S.; Maccalli, C. The Cross Talk between Cancer Stem Cells/Cancer Initiating Cells and Tumor Microenvironment: The Missing Piece of the Puzzle for the Efficient Targeting of these Cells with Immunotherapy. Cancer Microenviron. 2019, 12, 133–148. [Google Scholar] [CrossRef]
- Jiang, W.; Chan, C.K.; Weissman, I.L.; Kim, B.Y.; Hahn, S.M. Immune Priming of the Tumor Microenvironment by Radiation. Trends Cancer 2016, 2, 638–645. [Google Scholar] [CrossRef]
- Frey, B.; Rückert, M.; Deloch, L.; Rühle, P.F.; Derer, A.; Fietkau, R.; Gaipl, U.S. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol. Rev. 2017, 280, 231–248. [Google Scholar] [CrossRef]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Wong, A.L.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.; Yang, L.; Li, H.; Li, R.; Yu, J.; Yang, L.; Wei, F.; Yan, C.; Sun, Q.; et al. Anti-CD47 Antibody As a Targeted Therapeutic Agent for Human Lung Cancer and Cancer Stem Cells. Front. Immunol. 2017, 8, 404. [Google Scholar] [CrossRef]
- Han, D.; Yu, T.; Dong, N.; Wang, B.; Sun, F.; Jiang, D. Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 289. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, G.; Yuan, X.; Xu, M.; Wang, H.; Ji, J.; Konda, B.; Black, K.L.; Yu, J.S. Antigen-Specific T-Cell Response from Dendritic Cell Vaccination Using Cancer Stem-Like Cell-Associated Antigens. Stem Cells 2009, 27, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, K.; Wang, Y.; Han, W. Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: A potential and curable approach for cancer treatment. Protein Cell 2017, 9, 516–526. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Inhibitor | Mode of Action/Experimental Setting | Reference |
|---|---|---|
| HDAC11 (Histone deacetylase 11) | Reduced self-renewal property of NSCLC-derived CSCs; decreased SOX2 expression/in vitro | [94] |
| Salinomycin | Specifically targeted ALDH+ CSCs/in vitro | [95] |
| Disrupted ALDH+ cells in A549-derived tumorspheres by decreasing Oct4, NANOG, and SOX2 expression levels in vitro | [96] | |
| Eliminated CSCs in metastatic LLC mouse model/in vivo | [97,98] | |
| Aspirin (non-steroidal anti-inflammatory drug, NSAID) | Sensitized cisplatin-resistant NSCLC stem cells by targeting mTOR–AKT axis to repress cell migration/in vitro | [99] |
| Reduced ALDH+ and SP cells; chemoresistance and sphere formation in lung cancer cells/in vitro; inhibited tumor growth, metastasis, and prolonged survival via a reduction in KDM6A/B expression mediating histone methylation that suppressed gene expression in a COX-independent manner/in vivo | [100] | |
| Quercetin (Hsp 27 inhibitor) | Blunted activation of p38MAPK, MAPKAPK2, and Hsp27 after chemotoxic treatments; decreased survival of drug-resistant lung CSCs in combination with traditional chemotherapy/in vivo | [73] |
| Verrucarin J | Inhibited cell proliferation of CSCs; downregulates ALDH1, LGRs, Oct4, and CD133 via inhibition of the Wnt1/β-catenin and Notch1 pathways/in vitro | [101] |
| BBI608 (napabucasin) | Reduced ALDH+ CSC subpopulation by decreasing the mRNA levels of CSC-associated genes; had higher cytotoxic effects when combined with cisplatin; showed synergistic actions with paclitaxel/in vivo | [102] |
| Suppressed the STAT pathway/in vitro; Phase 1b dose-escalation study in advanced solid tumors with napabusin plus weekly paclitaxel showed good toleration; Phase II study with napabusin and weekly paclitaxel in pretreated advanced NSCLC patients resulted in tumor regression, durable disease control, and prolonged progression overall survival | [103] | |
| Nigericin | Inhibited cell viability of lung CSCs and resistance to anti-cancer agents; downregulated key proteins of the canonical Wnt signaling pathway/in vitro | [104] |
| MF-438 (SCDI inhibitor) | Restrained growth of cells with stem-like phenotype; reduced ALDH1 expression; impaired in vivo tumorigenicity/in vitro | [105] |
| In combination with cisplatin, downregulated CSC markers; inhibited sphere formation and induced apoptosis/in vitro | [106] | |
| Afatinib (EGFR inhibitor) | Ensued higher effectivity than cisplatin in enriched lung CSC subpopulations harboring EGFR mutations and in NSCLC primary cells expressing CD133/EpCAM/in vitro | [107] |
| AZD7762 (Chk1 inhibitor) | In combination with chemotherapy, significantly restrained NSCLC survival through modulation of premature cell cycle progression/in vitro; and reduced NSCLC CSCs in mouse xenografts/in vivo | [86] |
| GDC-0449 (Hedgehog inhibitor) | Reduced cell growth of HCC and H1339 lung cancer cells via suppression of SP cells/in vitro | [108] |
| DAPT (Notch1 inhibitor) | Inhibited cell growth of A549-derived CD44+/CD24− subpopulation expressing high Notch1/in vitro | [109] |
| CPTH6 (histone acetyltransferase inhibitor) | Suppressed cell growth of lung cancer stem-like cells via induction of apoptosis/in vitro; inhibited tumor growth and reduced CSCs and tubulin acetylation in tumor xenografts/in vivo | [110] |
| IOX-101 (arylidene derivative) | Inhibited cell proliferation of A549 CSCs by increasing the sub-G0 cell cycle phase and rate of apoptosis; reduced MDR-1 and LRP expression; deactivated Akt and sub-G0 cell cycle/in vitro | [111] |
| Shisa3 (regulator of WNT and FGF signaling) | Controlled the growth of TKI-resistant PC9/ER xenografts and CSCs via interaction with FGFR1/3 to regulate the AKT/mTOR pathway/in vitro and in vivo | [112] |
| VS-5584 (dual PI3-mTOR inhibitor) | More potent than cisplatin and paclitaxel in eliminating CSCs in human cancer xenograft models; eliminated CSCs and delayed tumor regrowth in SCLC xenograft model after chemotherapy/in vivo | [113] |
| Inhibitor | Mode of Action/Experimental Condition | Reference |
|---|---|---|
| VF166 (isoflavone derivative of soy daidzein) | Inhibited cell proliferation, migration, and invasion of lung CSCs; regulated genes promoting cell invasion-related pathways, including Wnt-β-catenin, Hedgehog, STAT3, and SPARC/in vitro | [114] |
| Genistein (4′ 5, 7-trihydroxyisoflavone) | Inhibited cell viability and sphere-forming capacity and decreased protein expression of CD133, CD44, Bmi1, and Nanog in lung CSCs via regulation of MnSOD and FoxM1 expression levels/in vitro | [115] |
| Curcumin (diferuloylmethane) derived from Curcuma longa | Reduced self-renewal and sphere-forming abilities of lung CSCs via inhibition of DNA repair mechanisms/in vitro | [116] |
| Suppressed colony and sphere-formation of lung CSCs through blockage of the JAK2/STAT pathway/in vitro | [117] | |
| Reduced CD133+ cells and other CSC markers; restrained cell proliferation and tumorsphere formation by inhibition of the Wnt/ß-catenin and Sonic Hedgehog pathways/in vitro | [118] | |
| Promoted sensitivity of CD166+/EpCAM+ lung CSC subpopulation to cisplatin through the p21 and cyclin D1-driven tumor cell inhibition/in vitro | [119] | |
| Gigantol (extract from Dendrobium draconis) | Reduced sphere-forming ability and expression of CD133 and ALDH1A1; suppressed Oct4 and Nanog levels via inhibition of protein kinase B (Akt) activation/in vitro | [120] |
| Chrysotoxine (extract from Dendrobium pulchenium) | Restrained CSC phenotypes in H460 and H23 lung cancer cells via downregulation of Src/protein kinase B (Akt) signaling, that, in turn, depleted Sox-2 mediated-CSC phenotype/in vitro | [121] |
| Casticin (derivative of Fructus viticis simplicifoliae) | Suppressed self-renewal and cell proliferation of A549-derived lung CSCs; lowered protein levels of CD133, CD44, and ALDH1; decreased MMP-9 activity/in vitro | [122] |
| Renieramycin M (derivative of sponge Xestospongia species) | Reduced colony and sphere formation abilities of H460 CSCs; lowered expression of CD133, CD44, and ALDH1A1 in CSC-enriched H460 cells/in vitro | [123] |
| Silibinin (extract of Silybum marianum) | Decreased the percentage of stem cell-like ALDHbright cells and self-renewal capacity of erlotinib-refractory NSCLC cells/in vitro | [124] |
| Vanillin (principal component of Vanilla planifolia seeds) | Restrained spheroid and colony formation; controlled CD133, ALDH1A1, Oct4, and Nanog at low levels in H460 lung cancer cells via induction of Akt-proteasomal degradation and reduction of downstream CSC transcription factors/in vitro | [125] |
| BRM270 (extract from seven herbal plants) | Regulated A549 CSCs’ self-renewal property and their ability to initiate tumor through regulation of the miRNA-128; decreased cell proliferation and mediated apoptosis in drug-refractory A549 through regulation of VEGF/PI3K/AKT signaling via miR-128/in vitro and in vivo | [126] |
| Chetomin (extract of Chaetonium globosum) | Decreased sphere-forming capacity and stem cell-like phenotypes of NSCLC CSCs by blocking the heat shock protein 90/hypoxia-inducible factor-alpha (Hsp90/HIF1α) signaling activity/in vitro | [127] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes-Dericks, L.; Galetta, D. Impact of Cancer Stem Cells and Cancer Stem Cell-Driven Drug Resiliency in Lung Tumor: Options in Sight. Cancers 2022, 14, 267. https://doi.org/10.3390/cancers14020267
Cortes-Dericks L, Galetta D. Impact of Cancer Stem Cells and Cancer Stem Cell-Driven Drug Resiliency in Lung Tumor: Options in Sight. Cancers. 2022; 14(2):267. https://doi.org/10.3390/cancers14020267
Chicago/Turabian StyleCortes-Dericks, Lourdes, and Domenico Galetta. 2022. "Impact of Cancer Stem Cells and Cancer Stem Cell-Driven Drug Resiliency in Lung Tumor: Options in Sight" Cancers 14, no. 2: 267. https://doi.org/10.3390/cancers14020267
APA StyleCortes-Dericks, L., & Galetta, D. (2022). Impact of Cancer Stem Cells and Cancer Stem Cell-Driven Drug Resiliency in Lung Tumor: Options in Sight. Cancers, 14(2), 267. https://doi.org/10.3390/cancers14020267