
Transcriptome Analysis of Human Endogenous Retroviruses at Locus-Specific Resolution in Non-Small Cell Lung Cancer
 , ,
, ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Pre-Processing of the RNA-Seq Data
2.2. Differential HERV Expression Analysis
2.3. Differential Gene Expression Analysis
2.4. Correlation Analyses
2.5. Pathway Analysis
2.6. Survival Analysis
3. Results
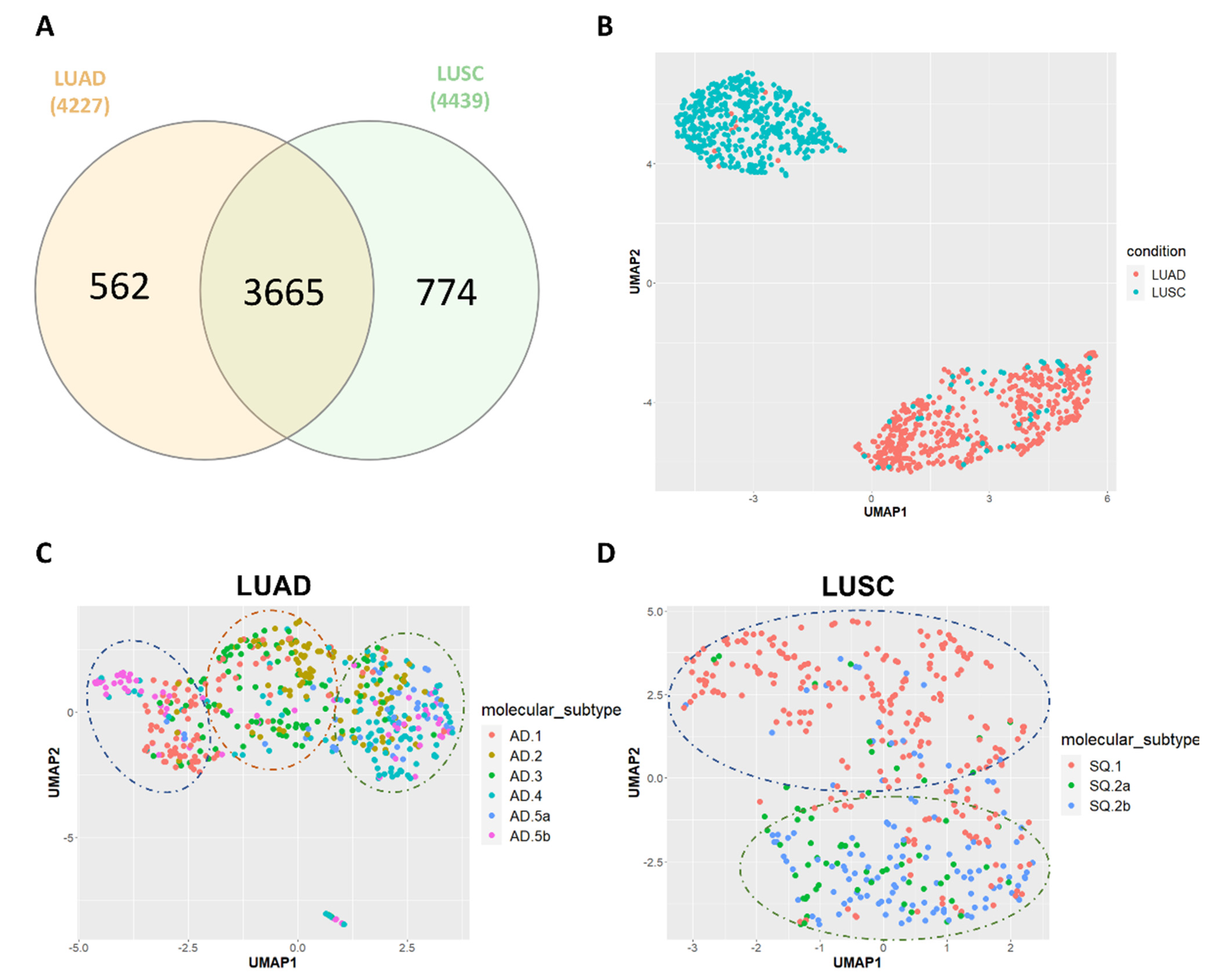
3.1. HERV Profiling in TCGA-LUAD and TCGA-LUSC
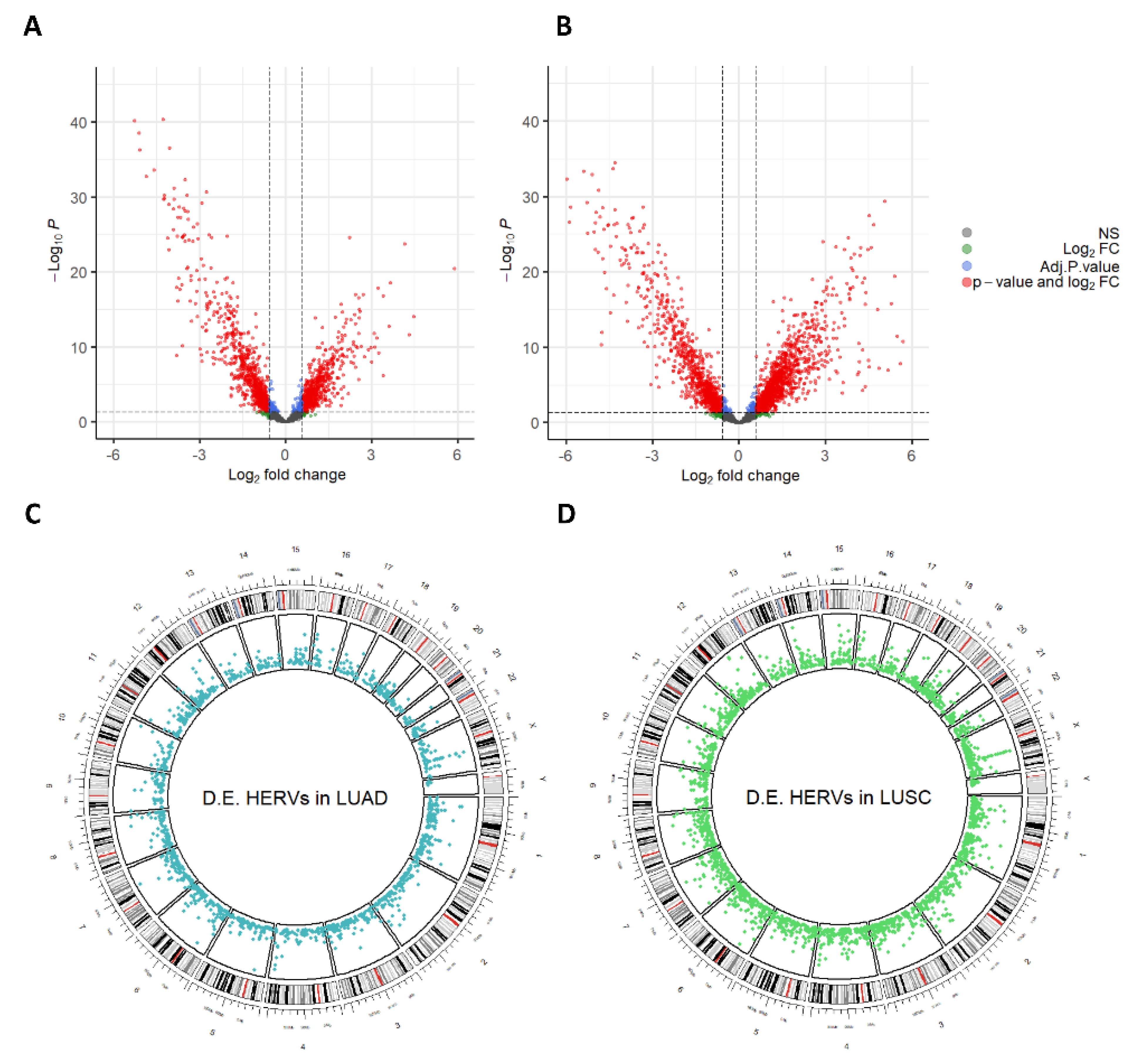
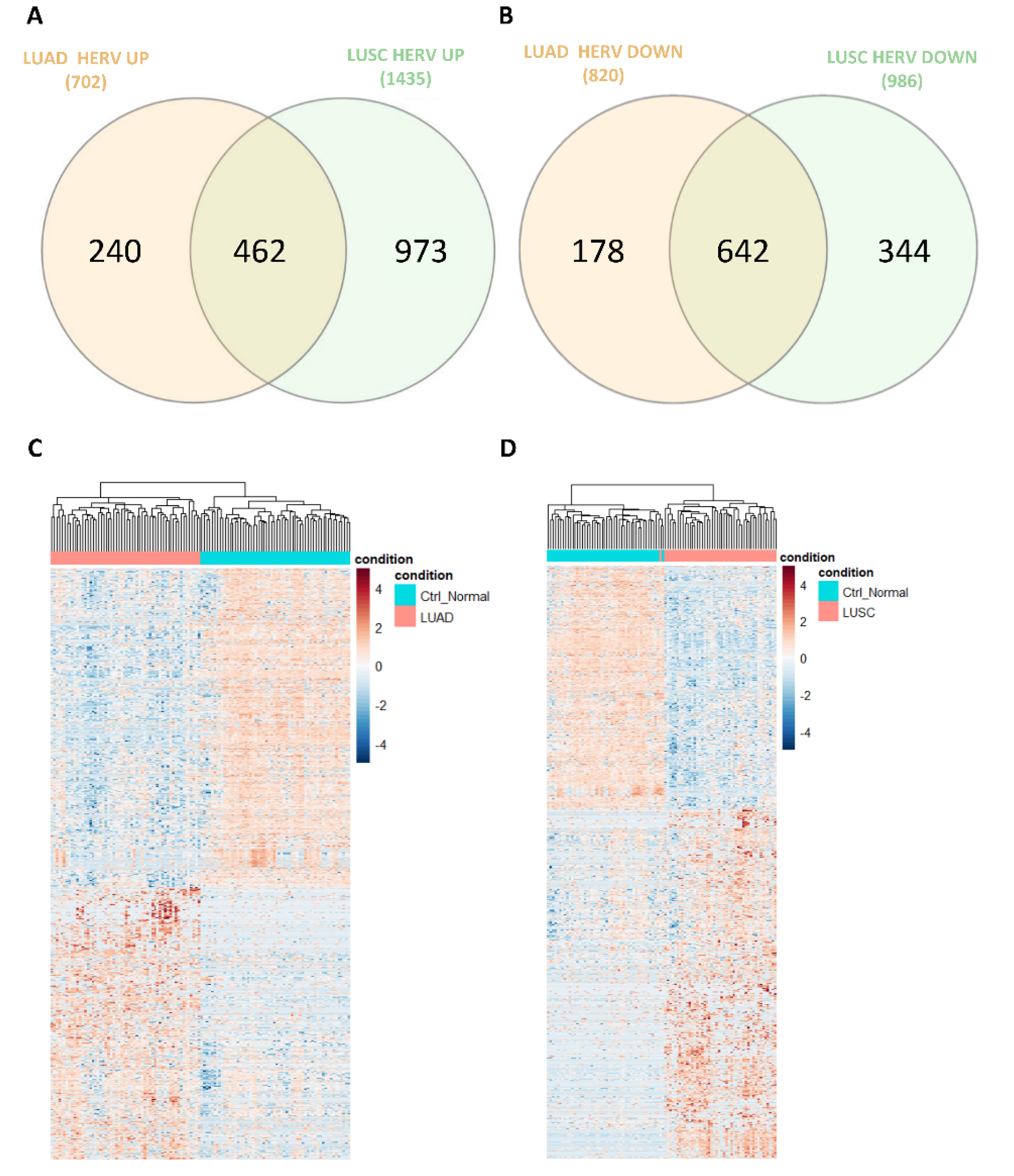
3.2. Dysregulation of HERVs in TCGA-LUAD and TCGA-LUSC
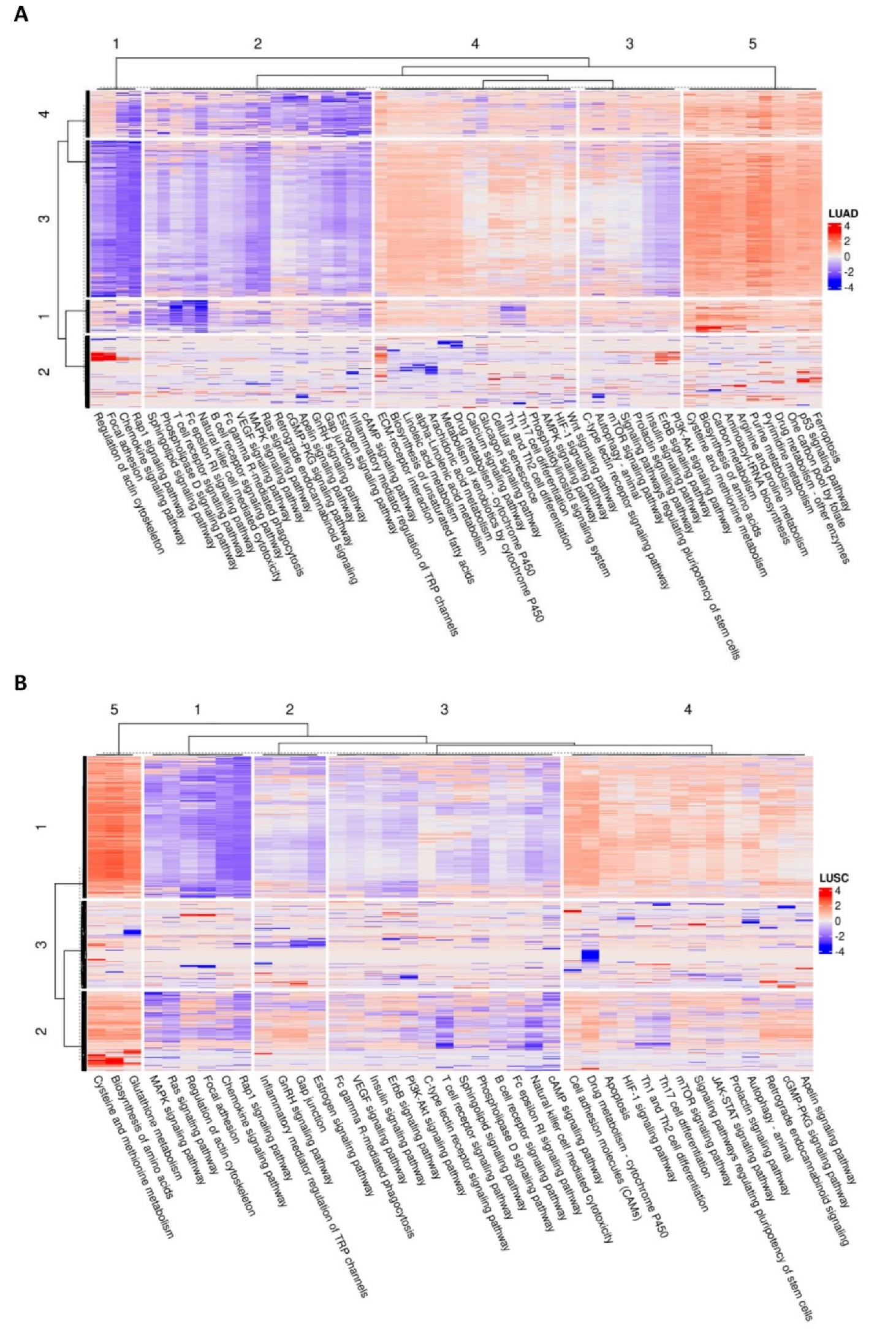
3.3. HERV-Specific Neural Network-Based Pathway Analysis
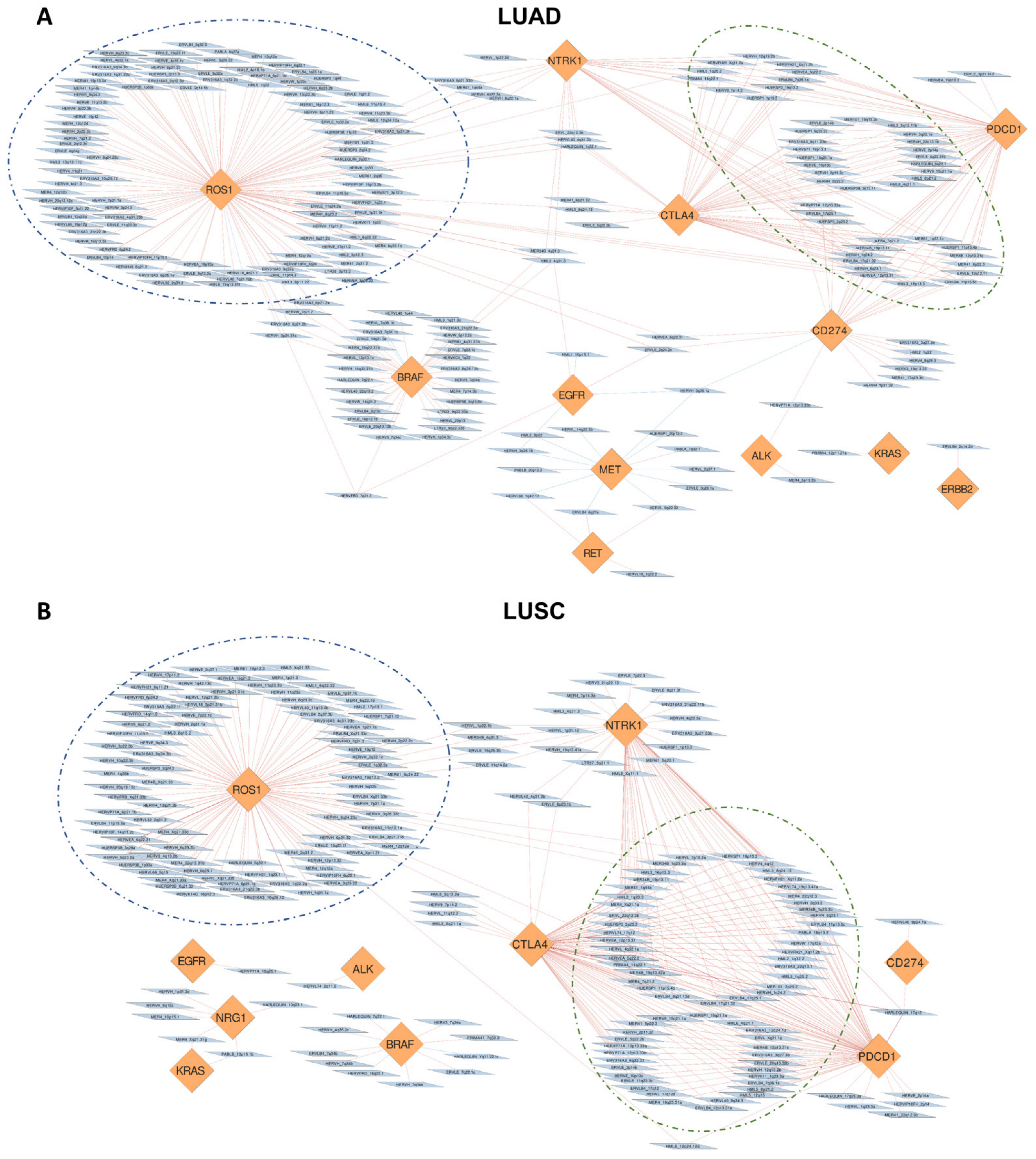
3.4. Correlation of HERVs with Oncogenes and Adaptive Immunity Regulators in TCGA-LUAD and TCGA-LUSC
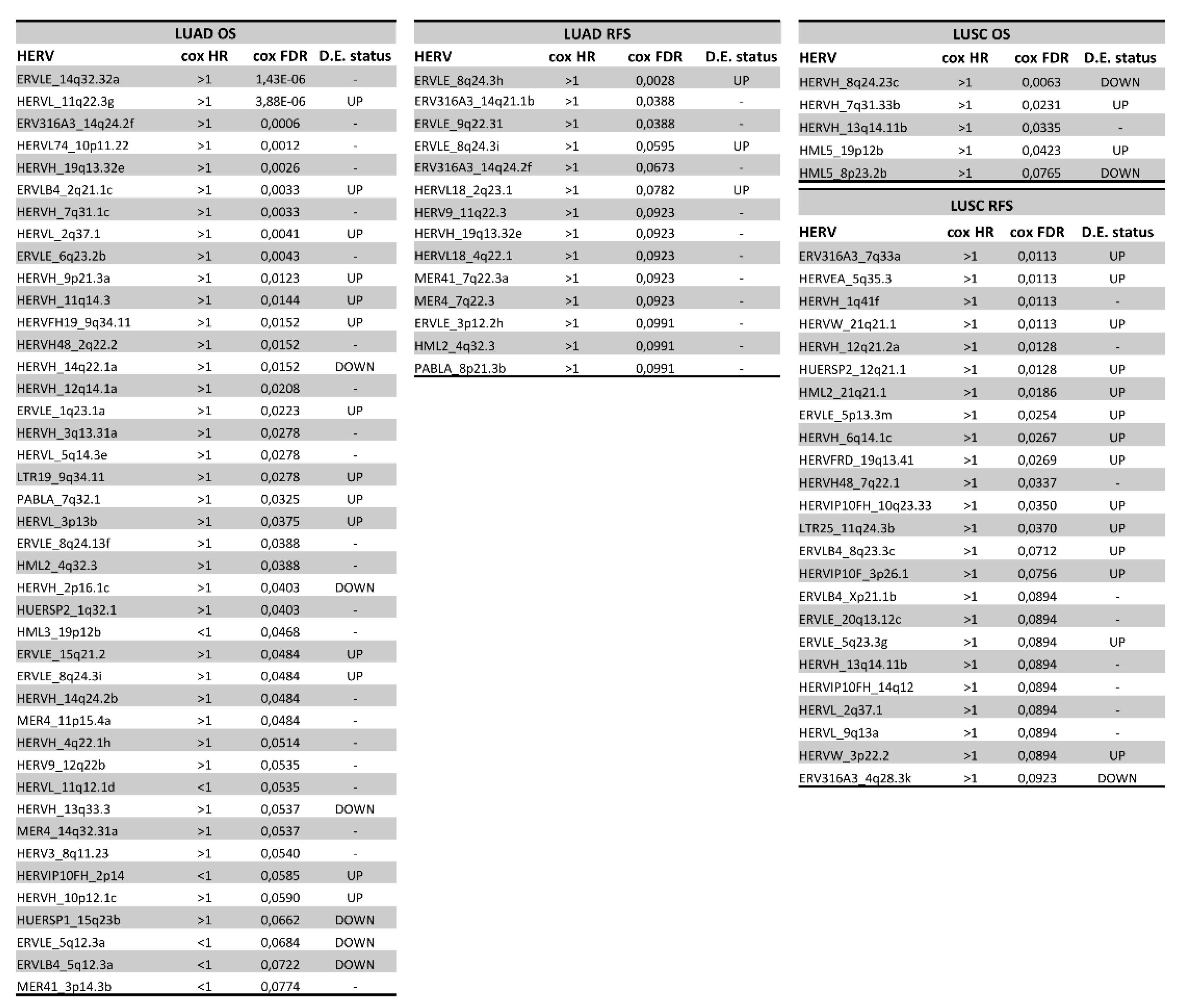
3.5. Association of HERVs with Survival in TCGA-LUAD and TCGA-LUSC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, A.G.; Cote, M.L. Epidemiology of Lung Cancer. Adv. Exp. Med. Biol. 2016, 893, 21–41. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Cagle, P.T.; Allen, T.C.; Olsen, R.J. Lung Cancer Biomarkers: Present Status and Future Developments. Arch. Pathol. Lab. Med. 2013, 137, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Patz, E.F., Jr.; Pinsky, P.; Gatsonis, C.; Sicks, J.D.; Kramer, B.S.; Tammemägi, M.C.; Chiles, C.; Black, W.C.; Aberle, D.R. NLST Overdiagnosis Manuscript Writing Team Overdiagnosis in Low-Dose Computed Tomography Screening for Lung Cancer. JAMA Intern. Med. 2014, 174, 269–274. [Google Scholar] [CrossRef]
- Konopka, K.E. Diagnostic Pathology of Lung Cancer. Semin. Respir. Crit. Care Med. 2016, 37, 681–688. [Google Scholar] [CrossRef]
- Xi, K.-X.; Zhang, X.-W.; Yu, X.-Y.; Wang, W.-D.; Xi, K.-X.; Chen, Y.-Q.; Wen, Y.-S.; Zhang, L.-J. The Role of Plasma miRNAs in the Diagnosis of Pulmonary Nodules. J. Thorac. Dis. 2018, 10, 4032–4041. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.-L.; Paz-Ares, L. Lung Cancer: Current Therapies and New Targeted Treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Santarpia, M.; Liguori, A.; D’Aveni, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffinà, M.G.; Lazzari, C.; Altavilla, G.; Rosell, R. Liquid Biopsy for Lung Cancer Early Detection. J. Thorac. Dis. 2018, 10, S882–S897. [Google Scholar] [CrossRef]
- Curty, G.; Marston, J.L.; de Mulder Rougvie, M.; Leal, F.E.; Nixon, D.F.; Soares, M.A. Human Endogenous Retrovirus K in Cancer: A Potential Biomarker and Immunotherapeutic Target. Viruses 2020, 12, 726. [Google Scholar] [CrossRef]
- Lecuelle, J.; Favier, L.; Fraisse, C.; Lagrange, A.; Kaderbhai, C.; Boidot, R.; Chevrier, S.; Joubert, P.; Routy, B.; Truntzer, C.; et al. MER4 Endogenous Retrovirus Correlated with Better Efficacy of Anti-PD1/PD-L1 Therapy in Non-Small Cell Lung Cancer. J. Immunother. Cancer 2022, 10, e004241. [Google Scholar] [CrossRef] [PubMed]
- Golkaram, M.; Salmans, M.L.; Kaplan, S.; Vijayaraghavan, R.; Martins, M.; Khan, N.; Garbutt, C.; Wise, A.; Yao, J.; Casimiro, S.; et al. HERVs Establish a Distinct Molecular Subtype in Stage II/III Colorectal Cancer with Poor Outcome. NPJ Genom Med 2021, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Tavakolian, S.; Goudarzi, H.; Faghihloo, E. Evaluating the Expression Level of HERV-K Env, np9, Rec and Gag in Breast Tissue. Infect. Agent. Cancer 2019, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wei, H.; Wei, Y.; Tan, A.; Chen, X.; Liao, X.; Xie, B.; Wei, X.; Li, L.; Liu, Z.; et al. Screening and Identification of Human Endogenous Retrovirus-K mRNAs for Breast Cancer Through Integrative Analysis of Multiple Datasets. Front. Oncol. 2022, 12, 820883. [Google Scholar] [CrossRef]
- Manca, M.A.; Solinas, T.; Simula, E.R.; Noli, M.; Ruberto, S.; Madonia, M.; Sechi, L.A. HERV-K and HERV-H Env Proteins Induce a Humoral Response in Prostate Cancer Patients. Pathogens 2022, 11, 95. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef]
- Geis, F.K.; Goff, S.P. Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview. Viruses 2020, 12, 884. [Google Scholar] [CrossRef]
- Petrizzo, A.; Ragone, C.; Cavalluzzo, B.; Mauriello, A.; Manolio, C.; Tagliamonte, M.; Buonaguro, L. Human Endogenous Retrovirus Reactivation: Implications for Cancer Immunotherapy. Cancers 2021, 13, 1999. [Google Scholar] [CrossRef]
- Gonzalez-Cao, M.; Iduma, P.; Karachaliou, N.; Santarpia, M.; Blanco, J.; Rosell, R. Human Endogenous Retroviruses and Cancer. Cancer Biol. Med. 2016, 13, 483–488. [Google Scholar]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Kolbe, A.R.; Bendall, M.L.; Pearson, A.T.; Paul, D.; Nixon, D.F.; Pérez-Losada, M.; Crandall, K.A. Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival. Viruses 2020, 12, 956. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.C.; Marston, J.L.; Iñiguez, L.P.; Bendall, M.L.; Chiappinelli, K.B.; Nixon, D.F.; Crandall, K.A. Locus-Specific Characterization of Human Endogenous Retrovirus Expression in Prostate, Breast, and Colon Cancers. Cancer Res. 2021, 81, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Bendall, M.L.; Francis, J.H.; Shoushtari, A.N.; Nixon, D.F. Specific Human Endogenous Retroviruses Predict Metastatic Potential in Uveal Melanoma. JCI Insight 2022, 7. [Google Scholar] [CrossRef] [PubMed]
- Johanning, G.L.; Malouf, G.G.; Zheng, X.; Esteva, F.J.; Weinstein, J.N.; Wang-Johanning, F.; Su, X. Expression of Human Endogenous Retrovirus-K Is Strongly Associated with the Basal-like Breast Cancer Phenotype. Sci. Rep. 2017, 7, 41960. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous Retroviral Signatures Predict Immunotherapy Response in Clear Cell Renal Cell Carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef]
- Panda, A.; de Cubas, A.A.; Stein, M.; Riedlinger, G.; Kra, J.; Mayer, T.; Smith, C.C.; Vincent, B.G.; Serody, J.S.; Beckermann, K.E.; et al. Endogenous Retrovirus Expression Is Associated with Response to Immune Checkpoint Blockade in Clear Cell Renal Cell Carcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.-J.; Blanchette, C.; Albert, M.L.; et al. Transposable Element Expression in Tumors Is Associated with Immune Infiltration and Increased Antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Bendall, M.L.; de Mulder, M.; Iñiguez, L.P.; Lecanda-Sánchez, A.; Pérez-Losada, M.; Ostrowski, M.A.; Jones, R.B.; Mulder, L.C.F.; Reyes-Terán, G.; Crandall, K.A.; et al. Telescope: Characterization of the Retrotranscriptome by Accurate Estimation of Transposable Element Expression. PLoS Comput. Biol. 2019, 15, e1006453. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Signal, B.; Kahlke, T. How_are_we_stranded_here: Quick Determination of RNA-Seq Strandedness. BMC Bioinformatics 2022, 23, 49. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; ScienceOpen, Inc.: Burlington, MA, USA, 2010. [Google Scholar]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/map (SAM) Format and SAMtools. Bioinformatics 2009, 1000, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S. Lewis EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. R package version 2019, 1. [Google Scholar]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinformatics 2015, 16, 169. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and Enhances Circular Visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic Driver Mutations in Non-Small Cell Lung Cancer: Past, Present and Future. World J. Clin. Oncol. 2021, 12, 217–237. [Google Scholar] [CrossRef]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current Landscape and Future of Dual Anti-CTLA4 and PD-1/PD-L1 Blockade Immunotherapy in Cancer; Lessons Learned from Clinical Trials with Melanoma and Non-Small Cell Lung Cancer (NSCLC). J Immunother Cancer 2018, 6, 39. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Alaimo, S.; Giugno, R.; Acunzo, M.; Veneziano, D.; Ferro, A.; Pulvirenti, A. Post-Transcriptional Knowledge in Pathway Analysis Increases the Accuracy of Phenotypes Classification. Oncotarget 2016, 7, 54572–54582. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, Y.; Parra, E.; Rodriguez, J.; Behrens, C.; Akbani, R.; Lu, Y.; Kurie, J.M.; Gibbons, D.L.; Mills, G.B.; et al. Multiplatform-Based Molecular Subtypes of Non-Small-Cell Lung Cancer. Oncogene 2017, 36, 1384–1393. [Google Scholar] [CrossRef]
- Zare, M.; Mostafaei, S.; Ahmadi, A.; Azimzadeh Jamalkandi, S.; Abedini, A.; Esfahani-Monfared, Z.; Dorostkar, R.; Saadati, M. Human Endogenous Retrovirus Env Genes: Potential Blood Biomarkers in Lung Cancer. Microb. Pathog. 2018, 115, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Guo, X.; Li, J.; Han, J.; Jia, L.; Wen, H.-L.; Sun, C.; Wang, X.; Zhang, B.; Li, J.; et al. Significant Upregulation of HERV-K (HML-2) Transcription Levels in Human Lung Cancer and Cancer Cells. Front. Microbiol. 2022, 13, 850444. [Google Scholar] [CrossRef]
- Arroyo, M.; Bautista, R.; Larrosa, R.; Cobo, M.Á.; Claros, M.G. Biomarker Potential of Repetitive-Element Transcriptome in Lung Cancer. PeerJ 2019, 7, e8277. [Google Scholar] [CrossRef]
- Konen, J.M.; Rodriguez, B.L.; Fradette, J.J.; Gibson, L.; Davis, D.; Minelli, R.; Peoples, M.D.; Kovacs, J.; Carugo, A.; Bristow, C.; et al. Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/p53 Mutant Lung Cancer. Cancers 2019, 11, 462. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Denne, M.; Sauter, M.; Armbruester, V.; Licht, J.D.; Roemer, K.; Mueller-Lantzsch, N. Physical and Functional Interactions of Human Endogenous Retrovirus Proteins Np9 and Rec with the Promyelocytic Leukemia Zinc Finger Protein. J. Virol. 2007, 81, 5607–5616. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.; Heyne, K.; Roemer, K.; Meese, E.; Mayer, J. HERV-K(HML-2) Rec and np9 Transcripts Not Restricted to Disease but Present in Many Normal Human Tissues. Mobile DNA 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Grandi, N.; Tramontano, E. HERV Envelope Proteins: Physiological Role and Pathogenic Potential in Cancer and Autoimmunity. Frontiers in Microbiology 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Alcazer, V.; Mutez, V.; Tonon, L.; Martin, J.; Chuvin, N.; Michel, E.; Boulos, R.E.; Estornes, Y.; Valladeau-Guilemond, J.; et al. Identification of Shared Tumor Epitopes from Endogenous Retroviruses Inducing High-Avidity Cytotoxic T Cells for Cancer Immunotherapy. Science Advances 2022, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Krishnamurthy, J.; Wei, Y.; Li, M.; Hunt, K.; Johanning, G.L.; Cooper, L.J.; Wang-Johanning, F. Chimeric Antigen Receptor T Cells Targeting HERV-K Inhibit Breast Cancer and Its Metastasis through Downregulation of Ras. Oncoimmunology 2015, 4, e1047582. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Rabinovich, B.A.; Mi, T.; Switzer, K.C.; Olivares, S.; Maiti, S.N.; Plummer, J.B.; Singh, H.; Kumaresan, P.R.; Huls, H.M.; et al. Genetic Engineering of T Cells to Target HERV-K, an Ancient Retrovirus on MelanomaAncient Retrovirus Targeted by Engineered T Cells in Melanoma. Clin. Cancer Res. 2015, 21, 3241–3251. [Google Scholar] [CrossRef]
- Vergara Bermejo, A.; Ragonnaud, E.; Daradoumis, J.; Holst, P. Cancer Associated Endogenous Retroviruses: Ideal Immune Targets for Adenovirus-Based Immunotherapy. Int. J. Mol. Sci. 2020, 21, 4843. [Google Scholar] [CrossRef]
- Weyerer, V.; Strissel, P.L.; Stöhr, C.; Eckstein, M.; Wach, S.; Taubert, H.; Brandl, L.; Geppert, C.I.; Wullich, B.; Cynis, H.; et al. Endogenous Retroviral–K Envelope Is a Novel Tumor Antigen and Prognostic Indicator of Renal Cell Carcinoma. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus Docetaxel for Patients with Previously Treated Non-Small-Cell Lung Cancer (POPLAR): A Multicentre, Open-Label, Phase 2 Randomised Controlled Trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus Docetaxel in Patients with Previously Treated Non-Small-Cell Lung Cancer (OAK): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Ferlita, A.; Distefano, R.; Alaimo, S.; Beane, J.D.; Ferro, A.; Croce, C.M.; Tsichlis, P.N.; Pulvirenti, A.; Nigita, G. Transcriptome Analysis of Human Endogenous Retroviruses at Locus-Specific Resolution in Non-Small Cell Lung Cancer. Cancers 2022, 14, 4433. https://doi.org/10.3390/cancers14184433
La Ferlita A, Distefano R, Alaimo S, Beane JD, Ferro A, Croce CM, Tsichlis PN, Pulvirenti A, Nigita G. Transcriptome Analysis of Human Endogenous Retroviruses at Locus-Specific Resolution in Non-Small Cell Lung Cancer. Cancers. 2022; 14(18):4433. https://doi.org/10.3390/cancers14184433
Chicago/Turabian StyleLa Ferlita, Alessandro, Rosario Distefano, Salvatore Alaimo, Joal D. Beane, Alfredo Ferro, Carlo M. Croce, Philip N. Tsichlis, Alfredo Pulvirenti, and Giovanni Nigita. 2022. "Transcriptome Analysis of Human Endogenous Retroviruses at Locus-Specific Resolution in Non-Small Cell Lung Cancer" Cancers 14, no. 18: 4433. https://doi.org/10.3390/cancers14184433
APA StyleLa Ferlita, A., Distefano, R., Alaimo, S., Beane, J. D., Ferro, A., Croce, C. M., Tsichlis, P. N., Pulvirenti, A., & Nigita, G. (2022). Transcriptome Analysis of Human Endogenous Retroviruses at Locus-Specific Resolution in Non-Small Cell Lung Cancer. Cancers, 14(18), 4433. https://doi.org/10.3390/cancers14184433