Simple Summary
Immunotherapy helps a person’s immune system to target tumor cells. Recent advances in cancer immunotherapy, including immune checkpoint inhibition, chimeric antigen receptor T-cell therapy and cancer vaccination, have changed the landscape of cancer treatment. These approaches have had profound success in certain cancer types but still fail in the majority of cases. This review will cover both successes and current challenges in cancer immunotherapy, as well as recent advances in the field of basic tumor immunology that will allow us to overcome resistance to existing treatments.
Abstract
Cancer immunotherapy has revolutionized the field of oncology in recent years. Harnessing the immune system to treat cancer has led to a large growth in the number of novel immunotherapeutic strategies, including immune checkpoint inhibition, chimeric antigen receptor T-cell therapy and cancer vaccination. In this review, we will discuss the current landscape of immuno-oncology research, with a focus on elements that influence immunotherapeutic outcomes. We will also highlight recent advances in basic aspects of tumor immunology, in particular, the role of the immunosuppressive cells within the tumor microenvironment in regulating antitumor immunity. Lastly, we will discuss how the understanding of basic tumor immunology can lead to the development of new immunotherapeutic strategies.
1. Introduction
Immunotherapy harnesses a patient’s immune system to target cancer and has resulted in novel therapeutic approaches and unprecedented clinical outcomes [1]. Although immunotherapeutic approaches have found success in a variety of cancer subtypes and clinical scenarios challenges still remain [2,3,4,5]. Thus, comprehensive knowledge of how these therapies function is essential to address these challenges [6,7]. Tumor–host immune system interactions are heterogenous and certain characteristics can predict immunotherapy responsiveness. The tumor microenvironment (TME), in particular, affects immunotherapeutic response and immune evasion [8,9]. A deep understanding of host–tumor interactions is critical to fostering the development of novel and more effective immunotherapies. In this review, we will summarize the current landscape of immune checkpoint inhibition (ICI), adoptive cellular therapy (CAR T-cell therapy) and cancer vaccination. We will also outline new standards of care and examine immuno-oncology trials, with a focus on factors that affect immunotherapeutic results. In particular, we will explore the TME and how it influences immunotherapy outcomes and how to potentially address host–tumor interactions to improve immunotherapy efficacy.
2. Current State of Cancer Immunotherapy
2.1. Immune Checkpoint Inhibitor Therapy
Immune checkpoint inhibitors (ICI) are one of the most promising types of cancer immunotherapy. Multiple drugs have received FDA approval for more than nine cancer types over the last decade [10]. ICI therapy is based on the premise that T cells contain evolutionarily conserved negative regulatory markers that act as “checkpoints” to regulate activation [9]. Early after activation, T cells upregulate inhibitory receptor cytotoxic T lymphocyte antigen 4 (CTLA4) and, later, programmed cell death 1 (PD-1), which then bind to co-stimulatory ligands B7-1, B7-2 and PD-L1 or PD-L2, respectively [11,12]. Ligands are presented by tumor cells, regulatory T cells (Tregs), myeloid cells and antigen-presenting cells (APCs), which dampen cytotoxic T-cell activation, resulting in immune suppression and tumor growth [13,14,15]. Upon treatment with immune checkpoint inhibitors, inhibition is released and cancer cells are targeted and destroyed by the primed and activated cytotoxic T cells [16,17]. ICI has resulted in successful treatment for a variety of recalcitrant cancers [18].
ICI has achieved marked success in patients with previously dismal outcomes treated with conventional cancer therapies, such as chemo, radiation and targeted therapy. Durable responses suggest long-lasting immunological memory can be seen, established in patients who respond to ICI. FDA-approved ICI therapies are outlined in Table 1.

Table 1.
FDA approved, combination and ongoing clinical trials with ICI.
Ipilimumab, a therapeutic anti-CTLA4 human IgG1 monoclonal antibody, was the first ICI approved by the FDA and has since been utilized for treatment in a number of diseases [19,20,21,22,23]. Though some ICIs have been successful, some have also failed. Another anti-CTLA antibody, Tremelimumab, is an IgG2 isotype that failed to meet its primary endpoint in late-stage trials for advanced metastatic melanoma [24,25]. Ipilimumab and tremelimumab appear to have different antibody-dependent cell-mediated cytotoxicity (ADCC) effects, though it is unclear why. These antibodies may mediate depletion of tumor-infiltrating regulatory T cells via ADCC, modulating their clinical outcome [26,27]. Pembrolizumab (pembro) and nivolumab, two human IgG4 anti-PD-1 checkpoint inhibitor antibodies, were the first PD-1-targeted, FDA-approved therapies for refractory and unresectable melanoma [23,28,29]. Pembro, in particular, has had marked success, demonstrating an increase in overall response rate (ORR) in NSCLC and in melanoma patients compared to standard-of-care chemo. In certain cases, for melanoma (nivolumab/pembro) and NSCLC (atezolizumab), ICI has become the first line of treatment over chemo [30,31,32,33]. Combination therapy with anti-CTLA4 + anti-PD-1 has demonstrated significantly improved ORR for metastatic melanoma at 58% [34]. However, increased toxicity to combined treatment is common and remains a challenge to address.
Increased PD-L1 expression has been found to be associated with improved responses to pembro and PD-L1 expression level is used as a biomarker to guide the indication for ICI in certain cancer subtypes [35,36,37]. The type of scoring system used, whether tumor-derived PD-L1, TME-derived PD-L1 or a combined score, is important to note.
Anti-PD-L1 antibody treatments have also been proven effective in multiple types of cancer. Atezolizumab was the first approved anti-PD-L1 ICI for the treatment of urothelial carcinoma, and avelumab and durvalumab are approved for multiple types of solid tumor cancers [18,23,33,38,39,40,41,42]. Tumor PD-L1 expression has also been used as a biomarker for treatment indication for anti-PD-L1 ICI.
Microsatellite instability in cancer results from defective DNA mismatch repair (dMMR) proteins. The presence of microsatellite instability at high levels (MSI-H) has been associated with markedly improved outcomes in multiple cancer subtypes, including ovarian and colorectal cancer (CRC) [43,44]. Dramatically improved outcomes seen with MSI-H and ICI resulted in pembro being the first treatment approved in the disease agnostic setting based on the presence of MSI-H, regardless of cancer subtype [45]. A recent clinical study with dostarlimab, an anti-PD-1 ICI, in dMMR rectal cancer recently reported 100% clinical ORR and no recurrence to date [46].
Though ICI results in dramatic results for responders, only 20–30% of patients achieve a clinical response to ICI. Thus, in order to address the majority of patients, a deep understanding of additional checkpoint pathways and how they may be targeted is critical. Table 1 outlines ongoing clinical trials to investigate novel ICI agents. T-cell immunoreceptor with Immunoglobulin (Ig) and ITIM domains (TIGIT) is an inhibitory molecule present on CD8+ and CD4+ T cells, Tregs and natural killer (NK) cells that regulates T-cell immunity via the CD226-PVR pathway. Approximately two dozen monoclonal antibodies targeting TIGIT have been developed as both single agents and to be used in conjunction with anti-PD-1 or anti-PD-L1 agents [47]. Tiragolumab, an anti-TIGIT IgG1 antibody, when combined with atezolizumab (Table 1) showed improved clinical efficacy in NSCLC vs. atezo plus placebo and is currently being investigated in multiple Phase III trials in solid tumors [48,49]. Other anti-TIGIT antibodies, vibostolimab and etigilimab, have also shown promising clinical activity in early phase studies [50,51]. V-domain Immunoglobulin T-cell activation suppressor (VISTA) is another potential target. VISTA maintains naive T-cell and myeloid cell quiescence through binding to P-selectin glycoprotein ligand-1 (PSGL-1). VISTA binding activity is greater in low-pH and hypoxic settings, such as the TME. VISTA suppresses T-cell activation and reprograms macrophages to an immunosuppressive phenotype in the TME [52,53]. Anti-VISTA antibodies are under development, including a pH-selective VISTA blocking antibody that suppressed tumor growth in a mouse model of colon cancer [54]. T-cell immunoglobulin and mucin-domain containing-3 (Tim-3) is another co-inhibitory molecule expressed on T cells and leukemic cells, such as AML. Multiple Tim-3 inhibitors are being studied and have showed promise in treating MDS and AML (Table 1). Anti-Tim-3 ICI functions to target Tim3-expressing AML cells and boosts antitumor T-cell activity via its ICI activity [55,56].
Although immune checkpoint blockade has achieved remarkable successes, there remain challenges. There is still a significant portion of patients that do not respond at all and adverse effects of ICI, particularly combination therapy, need to be addressed [9,25]. Further study of alternative checkpoint inhibitor pathways that allow tumor escape and the influence of the TME on ICI will also be critical to future success.
2.2. CAR T-Cell Therapy
Adoptive cellular therapy (ACT) traditionally referred to three different approaches: infusion of tumor-infiltrating lymphocytes (TIL), genetically modified T-cell receptor (TCR) therapies and chimeric antigen receptor (CAR)-modified T cells [57]. The use of other immune cell types, such as natural killer cells (NK), CAR-NK cells, as well as CAR-macrophages (CAR-M), is being studied, though no therapies have yet obtained FDA approval. The most successful ACT has been CAR T-cell therapy (CART), which now carries a multitude of FDA-approved indications and is being utilized as standard of care worldwide for a variety of hematologic malignancies (Table 2). Here, we focus on successes in CART, as well as challenges facing CART and future areas for research.
Table 2.
FDA-approved and ongoing clinical trials with CAR T.
CAR T cells were originally described by Eshhar et al., whereby a murine single-chain variable fragment antibody domain (svFC) was linked with a CD3ζ signaling chain and then inserted onto a human T cell. These first-generation CARs allowed for major histocompatibility complex (HLA)-independent activation of T cells when presented with a specific target antigen recognized by the svFC [58]. In 2011, costimulatory domains (most commonly CD28 or 4-1BB) were added to the CAR construct, resulting in much improved CAR T-cell expansion, persistence and pre-clinical efficacy [59,60]. Ultimately, these “2nd generation” CAR constructs have displayed unprecedented clinical efficacy, particularly when targeted against CD19 expressing B-cell lymphomas and leukemias and against B-cell maturation antigen (BCMA)-expressing multiple myeloma.
CART has found particularly profound success in diffuse large B-cell lymphoma (DLBCL), the most common subtype of non-Hodgkin’s B-cell lymphoma (NHL). In relapsed or refractory (R/R) DLBCL, outcomes were dismal. Patients unfit for or who relapsed after autologous stem cell transplant traditionally had an overall response rate (ORR) to next line of therapy of 20–30%, with a median overall survival (OS) of approximately 6 months [61,62,63]. ZUMA-1 (axicabtagene ciloleucel or axi-cel) and JULIET (tisagenlecleucel or tisa-cel) were pivotal trials for an autologous anti-CD19 CART (CAR19) product for patients with R/R DLBCL. Autologous mononuclear cells were collected by apheresis, purified to select for T cells, engineered with an Anti-CD19 scFV, expanded and reinfused into the patient. Both axi-cel (CD28 costimulatory domain) and tisa-cel (4-1BB co-stimulatory domain) had substantial post-infusion expansion in the majority of patients [64,65]. CAR19 provided an unprecedented ORR of 54–82%, a complete response (CR) rate of 40–54% and a median OS measured in years [64,65]. Real-world trials demonstrated comparable results and most patients who achieved a CR and 30–40% of all patients appear to remain in durable remission five years or longer post-CAR infusion [66,67,68]. CAR19 is now considered standard of care for R/R aggressive NHL after 2 or more lines of therapy and recent studies support shifting CART to the second line (and for which axi-cel was recently FDA approved) [69,70].
CART has also found success in R/R Multiple Myeloma (MM) by targeting BCMA. Idecabtegene vicleucel was approved in 2021, based on a phase 2 study showing that heavily pre-treated, relapsed refractory patients achieved 73% ORR and 33% CR rate [71]. Ciltacabtagene autoleucel is a second anti-BCMA CART that did well in heavily pre-treated patients with R/R MM, with a 97% ORR, 67% CR rate and 77% durable response at 1 year [72]. A retrospective study analyzed a similar cohort of patients who received non-CART therapy and found significantly worse outcomes compared to those seen with CART [73]. Continued clinical trials will be required to determine the optimal timing or CART in MM.
Four CAR19 agents and two anti-BMCM CART agents have now been FDA approved (Table 2). CART has transformed hematologic cancer treatment, yet challenges remain. The majority of patients who receive CART ultimately fail therapy, whether due to primary progression or response then relapse [64,65,74]. In acute lymphoblastic leukemia (ALL), lasting remissions have been demonstrated in pediatrics, but most adult patients require an allogeneic stem cell transplant post CAR19 due to high rates of relapse [75,76]. In order to address these challenges, more research into the mechanisms of CART resistance is required. CART failure includes either primary resistance to CART or response and then relapse post CART; each characteristically results from a different mechanism, though mechanisms of failure may not be mutually exclusive. Causes of CART failure can be grouped into tumor or disease-intrinsic factors, CAR T-cell product-specific mechanisms and CAR T-cell/host interactions. Loss or mutation of the target antigen (e.g., the CD19 extracellular epitope on leukemic/lymphoma cells) results in tumor intrinsic CAR T-cell failure [77,78]. Further, 10–20% of ALL patients will relapse with CD19 (-) leukemia [75,78,79]. However, in one study of patients who achieved a CR and then relapsed, 68% relapsed with CD19 (-) disease (with 27% not evaluable and only 4.5% with CD19+ disease). In the pivotal ZUMA-1 trial, 3/11 (27%) patients who failed CART who had evaluable tissue had lost CD19 expression at time of progression [64]. Alternative antigen and multi-antigen treatments are being developed to address antigen loss. Shah et al. found that anti-CD22-directed CAR T-cell treatment achieved 70% CR rates in ALL after failure of CAR19 [80]. Clinical trials of anti-CD22 following CAR19 failure in NHL have also been successful [81]. Though studies are promising, antigen loss of the new target (ex. CD22 or CD20) is also a potential drawback. Thus, several clinical trials integrating multiple antigen targets (ex. CD19/22) on a single CAR are underway, including a tri-specific CART targeting CD19/20/22, soon to open here at the OSUCCC [82,83].
Poor expansion and function are caused by CAR T-cell product intrinsic deficiencies. Clinical performance relies on CART viability, transduction efficiency and phenotype. Inadequate products can be due to manufacturing error, poor culture conditions or low-quality donor T cells due to past therapy and/or high disease burden at the time of apheresis [84]. Both patient status and the manufacturing process must be optimized to address CART product intrinsic mechanisms of failure [80,82]. Studies evaluating cell product collection pre- and post-bridging therapy and how cytoreduction may affect product quality are required. Point-of-care manufacturing, novel culture conditions and varying culture length are innovative CAR T-cell manufacturing methodologies. Point-of-care manufacturing reduces vein-to-vein time and may improve therapeutic results [82,85]. Recent studies utilizing IL7 and IL15 for the expansion phase of CART culture (instead of IL-2 utilized FDA-approved products) and a shorter 8-day manufacturing period have shown an increase in T stem-cell-like memory populations and have been hypothesized to improve expansion and overall effectiveness [86]. Commercial production of 4-1BB CAR19 using a 2-day, expansion-free approach has demonstrated encouraging results [87]. This shorter method boosted stemness and proliferative potential by expanding CAR T cells in vivo (in human), as opposed to ex vivo [87,88].
Tumor, host and CAR T-cell interactions result from interplay of the TME with the infused CAR T-cell product. Immunosuppressive TME interactions with CAR T cells can reduce expansion and increase exhaustion, with host systemic inflammation and tumor burden contributing as well [89,90]. Retrospective studies quantifying risk factors for CAR T-cell failure have identified extranodal disease sites ≥2, increased C-reactive protein and lactate dehydrogenase (inflammatory markers) and high metabolic tumor volume at the time of treatment as predictive of outcomes. [91] Jain et al. identified increased protumoral tumor-associated macrophage (TAM) markers, increased PD-L1 expression in the TME and increased mononuclear-myeloid-derived suppressor cells (MDSC’s) in circulation as factors that correlated with poorer response to CAR19 [92]. In CART for solid tumors, a more robust TME and increased tumor heterogeneity are hypothesized to be the primary reason for the lack of efficacy [93]. Both novel bridging and conditioning regimens, as well as radiation therapy pre-CART, are being studied to abrogate TME-mediated CART inhibition [94,95].
In CLL, Bruton’s Tyrosine Kinase inhibitor (BTKi) use prior to leukapheresis has been shown to improve CAR T-cell expansion and function, decrease T-cell exhaustion and also mitigate toxicity [96,97]. BTKi augments CART function via TME modulation, including reduced PD-1 and CTLA-4 expression, inhibition of Tregs, downregulation of B-cell chemokines and disruption of tumor cell adhesion/homing [98]. Here, at the Ohio State University Comprehensive Cancer Center (OSUCCC), we have achieved relative success by utilizing ibrutinib through leukapheresis and bridging prior to CAR T-cell therapy in patients with Richter’s Syndrome (DLBCL with antecedent CLL). Five of nine patients treated achieved a CR and four of these remained disease free long term [99].
Increased PD-L1 expression by TAMs is associated with poorer outcomes in CART [92]. Zuma-6 investigated the use of anti-PD-L1 atezolizumab post axi-cel infusion. Though atezolizumab was determined to be safe, clinical outcomes were similar to giving axi-cel alone [100].
Another approach to address mechanisms of CART failure is the use of novel salvage agents. The focus is on restoring CART proliferation and function to recapture a clinical response after failure. In a study of pembro given post-CART failure, 3/12 patients responded to therapy, with only 1 CR. Though outcomes were poor with pembro compared to results seen with other salvage agents, the responders to CAR T cells re-expanded and were more functionally active [101,102,103]. Thus, this is proof of concept that CART activity and proliferation can be recaptured post-CART failure with salvate therapy. In a point-of-care manufactured CAR19 product, TIGIT expression post CART was found to be associated with poorer outcomes. Anti-TIGIT ICI rescued CART function in vitro and in vivo and may be a promising agent post-CART failure. Lenalidomide is an immunomodulatory agent that enhances CAR T-cells’ antitumor efficacy by altering tumor cell receptor expression, via modification of the TME landscape and via direct effects on CAR T cells [104,105]. Multiple abstracts and studies have revealed the benefit of lenalidomide containing salvage regimens post CAR T-cell failure and clinical trials to study this further are warranted [101]. Continued research with robust clinical studies investigating the timing and appropriate population to use post-CAR immunomodulatory agents to enhance CAR T-cell function are required.
CART efficacy has also remained minimal in solid tumors, with no FDA-approved agents as of yet. This is due to both a lack of efficacy and increased toxicity seen in clinical trials [106]. Solid tumors are highly heterogeneous and CART requires a ubiquitously expressed target on tumor, with relatively low expression on normal tissue [106]. Further, solid tumors contain a highly robust and immunosuppressive TME, which impairs trafficking to and function within the tumor [107].
Though CAR T-cell therapy has found unprecedented success in CD19 and BCMA-expressing hematologic malignancies, it has still not been successful in the majority of patients nor in the majority of other hematologic and solid malignancies. In order to address this, further basic research into novel CART cell engineering strategies to enhance tumor recognition, TME infiltration, and improve anti-cancer activity is required.
2.3. Cancer Vaccines
Cancer vaccines aim to generate and amplify pre-existing T-cell and immune responses by providing tumor-specific or tumor-associated antigens (TAA) for elimination by the immune system [108,109]. Cancer vaccines purport to create persistent, targeted anticancer immunity. Developing tumor-specific antigens for cancer vaccines is challenging. TAAs are antigens selectively expressed or overexpressed in malignancies but also expressed in normal tissues. Targeting TAAs may induce autoreactive immune responses, resulting in organ toxicity and autoimmunity. Neoantigens result from carcinogenesis-related gene mutations. Neoantigens, which are not found in normal tissues, can be displayed on target cell surfaces, identified by T cells, and are not influenced by tolerance. Recently, the availability and affordability of next-generation sequencing has led to large-scale identification and the establishment of a plethora of neoantigens as potential immune system targets [108,109]. Cancer vaccine platforms are divided into four categories: cell-based, viral-based, peptide-based and nucleic-acid-based vaccines [110]. Table 3 outlines both FDA-approved cancer vaccines and ongoing trials. Sipuleucel-T (Provenge) was the first FDA-approved cancer vaccine for metastatic castration-resistant prostate cancer [111]. It is designed to elicit a prostate-cancer-specific immune response against prostatic acid phosphatase, an overexpressing TAA [112]. Other cell-based vaccinations include antigen-loaded dendritic cell (DC) vaccines produced from tumor lysates or mRNA, TAA peptides, TAA-coding mRNA or neoantigens. DC vaccines have shown promising results in clinical trials as monotherapy, including resulting in improved OS. Additional adjuvants, such as chemotherapy, could improve vaccine efficacy by inducing the release of danger signals by tumor cells and enhancing the immune response [113,114,115,116].
Table 3.
FDA approved and examples of ongoing clinical trials for cancer vaccines.
Oncolytic virus immunotherapy attacks tumor cells and stimulates antitumor responses. Herpes simplex virus and the adenovirus are used as vectors for specific genes and tumor antigen expression. Their mass-production speed and extensive host cell tropism have benefited clinical research [110]. The best clinical progress is T-VEC (Imlygic), a first-generation recombinant herpes simplex virus vector, which is FDA approved for treatment of recurrent unresectable melanoma [117,118]. Additionally, clinical trials with vaccines using adenovirus vectors, carrying immune-stimulating genes or providing TAAs, have induced strong antitumor immunity and been successful in HER2+ breast cancer and BCG-unresponsive non-muscular-invasive bladder cancer [119,120]. Peptide-based vaccines, including chemical and biosynthetic formulations of expected or known tumor antigens, generate a strong immune response against the tumor antigen. Polypeptide vaccine, DSP-788, induces cancer-cell-specific cytotoxic lymphocyte (CTL) and T-helper cell responses in a variety of solid and hematological tumor environments (Table 3) [121].
Nucleic acid vaccines also generate a robust MHC-I-mediated CD8+ T-cell response [110]. They can stimulate humoral and cellular immunity and encode full-length tumor antigens, allowing APCs to cross present several epitopes. Several DNA vaccines utilized in the treatment of cervical cancer have shown encouraging clinical efficacy. mRNA vaccines can encode immunostimulants, TAAs and tumor neoantigens. Immunostimulant-encoded mRNAs, such as TriMix, induce tumor cell death and release tumor antigens or are coupled with multiple-TAA-encoded mRNAs that produce robust CD8+ T-cell responses to improve response rates in patients [122]. mRNA vaccines encoding neoantigens lead to customizable vaccinations. Melanoma patients treated with personalized, multi-peptide NeoVax revealed long-term persistence of neoantigen-specific T cells exhibiting a memory phenotype, multiple TCR clones with distinct functional avidities and evidence of tumor infiltration [123,124]. Similar studies have been recapitulated in glioblastoma patients with comparable antitumor responses [125,126].
For a variety of immunotherapies (ex. ICI), increased tumor mutational burden has been linked to more potent immune responses and improved efficacy of treatments [127,128,129]. Neoantigens show strong individual heterogeneity; specific mutations across different types of tumors create different quantities of neoantigens. Thus, it is likely this will drive the application of this therapy to be more personalized [109]. The complexity of applying a personalized approach to cancer vaccination, with considerations to each individual TME, and further neoantigen study are areas that will benefit from further basic immunologic research in order to inform the development of effective cancer vaccines.
3. Basic Research in Cancer Immunology
3.1. The Tumor Microenvironment
Tumor and host interaction shapes local and systemic immunity to promote tumor development and the immunosuppressive tumor microenvironment [130]. The TME is heterogenous and varies by patient, cancer subtype and stage. TME composition influences cancer immunotherapy patient responses [8]. To improve immunotherapy efficacy, it is critical to understand TME cellularity and functionally. Tumor cells drive TME formation by forming physical barriers, inhibiting immune cells and recruiting immunosuppressive cells. Tumor cells secrete immunosuppressive cytokines (e.g., TGF-b, IL-10, VEGF), drive expression of inhibitory receptors and ligands (e.g., PD-L1/2, CTLA-4) and reduce tumor-specific MHC-I antigens. Tumor cells can generate chronic, weak antigen signals that drive T-cell reprogramming into an unresponsive, transcriptionally “exhausted” state. They also deplete nutrients and accumulate waste products, such as lactate and kynurenine, which inhibit T cells and create a hostile environment for effector cells [5,8] (Figure 1, created with BioRender.com, accessed on 15 August 2022).
Figure 1.
Tumor microenvironment.
The TME’s immunosuppressive function depends on the recruitment of stromal cells and immune cells (especially myeloid cells) and their re-direction towards pro-tumoral functions. Tumor stroma, composed of non-immune cells, such as carcinoma-associated fibroblasts (CAFs), tumor-associated vascular endothelial cells (TAECs) and extracellular matrix (ECM) components, forms a physical and immunosuppressive barrier, which allows angiogenesis to occur and spread [131,132]. Tumor cells, myeloid cells and CAFs have a complementary interaction in the TME. CAFs constitute over 80% of cells in pancreatic and breast tumors; these tumors also exhibit increased myeloid cell infiltration [132,133,134]. CAFs convert T cells into inducible Tregs and limit T- and NK-cell function. They increase myeloid infiltration, produce tolerogenic dendritic cells (tDCs), activate immunosuppressive M2-phenotypic macrophages by secreting TGF-b/IL-10 and remove APCs by inhibiting signal activity. TAECs influence immune cell movement, tumor cell intravasation and extravasation via angiogenesis [131,132]. TAECs can serve as non-professional APCs because they lack CD80 and CD86 co-stimulatory expression, triggering antigen-experienced T-cell effector capabilities but not naïve T cells. As non-professional APCs, TAECs reduce antitumor immunity and promote tumor growth [132].
Tumor-infiltrating lymphocytes (TILs) are various subsets of lymphocytes that are recruited into the TME. Their function depends greatly on TME composition and interacting pathways. TILs include CD8+ CTLs, CD4+ T-helper cells, Tregs, innate lymphoid cells (ILCs), NK cells and natural killer T (NKT) cells. CD8:CD4 TIL ratios have been studied as biomarkers in metastatic melanoma, NSCLC and colon cancer, with certain ratios correlating with increased ICI response predictions, diagnosis and survival rates in patients [1,8,135,136]. Immunologically “hot” tumors have heavy TIL infiltration. “Cold” tumors lack TILs and are unable to recruit/activate immune cells [137]. Hot tumors with inflammatory gene signatures respond better to ICI, while cold tumors are resistant [138]. These inflammatory gene signatures could be used as a clinical prognostic tool to evaluate ongoing trials and to help explain hot vs. cold TME status [137]. Tumor cells may induce cold TME by exhausting or functionally suppressing lymphocytes that enter the TME. Anti-angiogenic treatment combined with ICI to create high endothelial venules (HEVs) in the TME may enhance activated T-cell infiltration [139]. This was demonstrated in breast, pancreatic and glioblastoma tumor models, which are cold tumors, leading to sensitization of the TME for ICI and CART therapies [139,140]. Other autophagy-inhibiting targets may inhibit tumor growth in melanoma and CRC while enhancing ICI efficacy [141]. Continued research on novel methods to transform immune-cold environments into immune-hot environments is ongoing.
T-cell exhaustion is an area that has been extensively researched. Exhausted T cells can be “pre-exhausted” or “terminally exhausted” with increasing expression of immune checkpoints, such as CTLA-4, PD-1, Tim-3, lymphocyte activation gene-3 (LAG-3) and TIGIT, denoting a more terminally exhausted state [8]. Poor self-renewal, disrupted metabolism and gradual effector function loss characterize these cells [5,131]. In preclinical melanoma, ovarian, breast and bladder cancer models, targeting PI3K/Akt or Wnt, which control CD8+ T-cell infiltration and cytotoxicity and selectively inhibit Treg proliferation, enhances antitumor immunity and promotes tumor regression. Combinational therapies to normalize or sensitize the TME may improve ICI treatment efficacy [142,143,144,145,146,147,148].
3.2. Tumor-Associated Macrophages and Other Immunosuppressive Myeloid Cells
TAMs constitute most of the non-tumor stromal mass in solid tumors and modulate tumor growth and immunosuppression within the TME [149]. TAMs are protumoral and have M2-like functions [150,151]. They promote tumor growth and metastasis via anti-inflammatory cytokine secretion and immunosuppressive immune cell interactions and recruitment [152,153,154]. Similar to TAMs, myeloid-derived suppressor cells (MDSCs) are pathologically activated, immature, potent immunosuppressive cells at various stages in differentiation [155,156]. They are subdivided into mononuclear MDSCs (M-MDSCs), morphologically similar to blood monocytes, and polymorphonuclear (PMN-MDSCs), which are morphologically similar to neutrophils [157]. Their recruitment from bone marrow to secondary lymphoid organs and TME by cancer-cell-secreted growth factors promotes overall protumorgenic activity by inducing NK and T-cell inhibition, allowing tumor immunoevasion [158]. Poor prognosis and OS were correlated with solid tumor MDSC abundance [159,160]. Tumor-associated neutrophils (TANs) also have critical functions within the TME. N1s instruct effector T cells to reject tumor cells and N2s dampen the immune system by enlisting M2s and Tregs [132]. Shorter survival rates in HCC and poor prognosis for DLBCL have both been associated with TANs [161,162]. Due to their abundance in blood and their immediate reaction to inflammation and injury, their influence in the TME can set the tone for other immunosuppressive cells. DCs also infiltrate the TME but quickly adapt regulatory or tolerogenic phenotypes that promote tumor growth and immunoevasion [163,164,165]. Conventional DC types 1 (cDC1) and 2 (cDC2) play a key role as APCs and activate T cells for antitumor responses [166,167,168]. cDC1s and cDC2s in the TME are associated with a good prognosis in various cancers, while plasmacytoid DCs (pDC) frequencies are associated with a worse PFS and OS [169,170,171,172,173].
TAM functions are influenced by the TME, cancer type and stage [174]. Petty et al. described the role of hedgehog (Hh) signaling in polarizing TAMs towards an M2 phenotype and regulating CD8+ T-cell-mediated antitumor immunity [175,176]. Tumor-derived sonic hedgehog (SHH), a Hh ligand, drives TAM M2 polarization resulting in downregulation of CXCL9 and CXCL10 signaling and suppression of CD8+ infiltration into the TME [175]. TAMs also inhibit CD8+ T-cell activation by depleting essential metabolites for proliferation [174]. They inhibit T-cell function by producing IL-10, TGF-b and PGE-2 and upregulating PD-L1 [174,176,177]. TAMs are a major source of elevated CCL2 expression in human glioblastoma, which correlates with reduced OS, through promotion of CCR2+ M-MDSC infiltration [178,179].
TAMs correlate with poor outcomes in many cancer subtypes [180]. TAMs’ role as major carriers of checkpoint inhibitor ligands (e.g., PDL-1/2, B7-H4, VISTA) and as mediators of T-cell exhaustion has been well described [181]. Studies have also revealed how an increased concentration of M2-polarized TAMs mediate anti-PD-L1 ICI resistance in HNSCC and in prostate cancer [182,183].
Given their role in modulating immunotherapy, TAMs represent a promising target to augment immunotherapy. Reducing TAM presence in the TME by depletion or preventing trafficking and reprogramming TAMs to an immune-activating, M1-like phenotype are approaches being studied. Trabectedin is an FDA-approved agent for soft tissue sarcoma which targets TAMs to inhibit CCL2 and IL-6 production. When used in combination with an anti-PD-1 ICI in an ICI-resistant pre-clinical sarcoma model, trabectedin allowed for the recapture of response to anti-PD-1 therapy [184]. Targeting of PI3k-γ in myeloid cells using eganelisib (IPI-549) has also shown promise in pre-clinical models by restoring sensitivity to ICI [185,186].
Studies evaluating the repolarization of TAMs to a more M1-like, T-cell-activating phenotype are ongoing. Activation of toll-like receptors (TLR) on macrophages can lead to M1 polarization. Local delivery of a TLR7/8 agonist, telratolimod, in combination with ICI in murine melanoma boosted both local and systemic antitumor immunity [187]. Another novel approach utilizes low-pathogenicity influenza aviruses (IAVs), which have both oncolytic activity and also result in significant repolarization of TAMs to an M1-like state. When IAVs were combined with a novel B7-H3 ICI, responses were dramatic in a resistant NSCLC model [188]. Hh signaling blockade with vismogedib reduced TAM M2 polarization, increased CD8+ T-cell infiltration and suppressed tumor growth in murine lung and HCC [175]. Synergy between Hh inhibition and ICI was demonstrated as well with greater reduction in tumorigenesis than seen in either agent alone [175]. CART (and, in particular, CART targeting solid tumors with a more robust TME) and ICI may benefit from innovative approaches to abrogate TAMs’ immunosuppressive activity in the TME.
3.3. Approaches to Enhance ICI Therapy
Given the response rates seen thus far with currently FDA-approved ICI therapies, it is unlikely that one type of ICI will overcome the various mechanisms of resistance employed by different types of tumors. Targeting aspects of the TME to overcome tumor resistance has shown promise in enhancing ICI therapy [189].
Targeting the CXCR4/CXCL12 (SDF-1) signaling pathway can help overcome the physical barrier of the TME and enhance ICI. CXCR4 is upregulated on MDSC/TAMs, playing a role in intratumoral fibrosis, and is associated with poor prognosis in several types of cancer. Nanocomplex technology, polymer-based combinatory approaches and liposomal formation have been used to combine anti-PD-L1 agents and CXCR4 antagonists to overcome ICI resistance [190,191,192]. Increased effector T-cell infiltration, decreased Treg and MDSC populations and inhibition of primary tumor growth and metastasis were all observed in several tumor models. Nanoparticle technology applying a CSF1R inhibitor in combination with ICI allowed for the development of a sustained codelivery method that successfully reprogramed TAMs to an antitumoral M1-like phenotype and enhanced their phagocytic capabilities in a melanoma model [193]. In glioblastoma, overcoming the TME physical barrier is also being explored. One group combined brain-tumor-targeted peptide-coated extracellular vesicles, loaded with small interfering RNA (siRNA) against PD-L1, and then delivered them with bursts of radiation therapy [194]. Dual inhibition of PI3K/mTOR pathway combined with the microtubule targeting chemotherapy, paclitaxel, along with ICI, induced sustainable DC, T cell and NK responses, both locally in the TME and systemically [195]. Similar results targeting cancer stem cells with the polyketide antibiotic, Mithraymcin-A, and ICI in a CRC mouse model resulted in turning an immunologically cold tumor hot by increasing CD8+ T-cell infiltration and decreasing quantities of MDSC/TAMs in the TME [196].
New ICI targets are currently being studied as well. NKG2A is a checkpoint inhibitor expressed on subsets of cytotoxic T cells and NK cells. It binds to the MHC-I molecule HLA-E (Qa-1b mouse homolog) and is regulated in a TAP1-dependent manner [197]. Co-deletion of TAP-1/Qa-1b or blocking NKG2A with monalizumab unleashed effector cell activity and reversed resistance to ICI, resulting in tumor control [198,199]. Another target previously mentioned, used as both a monotherapy and in combination with ICI, is VISTA. Preclinical studies using L557-0155, an inhibitor for VSIG-8 (a VISTA receptor) promoted cytokine production by T cells and suppressed melanoma growth [200]. An orally administered combinatory small molecular inhibitor of VISTA and PD-L1, CA-170, showed similar antitumor efficacy in a number of mouse tumor models, prompting advancement towards clinical trials [201].
Diverse techniques to overcome mechanisms of resistance to ICI, including the immunosuppressive TME, as well as alternative ICI pathways that allow for tumor escape are ongoing. Continued focus on turning these basic discoveries into clinically applicable therapies to augment ICI is required.
3.4. Strategies to Improve CART Therapy
CART efficacy has achieved a high degree of success in hematologic malignancies. However, certain patients with risk factors, such as tumor bulk and a highly immunosuppressive TME, have worse outcomes. CART in solid tumors, in particular, has been characterized by a lack of efficacy due to the highly immunosuppressive TME present, resulting in impaired CART trafficking and suppressed proliferation and activation within tumors [107].
The TME inhibits trafficking and CART by producing suppressive soluble factors and overexpressing negative immune checkpoints. Strategies to deplete immunosuppressive elements of the TME, such as TAMs, to negate these effects are being studies. TAMs and PD-L1 expression reduce CART expansion and result in increased exhaustion, according to multiple studies [90,92]. PF-04136309, a small-molecule inhibitor of the CCL2-CCR2 axis responsible for TAM recruitment, has been used in pancreatic adenocarcinoma with chemo to reduce TAM and Treg infiltration and increase CD4+ and CD8+ effector cell presence in the tumor stroma [202]. This agent has been proposed for use as part of novel conditioning regimens prior to CART to help reduce TAM-mediated suppression of CART expansion and function [202]. Blockage of colony-stimulating factor 1 receptor (CSF1R) signaling, involved in TAM recruitment, with pexidartinib (PLX3397), improved the efficacy of adoptive cell transfer in murine melanoma through inhibition of TAM recruitment and activation [203]. Folate receptor beta (FR) is highly expressed in M2-polarized protumoral TAMs. Anti-FR targeting CAR T-cells was utilized to condition melanoma and colon cancer prior to antigen-specific CART therapy and improved outcomes in CART-resistant murine models [204].
New CAR T-cell engineering strategies have also been used to boost tumor infiltration and anti-cancer activity in the hostile TME [205,206]. A CCL19/IL7 secreting anti-mesothelin CART product was used increase CART and non-CAR T-cell infiltration into the TME, resulting in growth inhibition of xenografted pancreatic cancer. An IL-15/IL-21 secreting CAR targeting GPC3 in HCC was used and found to help maintain TCF1 expression (critical for T-cell development, proliferation and memory formation in CART). This resulted in robust proliferation and expansion as well as superior tumor control and survival in mice [207]. One promising method is the use of TGF-beta knockouts to engineer CARTs more resistant to immunosuppression. A TGF-beta 2 receptor knockout CART, developed using the CRISPR/Cas9 system, was found to reduce Treg-induced conversion, prevent CART exhaustion, improve in vivo elimination of tumor and improve CART memory subset formation to improve long-term efficacy [208]. One novel CART product was engineered to express CCR8 to improve homing to the site of the tumor, as well as a dominant negative TGF-beta 2 receptor to shield CART from TME-derived immunosuppression. This resulted in increased and more sustained infiltration of CARTs in xenografted cancer models [209]. Continued work developing these novel engineered CARTs into clinical-grade products is required to determine what strategies may provide the best outcomes and provide more insight into what is efficacious in solid tumors.
3.5. Neoantigens in Cancer Vaccination
Neoantigen vaccinations elicit a more powerful and specialized immune response than self-derived TAA vaccines [108]. Next-generation sequencing (NGS) of tumor DNA has led to customized clinical recombinant vaccines [210,211]. A patient’s HLA alleles might be used to identify a TAA or neoantigen for vaccine development and Sahin et al. demonstrated NGS-derived neoantigens elicit strong CD4+ and CD8+ responses [127,212,213]. New study domains include choosing and predicting how a vaccination strategy would function and utilizing aspects of the TME to assist these efforts.
Although early neoantigen vaccine trials have shown promising results, there remain failures and eliciting stronger antitumor responses is a continued goal. Coupling the binding properties of MHC-I with MHC-II has also presented challenges. A single MHC-I neoantigen is insufficient for antitumor immunity in murine models; thus, individualized vaccines should comprise neoepitopes expected to bind to MHC-I and MHC-II alleles [214,215]. MHC-II molecules have increased diversity and their open binding pockets make it difficult to predict a suitable binding motif [216]. Deep learning approaches that utilize artificial neural networks, inspired by biological neural networks, to predict ligand binding epitopes of MHC molecules are currently being investigated [217,218,219,220]. MARIA, a deep learning model, was used to analyze T-cell response data from a melanoma neoantigen vaccine study. This study demonstrated neoantigen candidates with high projected MARIA scores produced a CD4+ T-cell response post vaccination [221]. New AI-based applications and increased processing power help curb the complexity in predicting clinical outcomes and can begin closing knowledge gaps [215].
Although vaccines used as monotherapy show some potency, it is hypothesized that the vaccine-activated T cells are still suppressed by the TME. Combination approaches have been investigated clinically with current vaccines and also led to new insights in preclinical studies. Vaccines can reshape the TME, improving their effectiveness. Both combination and monotherapies used to enhance specific immune facets within the TME have shown potential to enable cytotoxic effects of vaccines in preclinical studies. Strategies, such as directly targeting TME vasculature (angiogenesis), targeting CAFs, and persistent cytokines, such as GM-CSF, IDO1, BAFF and other interleukins, with cancer vaccines have shown relatively consistent antitumoral effectiveness in preclinical models [222,223,224,225,226,227,228,229]. DNA vaccines utilizing novel nanobiomaterials include minimally invasive, injectable smart hydrogels [230]. These are scaffold-based cancer vaccines that allow for spatial and temporal control of antigen and other therapeutic agents and have shown strong antitumor effects via increased DC infiltration [231]. Targeting TAM infiltration, activity and polarization is also essential in the delivery of cancer vaccines. Combinatorial techniques have utilized biomimetic recombinant bacterial and viral vectors, nanoparticle and nanoemulsion technology for M1 agonists [232,233,234,235,236,237,238,239,240,241,242]. These have shown remarkable therapeutic efficacy through recruitment of lymphocytes, phenotypic transformation of macrophages from M2 to M1 and restoration of exhausted T lymphocytes, enhancing their ability to kill cancer cells in tumor models. With a deeper knowledge of bidirectional communication within the TME, future research will examine cancer vaccines that target immune pathways in the TME to boost vaccination efficacy. Neoantigen identification has developed greatly in the past decade, attributable to NGS, increasing computer capacity and better algorithms. Clinical studies that examine neoantigens as single or combinatorial immunotherapy targets are required to enhance this promising technology.
4. Conclusions and Future Directions
Over the last decade, cancer immunotherapy has markedly changed how we treat cancer patients. Immunotherapeutic modalities have found great success in a wide variety of settings and in patients with previously refractory disease. However, challenges remain. Further study of alternative checkpoint inhibitor pathways that allow for tumor escape and understanding the TME’s suppressive effect on ICI are critical areas of study required to develop more successful ICI therapies. CART has found good success in subsets of patients with hematologic malignancies but not for patients with a highly immunosuppressive TME nor with solid tumors. Targeting the immunosuppressive elements of the TME, including TAMs, has shown promise in improving both ICI and CART efficacy. Clinical studies utilizing these agents, either as pre-conditioning or in combination with ICI and CART, will be required to determine which approaches show the most promise. Targeting elements of alternative pathways with ICI has shown promise and will likely pave the way for new combination therapies. Novel engineering of CART to allow for improved CART trafficking and reduced immunosuppression within solid tumors may finally allow for improved outcomes to be obtained. Cancer vaccines have made great strides as well in recent years. In particular, studies utilizing neoantigen vaccines in combination with TME targeting have the potential to open up this exciting field.
Cancer immunotherapy is a modality that is rapidly becoming critical to the treatment of the majority of cancers. A continued focus on basic immuno-oncology research will drive our ability to develop novel therapies in the field and continue to drive successes on top of those we have achieved with cancer immunotherapy to date.
Author Contributions
Conceptualization: C.P., N.D. and Y.Y.; Writing—original draft: C.P. and N.D.; Writing—review and editing: C.P., N.D. and Y.Y.; Visualization: C.P.; Supervision: N.D. and Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by NIH grants CA193167, HL151195 and CA260858.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gonzales Carazas, M.M.; Pinto, J.A.; Casado, F.L. Biological bases of cancer immunotherapy. Expert Rev. Mol. Med. 2021, 23, e3. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Beckermann, K.E.; Dudzinski, S.O.; Rathmell, J.C. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. 2017, 35, 7–14. [Google Scholar] [CrossRef]
- Wang, S.; Xie, K.; Liu, T. Cancer Immunotherapies: From Efficacy to Resistance Mechanisms—Not Only Checkpoint Matters. Front. Immunol. 2021, 12, 690112. [Google Scholar] [CrossRef]
- Lagos, G.G.; Izar, B.; Rizvi, N.A. Beyond Tumor PD-L1: Emerging Genomic Biomarkers for Checkpoint Inhibitor Immunotherapy. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e47–e57. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, Q.; Xiang, Y.; Gou, X.; Li, W. Role of the tumor immune microenvironment in tumor immunotherapy (Review). Oncol. Lett. 2021, 23, 53. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Dobosz, P.; Dzieciatkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef]
- Hou, T.Z.; Qureshi, O.S.; Wang, C.J.; Baker, J.; Young, S.P.; Walker, L.S.; Sansom, D.M. A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J. Immunol. 2015, 194, 2148–2159. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Lenz, H.J.; Van Cutsem, E.; Luisa Limon, M.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Motzer, R.J.; McDermott, D.F.; Escudier, B.; Burotto, M.; Choueiri, T.K.; Hammers, H.J.; Barthelemy, P.; Plimack, E.R.; Porta, C.; George, S.; et al. Conditional survival and long-term efficacy with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma. Cancer 2022, 128, 2085–2097. [Google Scholar] [CrossRef]
- Naimi, A.; Mohammed, R.N.; Raji, A.; Chupradit, S.; Yumashev, A.V.; Suksatan, W.; Shalaby, M.N.; Thangavelu, L.; Kamrava, S.; Shomali, N.; et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun. Signal 2022, 20, 44. [Google Scholar] [CrossRef]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III Randomized Clinical Trial Comparing Tremelimumab With Standard-of-Care Chemotherapy in Patients With Advanced Melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef]
- Kubli, S.P.; Berger, T.; Araujo, D.V.; Siu, L.L.; Mak, T.W. Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov. 2021, 20, 899–919. [Google Scholar] [CrossRef]
- Sharma, N.; Vacher, J.; Allison, J.P. TLR1/2 ligand enhances antitumor efficacy of CTLA-4 blockade by increasing intratumoral Treg depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 10453–10462. [Google Scholar] [CrossRef]
- Furness, A.J.; Vargas, F.A.; Peggs, K.S.; Quezada, S.A. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol. 2014, 35, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; Mcneil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Shin, D.S.; Zaretsky, J.; Frederiksen, J.; Cornish, A.; Avramis, E.; Seja, E.; Kivork, C.; Siebert, J.; Kaplan-Lefko, P.; et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol. Res. 2016, 4, 194–203. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Lee, H.J.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Pembrolizumab in relapsed or refractory Hodgkin lymphoma: 2-year follow-up of KEYNOTE-087. Blood 2019, 134, 1144–1153. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulieres, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.J.; Soria, A.; Machiels, J.P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Lebbe, C.; Mortier, L.; Brohl, A.S.; Fazio, N.; Grob, J.J.; Prinzi, N.; Hanna, G.J.; Hassel, J.C.; Kiecker, F.; et al. First-line avelumab in a cohort of 116 patients with metastatic Merkel cell carcinoma (JAVELIN Merkel 200): Primary and biomarker analyses of a phase II study. J. Immunother Cancer 2021, 9, e002646. [Google Scholar] [CrossRef]
- Apolo, A.B.; Infante, J.R.; Balmanoukian, A.; Patel, M.R.; Wang, D.; Kelly, K.; Mega, A.E.; Britten, C.D.; Ravaud, A.; Mita, A.C.; et al. Avelumab, an Anti-Programmed Death-Ligand 1 Antibody, In Patients With Refractory Metastatic Urothelial Carcinoma: Results From a Multicenter, Phase Ib Study. J. Clin. Oncol. 2017, 35, 2117–2124. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Fraune, C.; Rosebrock, J.; Simon, R.; Hube-Magg, C.; Makrypidi-Fraune, G.; Kluth, M.; Buscheck, F.; Hoflmayer, D.; Schmalfeldt, B.; Muller, V.; et al. High homogeneity of MMR deficiency in ovarian cancer. Gynecol. Oncol. 2020, 156, 669–675. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Boyiadzis, M.M.; Kirkwood, J.M.; Marshall, J.L.; Pritchard, C.C.; Azad, N.S.; Gulley, J.L. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J. Immunother. Cancer 2018, 6, 35. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair–Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Chiang, E.Y.; Mellman, I. TIGIT-CD226-PVR axis: Advancing immune checkpoint blockade for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004711. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): Primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef]
- Rodriguez-Abreu, D.; Johnson, M.L.; Hussein, M.A.; Cobo, M.; Patel, A.J.; Secen, N.M.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.-H.; et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J. Clin. Oncol. 2020, 38, 9503. [Google Scholar] [CrossRef]
- Niu, J.; Maurice-Dror, C.; Lee, D.H.; Kim, D.W.; Nagrial, A.; Voskoboynik, M.; Chung, H.C.; Mileham, K.; Vaishampayan, U.; Rasco, D.; et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer(☆). Ann. Oncol. 2022, 33, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Mettu, N.B.; Ulahannan, S.V.; Bendell, J.C.; Garrido-Laguna, I.; Strickler, J.H.; Moore, K.N.; Stagg, R.; Kapoun, A.M.; Faoro, L.; Sharma, S. A Phase 1a/b Open-Label, Dose-Escalation Study of Etigilimab Alone or in Combination with Nivolumab in Patients with Locally Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2022, 28, 882–892. [Google Scholar] [CrossRef]
- Archilla-Ortega, A.; Domuro, C.; Martin-Liberal, J.; Munoz, P. Blockade of novel immune checkpoints and new therapeutic combinations to boost antitumor immunity. J. Exp. Clin. Cancer Res. 2022, 41, 62. [Google Scholar] [CrossRef]
- Yuan, L.; Tatineni, J.; Mahoney, K.M.; Freeman, G.J. VISTA: A Mediator of Quiescence and a Promising Target in Cancer Immunotherapy. Trends Immunol. 2021, 42, 209–227. [Google Scholar] [CrossRef]
- Johnston, R.J.; Su, L.J.; Pinckney, J.; Critton, D.; Boyer, E.; Krishnakumar, A.; Corbett, M.; Rankin, A.L.; Dibella, R.; Campbell, L.; et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019, 574, 565–570. [Google Scholar] [CrossRef]
- Rezaei, M.; Tan, J.; Zeng, C.; Li, Y.; Ganjalikhani-Hakemi, M. TIM-3 in Leukemia; Immune Response and Beyond. Front. Oncol. 2021, 11, 753677. [Google Scholar] [CrossRef]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef]
- van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Martin, P.; Ostrow, K.; Barrientos, J.; Chadburn, A.; Furman, R.; Ruan, J.; Shore, T.; Schuster, M.; Cerchietti, L.; et al. Response to second-line therapy defines the potential for cure in patients with recurrent diffuse large B-cell lymphoma: Implications for the development of novel therapeutic strategies. Clin. Lymphoma Myeloma Leuk. 2010, 10, 192–196. [Google Scholar] [CrossRef]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef]
- Hamadani, M.; Hari, P.N.; Zhang, Y.; Carreras, J.; Akpek, G.; Aljurf, M.D.; Ayala, E.; Bachanova, V.; Chen, A.I.; Chen, Y.B.; et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol. Blood Marrow. Transpl. 2014, 20, 1729–1736. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Schuster, S.J.; Tam, C.S.; Borchmann, P.; Worel, N.; McGuirk, J.P.; Holte, H.; Waller, E.K.; Jaglowski, S.; Bishop, M.R.; Damon, L.E.; et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 1403–1415. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Jacobsen, E.D.; Miklos, D.B.; Lekakis, L.J.; Braunschweig, I.; Oluwole, O.O.; Lin, Y.; Siddiqi, T.; et al. Durability of response in ZUMA-1, the pivotal phase 2 study of axicabtagene ciloleucel (Axi-Cel) in patients (Pts) with refractory large B-cell lymphoma. J. Clin. Oncol. 2018, 36, 3003. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- NCCN. NCCN Clinical Practice Guidelines in Oncology—B-Cell Lymphomas Version 5. 2021—21 September 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/b-cell.pdf (accessed on 10 March 2021).
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Costa, L.J.; Lin, Y.; Cornell, R.F.; Martin, T.; Chhabra, S.; Usmani, S.Z.; Jagannath, S.; Callander, N.S.; Berdeja, J.G.; Kang, Y.; et al. Comparison of Cilta-cel, an Anti-BCMA CAR-T Cell Therapy, Versus Conventional Treatment in Patients With Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2022, 22, 326–335. [Google Scholar] [CrossRef]
- Hunter, B.D.; Rogalski, M.; Jacobson, C.A. Chimeric antigen receptor T-cell therapy for the treatment of aggressive B-cell non-Hodgkin lymphomas: Efficacy, toxicity, and comparative chimeric antigen receptor products. Expert Opin. Biol. 2019, 19, 1157–1164. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef]
- Maude, S.L.; Pulsipher, M.A.; Boyer, M.W.; Grupp, S.A.; Davies, S.M.; Phillips, C.L.; Verneris, M.R.; August, K.J.; Schlis, K.; Driscoll, T.A.; et al. Efficacy and Safety of CTL019 in the First US Phase II Multicenter Trial in Pediatric Relapsed/Refractory Acute Lymphoblastic Leukemia: Results of an Interim Analysis. Blood 2016, 128, 2801. [Google Scholar] [CrossRef]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef]
- Baird, J.H.; Frank, M.J.; Craig, J.; Patel, S.; Spiegel, J.Y.; Sahaf, B.; Oak, J.S.; Younes, S.F.; Ozawa, M.G.; Yang, E.; et al. CD22-directed CAR T-cell therapy induces complete remissions in CD19-directed CAR-refractory large B-cell lymphoma. Blood 2021, 137, 2321–2325. [Google Scholar] [CrossRef]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: A phase 1 dose escalation and expansion trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef]
- Schneider, D.; Xiong, Y.; Wu, D.; Hu, P.; Alabanza, L.; Steimle, B.; Mahmud, H.; Anthony-Gonda, K.; Krueger, W.; Zhu, Z.; et al. Trispecific CD19-CD20-CD22-targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Hunter, B.D.; Redd, R.; Rodig, S.J.; Chen, P.H.; Wright, K.; Lipschitz, M.; Ritz, J.; Kamihara, Y.; Armand, P.; et al. Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity. J. Clin. Oncol. 2020, 38, 3095–3106. [Google Scholar] [CrossRef] [PubMed]
- Jackson, Z.; Roe, A.; Sharma, A.A.; Lopes, F.B.T.P.; Talla, A.; Kleinsorge-Block, S.; Zamborsky, K.; Schiavone, J.; Manjappa, S.; Schauner, R.; et al. Automated Manufacture of Autologous CD19 CAR-T Cells for Treatment of Non-hodgkin Lymphoma. Front. Immunol. 2020, 11, 1941. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Zurko, J.C.; Schneider, D.; Yim, S.; Hamadani, M.; Fenske, T.S.; Johnson, B.; Hari, P. Phase 1/2 Trial of IL7/IL15-Expanded Bispecific LV20.19 CAR T-Cells for Relapsed, Refractory B-Cell Non-Hodgkin Lymphoma. Blood 2021, 138, 95. [Google Scholar] [CrossRef]
- Flinn, I.W.; Jaeger, U.; Shah, N.N.; Blaise, D.; Briones, J.; Shune, L.; Boissel, N.; Bondanza, A.; Lu, D.; Zhu, X.; et al. A First-in-Human Study of YTB323, a Novel, Autologous CD19-Directed CAR-T Cell Therapy Manufactured Using the Novel T-Charge TM platform, for the Treatment of Patients (Pts) with Relapsed/Refractory (r/r) Diffuse Large B-Cell Lymphoma (DLBCL). Blood 2021, 138, 740. [Google Scholar] [CrossRef]
- Engels, B.; Zhu, X.; Yang, J.; Price, A.; Sohoni, A.; Stein, A.M.; Parent, L.; Greene, M.; Niederst, M.; Whalen, J.; et al. Preservation of T-Cell Stemness with a Novel Expansionless CAR-T Manufacturing Process, Which Reduces Manufacturing Time to Less Than Two Days, Drives Enhanced CAR-T Cell Efficacy. Blood 2021, 138, 2848. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Melief, J.; Pico de Coana, Y.; Wickström, S.L.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T cell dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef]
- Locke, F.L.; Rossi, J.M.; Neelapu, S.S.; Jacobson, C.A.; Miklos, D.B.; Ghobadi, A.; Oluwole, O.O.; Reagan, P.M.; Lekakis, L.J.; Lin, Y.; et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020, 4, 4898–4911. [Google Scholar] [CrossRef]
- Vercellino, L.; Di Blasi, R.; Kanoun, S.; Tessoulin, B.; Rossi, C.; D’Aveni-Piney, M.; Oberic, L.; Bodet-Milin, C.; Bories, P.; Olivier, P.; et al. Predictive factors of early progression after CAR T-cell therapy in relapsed/refractory diffuse large B-cell lymphoma. Blood Adv. 2020, 4, 5607–5615. [Google Scholar] [CrossRef]
- Jain, M.D.; Zhao, H.; Wang, X.; Atkins, R.; Menges, M.; Reid, K.; Spitler, K.; Faramand, R.; Bachmeier, C.; Dean, E.A.; et al. Tumor interferon signaling and suppressive myeloid cells are associated with CAR T-cell failure in large B-cell lymphoma. Blood 2021, 137, 2621–2633. [Google Scholar] [CrossRef]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef]
- Gauthier, J.; Bezerra, E.D.; Hirayama, A.V.; Fiorenza, S.; Sheih, A.; Chou, C.K.; Kimble, E.L.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell malignancies. Blood 2021, 137, 323–335. [Google Scholar] [CrossRef]
- Fang, P.Q.; Gunther, J.R.; Wu, S.Y.; Dabaja, B.S.; Nastoupil, L.J.; Ahmed, S.; Neelapu, S.S.; Pinnix, C.C. Radiation and CAR T-cell Therapy in Lymphoma: Future Frontiers and Potential Opportunities for Synergy. Front. Oncol. 2021, 11, 648655. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef]
- Ruella, M.; Kenderian, S.S.; Shestova, O.; Klichinsky, M.; Melenhorst, J.J.; Wasik, M.A.; Lacey, S.F.; June, C.H.; Gill, S. Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia 2017, 31, 246–248. [Google Scholar] [CrossRef]
- Zou, Y.X.; Zhu, H.Y.; Li, X.T.; Xia, Y.; Miao, K.R.; Zhao, S.S.; Wu, Y.J.; Wang, L.; Xu, W.; Li, J.Y. The impacts of zanubrutinib on immune cells in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Hematol. Oncol. 2019, 37, 392–400. [Google Scholar] [CrossRef]
- Kittai, A.S.; Bond, D.A.; William, B.; Saad, A.; Penza, S.; Efebera, Y.; Larkin, K.; Wall, S.A.; Choe, H.K.; Bhatnagar, B.; et al. Clinical activity of axicabtagene ciloleucel in adult patients with Richter syndrome. Blood Adv. 2020, 4, 4648–4652. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Westin, J.R.; Miklos, D.B.; Herrera, A.F.; Lee, J.; Seng, J.; Rossi, J.M.; Sun, J.; Dong, J.; Roberts, Z.J.; et al. Abstract CT055: Phase 1/2 primary analysis of ZUMA-6: Axicabtagene ciloleucel (Axi-Cel) in combination With atezolizumab (Atezo) for the treatment of patients (Pts) with refractory diffuse large B cell lymphoma (DLBCL). Cancer Res. 2020, 80, CT055. [Google Scholar] [CrossRef]
- Sigmund, A.M.; Denlinger, N.; Bajwa, A.; Elder, P.; Bond, D.A.; Brammer, J.E.; Saad, A.; Penza, S.; de Lima, M.J.G.; Jaglowski, S.; et al. Outcomes of Large B-Cell Lymphoma Patients By Post CAR-T Salvage Regimen at a Single Institution. Blood 2021, 138, 3851. [Google Scholar] [CrossRef]
- Alarcon Tomas, A.; Fein, J.A.; Fried, S.; Fingrut, W.; Anagnostou, T.; Alperovich, A.; Shah, N.; Fraint, E.; Lin, R.J.; Scordo, M.; et al. Novel Agents May be Preferable to Chemotherapy for Large B-Cell Lymphoma Progressing after CD19-CAR-T: A Multicenter Observational Study. Blood 2021, 138, 883. [Google Scholar] [CrossRef]
- Chong, E.A.; Alanio, C.; Svoboda, J.; Nasta, S.D.; Landsburg, D.J.; Lacey, S.F.; Ruella, M.; Bhattacharyya, S.; Wherry, E.J.; Schuster, S.J. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood 2022, 139, 1026–1038. [Google Scholar] [CrossRef]
- Jackson, Z.; Hong, C.; Schauner, R.; Dropulic, B.; Caimi, P.F.; de Lima, M.; Giraudo, M.F.; Gupta, K.; Reese, J.S.; Hwang, T.H.; et al. Sequential single cell transcriptional and protein marker profiling reveals TIGIT as a marker of CD19 CAR-T cell dysfunction in patients with non-Hodgkin’s lymphoma. Cancer Discov. 2022, 138, 164. [Google Scholar] [CrossRef]
- Otahal, P.; Prukova, D.; Kral, V.; Fabry, M.; Vockova, P.; Lateckova, L.; Trneny, M.; Klener, P. Lenalidomide enhances antitumor functions of chimeric antigen receptor modified T cells. Oncoimmunology 2016, 5, e1115940. [Google Scholar] [CrossRef] [PubMed]
- The Lancet, O. CAR T-cell therapy for solid tumours. Lancet. Oncol. 2021, 22, 893. [Google Scholar] [CrossRef]
- Murad, J.P.; Tilakawardane, D.; Park, A.K.; Lopez, L.S.; Young, C.A.; Gibson, J.; Yamaguchi, Y.; Lee, H.J.; Kennewick, K.T.; Gittins, B.J.; et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol. Ther. 2021, 29, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Shetty, K.; Ott, P.A. Personal Neoantigen Vaccines for the Treatment of Cancer. Annu. Rev. Cancer Biol. 2021, 5, 259–276. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Wei, X.X.; Perry, J.; Chang, E.; Zhang, L.; Hiatt, R.A.; Ryan, C.J.; Small, E.J.; Fong, L. Clinical Variables Associated With Overall Survival in Metastatic Castration-Resistant Prostate Cancer Patients Treated with Sipuleucel-T Immunotherapy. Clin. Genitourin. Cancer 2018, 16, 184–190.e2. [Google Scholar] [CrossRef]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef]
- Vreeland, T.J.; Clifton, G.T.; Hale, D.F.; Chick, R.C.; Hickerson, A.T.; Cindass, J.L.; Adams, A.M.; Bohan, P.M.K.; Andtbacka, R.H.I.; Berger, A.C.; et al. A Phase IIb Randomized Controlled Trial of the TLPLDC Vaccine as Adjuvant Therapy After Surgical Resection of Stage III/IV Melanoma: A Primary Analysis. Ann. Surg. Oncol. 2021, 28, 6126–6137. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Truxova, I.; Hensler, M.; Skapa, P.; Halaska, M.J.; Laco, J.; Ryska, A.; Spisek, R.; Fucikova, J. Rationale for the Combination of Dendritic Cell-Based Vaccination Approaches With Chemotherapy Agents. Int. Rev. Cell Mol. Biol. 2017, 330, 115–156. [Google Scholar] [CrossRef]
- Eisendle, K.; Weinlich, G.; Ebner, S.; Forstner, M.; Reider, D.; Zelle-Rieser, C.; Tripp, C.H.; Fritsch, P.; Stoitzner, P.; Romani, N.; et al. Combining chemotherapy and autologous peptide-pulsed dendritic cells provides survival benefit in stage IV melanoma patients. J. Dtsch. Derm. Ges. 2020, 18, 1270–1277. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Hrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef]
- Crosby, E.J.; Acharya, C.R.; Haddad, A.F.; Rabiola, C.A.; Lei, G.; Wei, J.P.; Yang, X.Y.; Wang, T.; Liu, C.X.; Wagner, K.U.; et al. Stimulation of Oncogene-Specific Tumor-Infiltrating T Cells through Combined Vaccine and alphaPD-1 Enable Sustained Antitumor Responses against Established HER2 Breast Cancer. Clin. Cancer Res. 2020, 26, 4670–4681. [Google Scholar] [CrossRef]
- Boorjian, S.A.; Alemozaffar, M.; Konety, B.R.; Shore, N.D.; Gomella, L.G.; Kamat, A.M.; Bivalacqua, T.J.; Montgomery, J.S.; Lerner, S.P.; Busby, J.E.; et al. Intravesical nadofaragene firadenovec gene therapy for BCG-unresponsive non-muscle-invasive bladder cancer: A single-arm, open-label, repeat-dose clinical trial. Lancet Oncol. 2021, 22, 107–117. [Google Scholar] [CrossRef]
- Spira, A.; Hansen, A.R.; Harb, W.A.; Curtis, K.K.; Koga-Yamakawa, E.; Origuchi, M.; Li, Z.; Ertik, B.; Shaib, W.L. Multicenter, Open-Label, Phase I Study of DSP-7888 Dosing Emulsion in Patients with Advanced Malignancies. Target Oncol. 2021, 16, 461–469. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Hu, Z.; Leet, D.E.; Allesoe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27, 515–525. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Warren, R.L.; Gibb, E.A.; Martin, S.D.; Spinelli, J.J.; Nelson, B.H.; Holt, R.A. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014, 24, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef]
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Uryvaev, A.; Passhak, M.; Hershkovits, D.; Sabo, E.; Bar-Sela, G. The role of tumor-infiltrating lymphocytes (TILs) as a predictive biomarker of response to anti-PD1 therapy in patients with metastatic non-small cell lung cancer or metastatic melanoma. Med. Oncol. 2018, 35, 25. [Google Scholar] [CrossRef]
- Zhou, R.; Zhang, J.; Zeng, D.; Sun, H.; Rong, X.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Immune cell infiltration as a biomarker for the diagnosis and prognosis of stage I–III colon cancer. Cancer Immunol. Immunother. 2019, 68, 433–442. [Google Scholar] [CrossRef]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T Cell–Inflamed versus Non-T Cell–Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef]
- Cui, X.; Jia, H.; Xin, H.; Zhang, L.; Chen, S.; Xia, S.; Li, X.; Xu, W.; Chen, X.; Feng, Y.; et al. A Novel Bispecific Antibody Targeting PD-L1 and VEGF With Combined Anti-Tumor Activities. Front. Immunol. 2021, 12, 778978. [Google Scholar] [CrossRef]
- Noman, M.Z.; Parpal, S.; Van Moer, K.; Xiao, M.; Yu, Y.; Viklund, J.; De Milito, A.; Hasmim, M.; Andersson, M.; Amaravadi, R.K.; et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Greshock, J.; Colligon, T.A.; Wang, Y.; Ward, R.; Katsaros, D.; Lassus, H.; Butzow, R.; Godwin, A.K.; et al. Frequent genetic abnormalities of the PI3K/AKT pathway in primary ovarian cancer predict patient outcome. Genes Chromosomes Cancer 2011, 50, 606–618. [Google Scholar] [CrossRef]
- Sai, J.; Owens, P.; Novitskiy, S.V.; Hawkins, O.E.; Vilgelm, A.E.; Yang, J.; Sobolik, T.; Lavender, N.; Johnson, A.C.; McClain, C.; et al. PI3K Inhibition Reduces Mammary Tumor Growth and Facilitates Antitumor Immunity and Anti-PD1 Responses. Clin. Cancer Res. 2017, 23, 3371–3384. [Google Scholar] [CrossRef]
- Borcoman, E.; De La Rochere, P.; Richer, W.; Vacher, S.; Chemlali, W.; Krucker, C.; Sirab, N.; Radvanyi, F.; Allory, Y.; Pignot, G.; et al. Inhibition of PI3K pathway increases immune infiltrate in muscle-invasive bladder cancer. Oncoimmunology 2019, 8, e1581556. [Google Scholar] [CrossRef]
- Carnevalli, L.S.; Sinclair, C.; Taylor, M.A.; Gutierrez, P.M.; Langdon, S.; Coenen-Stass, A.M.L.; Mooney, L.; Hughes, A.; Jarvis, L.; Staniszewska, A.; et al. PI3Kalpha/delta inhibition promotes anti-tumor immunity through direct enhancement of effector CD8(+) T-cell activity. J. Immunother. Cancer 2018, 6, 158. [Google Scholar] [CrossRef]
- Abu-Eid, R.; Samara, R.N.; Ozbun, L.; Abdalla, M.Y.; Berzofsky, J.A.; Friedman, K.M.; Mkrtichyan, M.; Khleif, S.N. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol. Res. 2014, 2, 1080–1089. [Google Scholar] [CrossRef]
- Leary, A.; Tan, D.; Ledermann, J. Immune checkpoint inhibitors in ovarian cancer: Where do we stand? Ther. Adv. Med. Oncol. 2021, 13, 17588359211039899. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Gaggar, S.; Gogenur, I. Cancer-Associated Fibroblasts and Tumor-Associated Macrophages in Cancer and Cancer Immunotherapy. Front. Oncol. 2021, 11, 668731. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Ma, T.; Renz, B.W.; Ilmer, M.; Koch, D.; Yang, Y.; Werner, J.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Solid Tumors. Cells 2022, 11, 310. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Gaggar, S.; Gogenur, I. Neutrophils and polymorphonuclear myeloid-derived suppressor cells: An emerging battleground in cancer therapy. Oncogenesis 2022, 11, 22. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef]
- Ai, L.; Mu, S.; Wang, Y.; Wang, H.; Cai, L.; Li, W.; Hu, Y. Prognostic role of myeloid-derived suppressor cells in cancers: A systematic review and meta-analysis. BMC Cancer 2018, 18, 1220. [Google Scholar] [CrossRef]
- Wang, P.F.; Song, S.Y.; Wang, T.J.; Ji, W.J.; Li, S.W.; Liu, N.; Yan, C.X. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology 2018, 7, e1494113. [Google Scholar] [CrossRef]
- Zhou, S.L.; Yin, D.; Hu, Z.Q.; Luo, C.B.; Zhou, Z.J.; Xin, H.Y.; Yang, X.R.; Shi, Y.H.; Wang, Z.; Huang, X.W.; et al. A Positive Feedback Loop Between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology 2019, 70, 1214–1230. [Google Scholar] [CrossRef]
- Manfroi, B.; Moreaux, J.; Righini, C.; Ghiringhelli, F.; Sturm, N.; Huard, B. Tumor-associated neutrophils correlate with poor prognosis in diffuse large B-cell lymphoma patients. Blood Cancer J. 2018, 8, 66. [Google Scholar] [CrossRef]
- Hubert, M.; Gobbini, E.; Bendriss-Vermare, N.; Caux, C.; Valladeau-Guilemond, J. Human Tumor-Infiltrating Dendritic Cells: From in Situ Visualization to High-Dimensional Analyses. Cancers 2019, 11, 1082. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Mahari, A.; Salahlou, R.; Khalili, M.; Azizi, M.; Sadeghzadeh, H. Immune evader cancer stem cells direct the perspective approaches to cancer immunotherapy. Stem. Cell Res. 2022, 13, 150. [Google Scholar] [CrossRef]
- Zhong, M.; Zhong, C.; Cui, W.; Wang, G.; Zheng, G.; Li, L.; Zhang, J.; Ren, R.; Gao, H.; Wang, T.; et al. Induction of tolerogenic dendritic cells by activated TGF-beta/Akt/Smad2 signaling in RIG-I-deficient stemness-high human liver cancer cells. BMC Cancer 2019, 19, 439. [Google Scholar] [CrossRef]
- Murphy, T.L.; Murphy, K.M. Dendritic cells in cancer immunology. Cell Mol. Immunol. 2022, 19, 3–13. [Google Scholar] [CrossRef]
- Ghorbaninezhad, F.; Asadzadeh, Z.; Masoumi, J.; Mokhtarzadeh, A.; Kazemi, T.; Aghebati-Maleki, L.; Shotorbani, S.S.; Shadbad, M.A.; Baghbanzadeh, A.; Hemmat, N.; et al. Dendritic cell-based cancer immunotherapy in the era of immune checkpoint inhibitors: From bench to bedside. Life Sci. 2022, 297, 120466. [Google Scholar] [CrossRef]
- Kvedaraite, E.; Ginhoux, F. Human dendritic cells in cancer. Sci. Immunol. 2022, 7, eabm9409. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Zhang, Z.; Liu, S.; Ow, A.; Ruan, M.; Yang, W.; Zhang, C. Increased tumor-infiltrating plasmacytoid dendritic cells predicts poor prognosis in oral squamous cell carcinoma. Arch. Oral Biol. 2017, 78, 129–134. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Moller, H.J.; Donskov, F.; Hoyer, M.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer 2012, 118, 2476–2485. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Petty, A.J.; Li, A.; Wang, X.; Dai, R.; Heyman, B.; Hsu, D.; Huang, X.; Yang, Y. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J. Clin. Investig. 2019, 129, 5151–5162. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef]
- Petty, A.J.; Dai, R.; Lapalombella, R.; Baiocchi, R.A.; Benson, D.M.; Li, Z.; Huang, X.; Yang, Y. Hedgehog-induced PD-L1 on tumor-associated macrophages is critical for suppression of tumor-infiltrating CD8+ T cell function. JCI Insight 2021, 6, e146707. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target Ther. 2021, 6, 75. [Google Scholar] [CrossRef]
- Zhou, J.; Dou, C.; Liu, C.; Liu, Y.; Yang, J.; Duan, H.; Yang, C.; Huang, Z.; Wang, H.; Liao, D.; et al. M2 subtype tumor associated macrophages (M2-TAMs) infiltration predicts poor response rate of immune checkpoint inhibitors treatment for prostate cancer. Ann. Med. 2021, 53, 730–740. [Google Scholar] [CrossRef]
- Chen, S.M.Y.; Popolizio, V.; Woolaver, R.A.; Ge, H.; Krinsky, A.L.; John, J.; Danis, E.; Ke, Y.; Kramer, Y.; Bian, L.; et al. Differential responses to immune checkpoint inhibitor dictated by pre-existing differential immune profiles in squamous cell carcinomas caused by same initial oncogenic drivers. J. Exp. Clin. Cancer Res. 2022, 41, 123. [Google Scholar] [CrossRef]
- Belgiovine, C.; Frapolli, R.; Liguori, M.; Digifico, E.; Colombo, F.S.; Meroni, M.; Allavena, P.; D’Incalci, M. Inhibition of tumor-associated macrophages by trabectedin improves the antitumor adaptive immunity in response to anti-PD-1 therapy. Eur. J. Immunol. 2021, 51, 2677–2686. [Google Scholar] [CrossRef]
- Luo, Q.; Zheng, N.; Jiang, L.; Wang, T.; Zhang, P.; Liu, Y.; Zheng, P.; Wang, W.; Xie, G.; Chen, L.; et al. Lipid accumulation in macrophages confers protumorigenic polarization and immunity in gastric cancer. Cancer Sci. 2020, 111, 4000–4011. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Khong, H.; Dai, Z.; Huang, X.F.; Wargo, J.A.; Cooper, Z.A.; Vasilakos, J.P.; Hwu, P.; Overwijk, W.W. Effective innate and adaptive antimelanoma immunity through localized TLR7/8 activation. J. Immunol. 2014, 193, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Masemann, D.; Meissner, R.; Schied, T.; Lichty, B.D.; Rapp, U.R.; Wixler, V.; Ludwig, S. Synergistic anti-tumor efficacy of oncolytic influenza viruses and B7-H3 immune- checkpoint inhibitors against IC-resistant lung cancers. Oncoimmunology 2021, 10, 1885778. [Google Scholar] [CrossRef] [PubMed]
- Vafaei, S.; Zekiy, A.O.; Khanamir, R.A.; Zaman, B.A.; Ghayourvahdat, A.; Azimizonuzi, H.; Zamani, M. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int. 2022, 22, 2. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Shen, Y.; Qian, C.; Oupicky, D.; Sun, M. Targeting pulmonary tumor microenvironment with CXCR4-inhibiting nanocomplex to enhance anti-PD-L1 immunotherapy. Sci. Adv. 2020, 6, eaaz9240. [Google Scholar] [CrossRef]
- Zhou, M.; Luo, C.; Zhou, Z.; Li, L.; Huang, Y. Improving anti-PD-L1 therapy in triple negative breast cancer by polymer-enhanced immunogenic cell death and CXCR4 blockade. J. Control. Release 2021, 334, 248–262. [Google Scholar] [CrossRef]
- Lu, G.; Qiu, Y.; Su, X. Targeting CXCL12-CXCR4 Signaling Enhances Immune Checkpoint Blockade Therapy Against Triple Negative Breast Cancer. Eur. J. Pharm. Sci. 2021, 157, 105606. [Google Scholar] [CrossRef]
- Ramesh, A.; Malik, V.; Ranjani, H.A.; Smith, H.; Kulkarni, A.A. Rational combination of an immune checkpoint inhibitor with CSF1R inhibitor-loaded nanoparticle enhances anticancer efficacy. Drug Deliv. Transl. Res. 2021, 11, 2317–2327. [Google Scholar] [CrossRef]
- Tian, T.; Liang, R.; Erel-Akbaba, G.; Saad, L.; Obeid, P.J.; Gao, J.; Chiocca, E.A.; Weissleder, R.; Tannous, B.A. Immune Checkpoint Inhibition in GBM Primed with Radiation by Engineered Extracellular Vesicles. ACS Nano 2022, 16, 1940–1953. [Google Scholar] [CrossRef]
- Yan, C.; Yang, J.; Saleh, N.; Chen, S.C.; Ayers, G.D.; Abramson, V.G.; Mayer, I.A.; Richmond, A. Inhibition of the PI3K/mTOR Pathway in Breast Cancer to Enhance Response to Immune Checkpoint Inhibitors in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 5207. [Google Scholar] [CrossRef]
- Dutta, R.; Khalil, R.; Mayilsamy, K.; Green, R.; Howell, M.; Bharadwaj, S.; Mohapatra, S.S.; Mohapatra, S. Combination Therapy of Mithramycin A and Immune Checkpoint Inhibitor for the Treatment of Colorectal Cancer in an Orthotopic Murine Model. Front. Immunol. 2021, 12, 706133. [Google Scholar] [CrossRef]
- Habif, G.; Crinier, A.; Andre, P.; Vivier, E.; Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell Mol. Immunol. 2019, 16, 415–422. [Google Scholar] [CrossRef]
- Zhang, X.; Sabio, E.; Krishna, C.; Ma, X.; Wang, J.; Jiang, H.; Havel, J.J.; Chan, T.A. Qa-1(b) Modulates Resistance to Anti-PD-1 Immune Checkpoint Blockade in Tumors with Defects in Antigen Processing. Mol. Cancer Res. 2021, 19, 1076–1084. [Google Scholar] [CrossRef]
- van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Chen, W.; Qie, C.; Hu, X.; Wang, L.; Jiang, J.; Liu, W.; Liu, J. A small molecule inhibitor of VSIG-8 prevents its binding to VISTA. Investig. New Drugs 2022, 40, 690–699. [Google Scholar] [CrossRef]
- Sasikumar, P.G.; Sudarshan, N.S.; Adurthi, S.; Ramachandra, R.K.; Samiulla, D.S.; Lakshminarasimhan, A.; Ramanathan, A.; Chandrasekhar, T.; Dhudashiya, A.A.; Talapati, S.R.; et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun. Biol. 2021, 4, 699. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-aniel, dose-finding, non-randomised, phase 1b trial. Lancet. Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Mok, S.; Koya, R.C.; Tsui, C.; Xu, J.; Robert, L.; Wu, L.; Graeber, T.; West, B.L.; Bollag, G.; Ribas, A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef]
- Andrea, A.E.; Chiron, A.; Mallah, S.; Bessoles, S.; Sarrabayrouse, G.; Hacein-Bey-Abina, S. Advances in CAR-T Cell Genetic Engineering Strategies to Overcome Hurdles in Solid Tumors Treatment. Front. Immunol. 2022, 13, 830292. [Google Scholar] [CrossRef]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.F.; Bulsara, S.; et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef] [PubMed]
- Cadilha, B.L.; Benmebarek, M.R.; Dorman, K.; Oner, A.; Lorenzini, T.; Obeck, H.; Vanttinen, M.; Di Pilato, M.; Pruessmann, J.N.; Stoiber, S.; et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci. Adv. 2021, 7, eabi5781. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Muller, M.; Pak, H.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Lang, F.; Schrors, B.; Lower, M.; Tureci, O.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef]
- Nielsen, M.; Lund, O.; Buus, S.; Lundegaard, C. MHC class II epitope predictive algorithms. Immunology 2010, 130, 319–328. [Google Scholar] [CrossRef]
- Sarkizova, S.; Klaeger, S.; Le, P.M.; Li, L.W.; Oliveira, G.; Keshishian, H.; Hartigan, C.R.; Zhang, W.; Braun, D.A.; Ligon, K.L.; et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020, 38, 199–209. [Google Scholar] [CrossRef]
- Riley, T.P.; Keller, G.L.J.; Smith, A.R.; Davancaze, L.M.; Arbuiso, A.G.; Devlin, J.R.; Baker, B.M. Structure Based Prediction of Neoantigen Immunogenicity. Front. Immunol. 2019, 10, 2047. [Google Scholar] [CrossRef]
- Wu, J.; Wang, W.; Zhang, J.; Zhou, B.; Zhao, W.; Su, Z.; Gu, X.; Wu, J.; Zhou, Z.; Chen, S. DeepHLApan: A Deep Learning Approach for Neoantigen Prediction Considering Both HLA-Peptide Binding and Immunogenicity. Front. Immunol. 2019, 10, 2559. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2018, 37, 55–63. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Pierini, S.; Mishra, A.; Perales-Linares, R.; Uribe-Herranz, M.; Beghi, S.; Giglio, A.; Pustylnikov, S.; Costabile, F.; Rafail, S.; Amici, A.; et al. Combination of vasculature targeting, hypofractionated radiotherapy, and immune checkpoint inhibitor elicits potent antitumor immune response and blocks tumor progression. J. Immunother. Cancer 2021, 9, e001636. [Google Scholar] [CrossRef]
- Shu, C.; Sun, P.; Xie, H.; Huang, W.; Qi, J.; Ma, Y. Virus-Like Particles Presenting the FGF-2 Protein or Identified Antigenic Peptides Promoted Antitumor Immune Responses in Mice. Int. J. Nanomed. 2020, 15, 1983–1996. [Google Scholar] [CrossRef]
- Geng, F.; Guo, J.; Guo, Q.Q.; Xie, Y.; Dong, L.; Zhou, Y.; Liu, C.L.; Yu, B.; Wu, H.; Wu, J.X.; et al. A DNA vaccine expressing an optimized secreted FAPalpha induces enhanced anti-tumor activity by altering the tumor microenvironment in a murine model of breast cancer. Vaccine 2019, 37, 4382–4391. [Google Scholar] [CrossRef]
- Lemdani, K.; Mignet, N.; Boudy, V.; Seguin, J.; Oujagir, E.; Bawa, O.; Peschaud, F.; Emile, J.F.; Capron, C.; Malafosse, R. Local immunomodulation combined to radiofrequency ablation results in a complete cure of local and distant colorectal carcinoma. Oncoimmunology 2019, 8, 1550342. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Sutanto-Ward, E.; Kopp, K.L.; DuHadaway, J.; Mondal, A.; Ghaban, D.; Lecoq, I.; Zocca, M.B.; Merlo, L.M.F.; Mandik-Nayak, L.; et al. Peptide vaccination directed against IDO1-expressing immune cells elicits CD8(+) and CD4(+) T-cell-mediated antitumor immunity and enhanced anti-PD1 responses. J. Immunother. Cancer 2020, 8, e000605. [Google Scholar] [CrossRef] [PubMed]
- Shurin, M.R.; Ma, Y.; Keskinov, A.A.; Zhao, R.; Lokshin, A.; Agassandian, M.; Shurin, G.V. BAFF and APRIL from Activin A-Treated Dendritic Cells Upregulate the Antitumor Efficacy of Dendritic Cells In Vivo. Cancer Res. 2016, 76, 4959–4969. [Google Scholar] [CrossRef]
- Gordy, J.T.; Luo, K.; Francica, B.; Drake, C.; Markham, R.B. Anti-IL-10-mediated Enhancement of Antitumor Efficacy of a Dendritic Cell-targeting MIP3alpha-gp100 Vaccine in the B16F10 Mouse Melanoma Model Is Dependent on Type I Interferons. J. Immunother. 2018, 41, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.P.; Authie, P.; Karolina Palucka, A.; Zurawski, G. Targeting interferon-alpha to dendritic cells enhances a CD8(+) T cell response to a human CD40-targeted cancer vaccine. Vaccine 2017, 35, 4532–4539. [Google Scholar] [CrossRef]
- Jiang, J.; Mei, J.; Yi, S.; Feng, C.; Ma, Y.; Liu, Y.; Liu, Y.; Chen, C. Tumor associated macrophage and microbe: The potential targets of tumor vaccine delivery. Adv. Drug Deliv. Rev. 2022, 180, 114046. [Google Scholar] [CrossRef]
- Duong, H.T.T.; Thambi, T.; Yin, Y.; Kim, S.H.; Nguyen, T.L.; Phan, V.H.G.; Kim, J.; Jeong, J.H.; Lee, D.S. Degradation-regulated architecture of injectable smart hydrogels enhances humoral immune response and potentiates antitumor activity in human lung carcinoma. Biomaterials 2020, 230, 119599. [Google Scholar] [CrossRef]
- Jarosz-Biej, M.; Kaminska, N.; Matuszczak, S.; Cichon, T.; Pamula-Pilat, J.; Czapla, J.; Smolarczyk, R.; Skwarzynska, D.; Kulik, K.; Szala, S. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS ONE 2018, 13, e0191012. [Google Scholar] [CrossRef]
- Xu, G.; Feng, D.; Yao, Y.; Li, P.; Sun, H.; Yang, H.; Li, C.; Jiang, R.; Sun, B.; Chen, Y. Listeria-based hepatocellular carcinoma vaccine facilitates anti-PD-1 therapy by regulating macrophage polarization. Oncogene 2020, 39, 1429–1444. [Google Scholar] [CrossRef]
- Wang, C.; Steinmetz, N.F. CD47 Blockade and Cowpea Mosaic Virus Nanoparticle In Situ Vaccination Triggers Phagocytosis and Tumor Killing. Adv. Healthc. Mater 2019, 8, e1801288. [Google Scholar] [CrossRef]
- Shukla, S.; Wang, C.; Beiss, V.; Steinmetz, N.F. Antibody Response against Cowpea Mosaic Viral Nanoparticles Improves In Situ Vaccine Efficacy in Ovarian Cancer. ACS Nano 2020, 14, 2994–3003. [Google Scholar] [CrossRef]
- Inoue, H.; Iga, M.; Nabeta, H.; Yokoo, T.; Suehiro, Y.; Okano, S.; Inoue, M.; Kinoh, H.; Katagiri, T.; Takayama, K.; et al. Non-transmissible Sendai virus encoding granulocyte macrophage colony-stimulating factor is a novel and potent vector system for producing autologous tumor vaccines. Cancer Sci. 2008, 99, 2315–2326. [Google Scholar] [CrossRef]
- Sakamoto, C.; Kohara, H.; Inoue, H.; Narusawa, M.; Ogawa, Y.; Hirose-Yotsuya, L.; Miyamoto, S.; Matsumura, Y.; Yamada, K.; Takahashi, A.; et al. Therapeutic vaccination based on side population cells transduced by the granulocyte-macrophage colony-stimulating factor gene elicits potent antitumor immunity. Cancer Gene 2017, 24, 165–174. [Google Scholar] [CrossRef]
- Che, Y.; Yang, Y.; Suo, J.; An, Y.; Wang, X. Induction of systemic immune responses and reversion of immunosuppression in the tumor microenvironment by a therapeutic vaccine for cervical cancer. Cancer Immunol. Immunother. 2020, 69, 2651–2664. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, R.; Guo, M.; Zhou, H.; Zhao, Y.; Liu, Y.; Wu, Y.; Chen, C. Gd-metallofullerenol drug delivery system mediated macrophage polarization enhances the efficiency of chemotherapy. J. Control. Release 2020, 320, 293–303. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, S.; Kim, J.E.; Lee, S.N.; Shin, I.W.; Shin, H.S.; Jin, S.M.; Noh, Y.W.; Kang, Y.J.; Kim, Y.S.; et al. Lyophilizable and Multifaceted Toll-like Receptor 7/8 Agonist-Loaded Nanoemulsion for the Reprogramming of Tumor Microenvironments and Enhanced Cancer Immunotherapy. ACS Nano 2019, 13, 12671–12686. [Google Scholar] [CrossRef]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef]
- Kim, H.; Khanna, V.; Kucaba, T.A.; Zhang, W.; Ferguson, D.M.; Griffith, T.S.; Panyam, J. Combination of Sunitinib and PD-L1 Blockade Enhances Anticancer Efficacy of TLR7/8 Agonist-Based Nanovaccine. Mol. Pharm. 2019, 16, 1200–1210. [Google Scholar] [CrossRef]
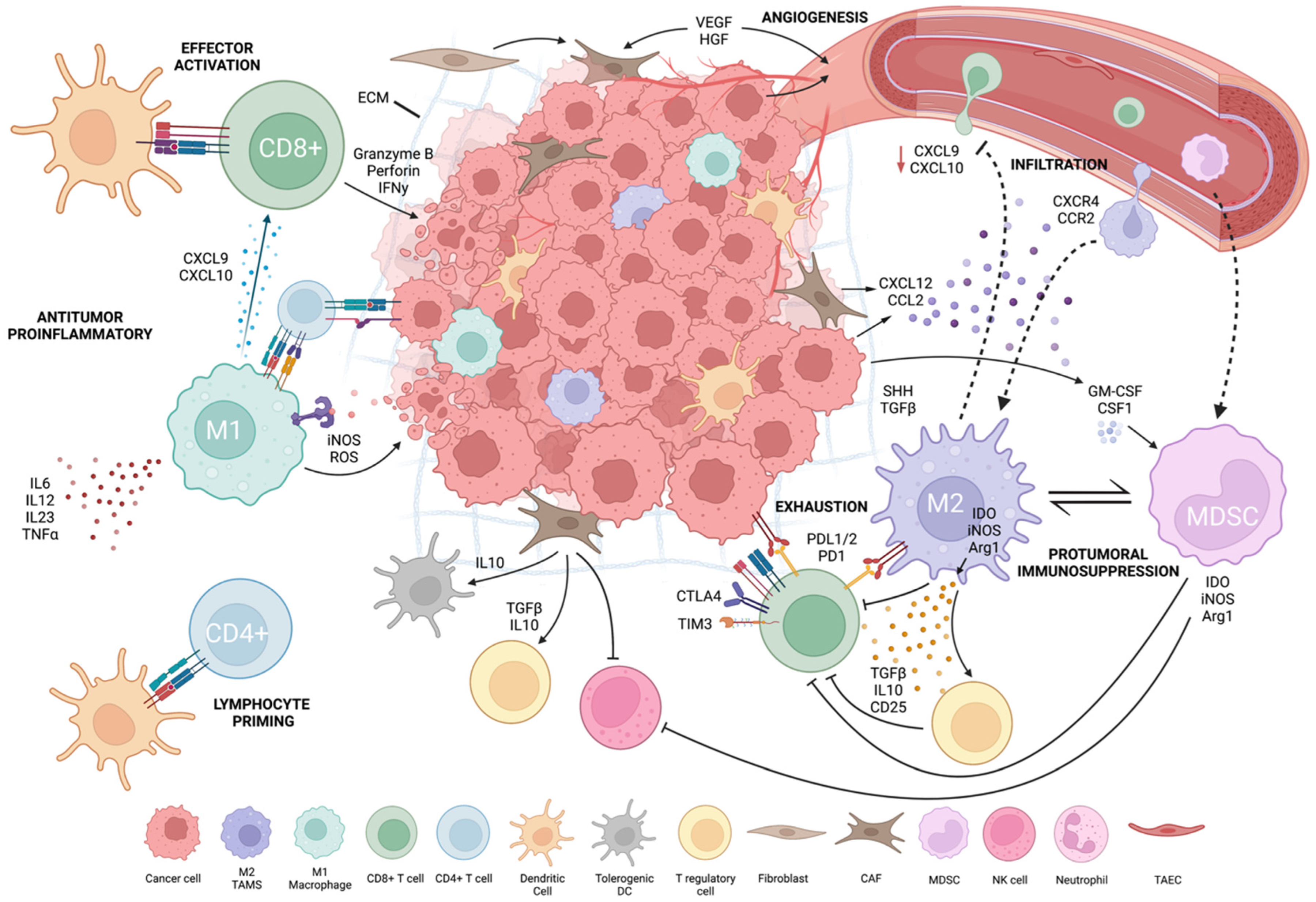
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).