
Local Breast Microbiota: A “New” Player on the Block

 ,
,  , ,
, , {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Human Microbiota and the Relation with the Host
3. Tumour Microenvironment
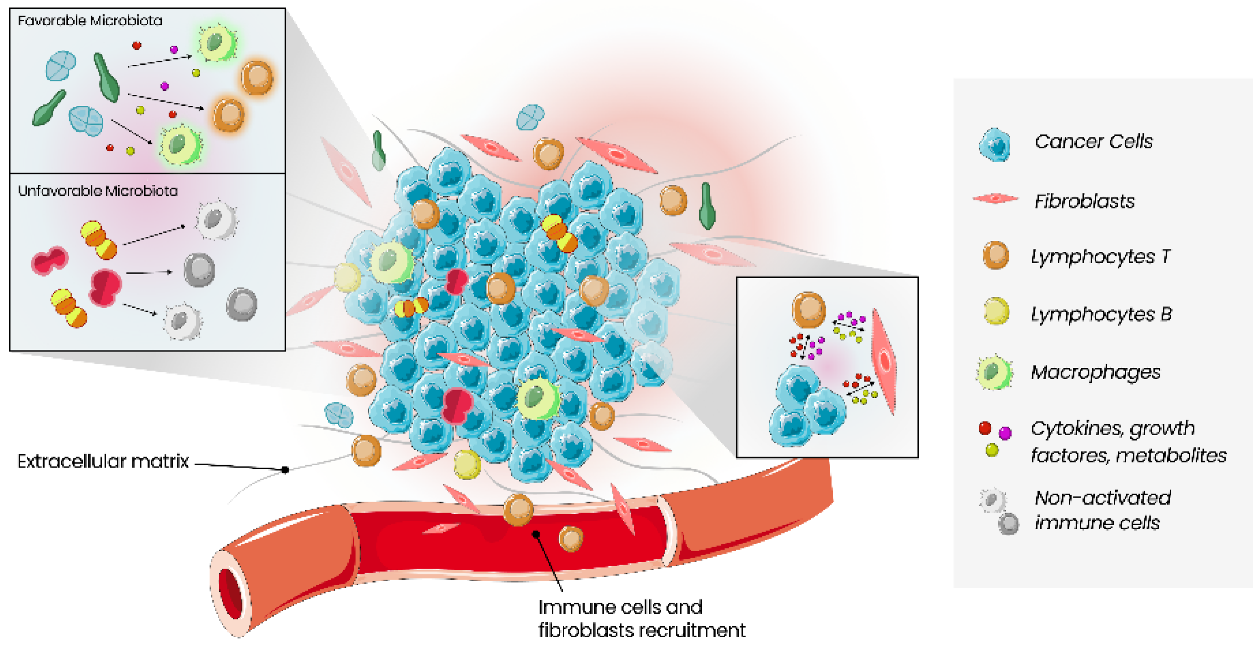
3.1. Tumour Immune Microenvironment
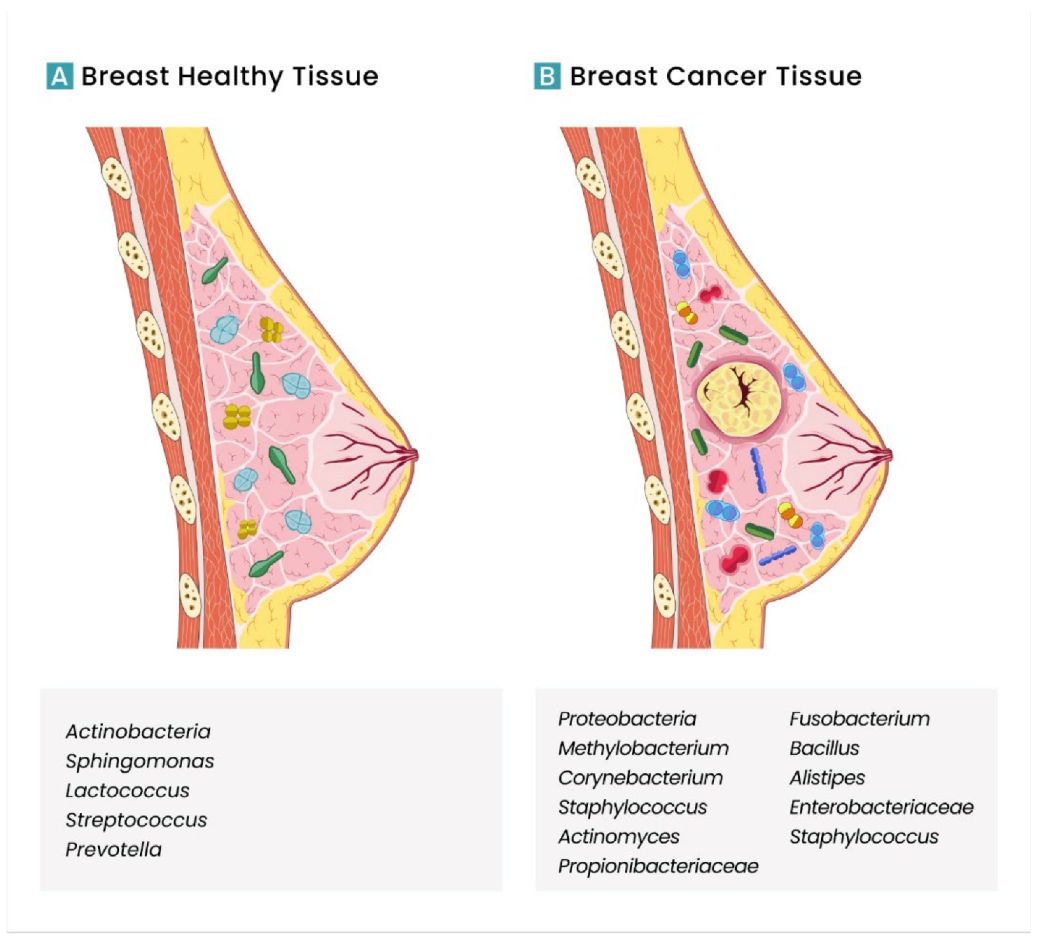
3.2. Tumour Microbe Microenvironment
4. Therapeutic Implications of the Tumour Microenvironment
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bagheri, Z.; Moeinzadeh, L.; Razmkhah, M. Roles of Microbiota in Cancer: From Tumor Development to Treatment. J. Oncol. 2022, 2022, 3845104. [Google Scholar] [CrossRef] [PubMed]
- Shui, L.; Yang, X.; Li, J.; Yi, C.; Sun, Q.; Zhu, H. Gut Microbiome as a Potential Factor for Modulating Resistance to Cancer Immunotherapy. Front. Immunol. 2020, 10, 2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552. [Google Scholar] [CrossRef] [PubMed]
- Doocey, C.M.; Finn, K.; Murphy, C.; Guinane, C.M. The impact of the human microbiome in tumorigenesis, cancer progression, and biotherapeutic development. BMC Microbiol. 2022, 22, 53. [Google Scholar] [CrossRef] [PubMed]
- Deepak, K.; Vempati, R.; Nagaraju, G.P.; Dasari, V.R.; Nagini, S.; Rao, D.; Malla, R.R. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 2020, 153, 104683. [Google Scholar] [CrossRef]
- Yang, X.; Guo, Y.; Chen, C.; Shao, B.; Zhao, L.; Zhou, Q.; Liu, J.; Wang, G.; Yuan, W.; Sun, Z. Interaction between intestinal microbiota and tumour immunity in the tumour microenvironment. Immunology 2021, 164, 476–493. [Google Scholar] [CrossRef]
- Qiu, Q.; Lin, Y.; Ma, Y.; Li, X.; Liang, J.; Chen, Z.; Liu, K.; Huang, Y.; Luo, H.; Huang, R.; et al. Exploring the Emerging Role of the Gut Microbiota and Tumor Microenvironment in Cancer Immunotherapy. Front. Immunol. 2021, 11, 612202. [Google Scholar] [CrossRef]
- Vitorino, M.; de Almeida, S.B.; Costa, D.A.; Faria, A.; Calhau, C.; Braga, S.A. Human Microbiota and Immunotherapy in Breast Cancer—A Review of Recent Developments. Front. Oncol. 2022, 11, 815772. [Google Scholar] [CrossRef]
- Costa, D.A.; Nobre, J.G.; Batista, M.V.; Ribeiro, C.; Calle, C.; Cortes, A.; Marhold, M.; Negreiros, I.; Borralho, P.; Brito, M.; et al. Human Microbiota and Breast Cancer—Is There Any Relevant Link?—A Literature Review and New Horizons Toward Personalised Medicine. Front. Microbiol. 2021, 12, 584332. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The Placenta Harbors a Unique Microbiome. Sci. Transl. Med. 2014, 6, 237ra65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mégraud, F. A Humble Bacterium Sweeps This Year’s Nobel Prize. Cell 2005, 123, 975–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Chervin, C.S.; Gajewski, T.F. Cancer and the Microbiome—Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology 2021, 160, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Sinha, R.; Pei, Z.; Dominianni, C.; Wu, J.; Shi, J.; Goedert, J.J.; Hayes, R.; Yang, L. Human Gut Microbiome and Risk for Colorectal Cancer. J. Natl. Cancer Inst. 2013, 105, 1907–1911. [Google Scholar] [CrossRef] [Green Version]
- Hibberd, A.A.; Lyra, A.; Ouwehand, A.C.; Rolny, P.; Lindegren, H.; Cedgård, L.; Wettergren, Y. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017, 4, e000145. [Google Scholar] [CrossRef] [Green Version]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Simeone, P.; Damato, M.; Maffia, M.; Lanuti, P.; Trerotola, M. The Cancer Microbiota: EMT and Inflammation as Shared Molecular Mechanisms Associated with Plasticity and Progression. J. Oncol. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rossi, T.; Vergara, D.; Fanini, F.; Maffia, M.; Bravaccini, S.; Pirini, F. Microbiota-Derived Metabolites in Tumor Progression and Metastasis. Int. J. Mol. Sci. 2020, 21, 5786. [Google Scholar] [CrossRef] [PubMed]
- Mangerich, A.; Knutson, C.G.; Parry, N.M.; Muthupalani, S.; Ye, W.; Prestwich, E.; Cui, L.; McFaline, J.L.; Mobley, M.; Ge, Z.; et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E1820–E1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, R.V.; Pearson, J.; Aitchison, A.; Dixon, L.; Frizelle, F.A.; Keenan, J.I. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS ONE 2017, 12, e0171602. [Google Scholar] [CrossRef] [Green Version]
- Gur, C.; Maalouf, N.; Gerhard, M.; Singer, B.B.; Emgård, J.; Temper, V.; Neuman, T.; Mandelboim, O.; Bachrach, G. The Helicobacter pylori HopQ outermembrane protein inhibits immune cell activities. OncoImmunology 2019, 8, e1553487. [Google Scholar] [CrossRef] [PubMed]
- Negi, S.; Das, D.K.; Pahari, S.; Nadeem, S.; Agrewala, J.N. Potential Role of Gut Microbiota in Induction and Regulation of Innate Immune Memory. Front. Immunol. 2019, 10, 2441. [Google Scholar] [CrossRef] [Green Version]
- Zerdan, M.B.; Niforatos, S.; Nasr, S.; Nasr, D.; Ombada, M.; John, S.; Dutta, D.; Lim, S.H. Fecal Microbiota Transplant for Hematologic and Oncologic Diseases: Principle and Practice. Cancers 2022, 14, 691. [Google Scholar] [CrossRef]
- Routy, B.; le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef]
- Pinato, D.J.; Howlett, S.; Ottaviani, D.; Urus, H.; Patel, A.; Mineo, T.; Brock, C.; Power, D.; Hatcher, O.; Falconer, A.; et al. Association of Prior Antibiotic Treatment with Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol. 2019, 5, 1774. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, J.; Kumar, A.; Parikh, K.; Anwar, A.; Knoll, B.M.; Puccio, C.; Chun, H.; Fanucchi, M.; Lim, S.H. Use of broad-spectrum antibiotics impacts outcome in patients treated with immune checkpoint inhibitors. OncoImmunology 2018, 7, e1507670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.-M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [Green Version]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Pottier, C.; Wheatherspoon, A.; Roncarati, P.; Longuespée, R.; Herfs, M.; Duray, A.; Delvenne, P.; Quatresooz, P. The importance of the tumor microenvironment in the therapeutic management of cancer. Expert Rev. Anticancer Ther. 2015, 15, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Lança, T.; Silva-Santos, B. The split nature of tumor-infiltrating leukocytes: Implications for cancer surveillance and immunotherapy. OncoImmunology 2012, 1, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2020, 18, 842–859. [Google Scholar] [CrossRef]
- Cózar, B.; Greppi, M.; Carpentier, S.; Narni-Mancinelli, E.; Chiossone, L.; Vivier, E. Tumor-Infiltrating Natural Killer Cells. Cancer Discov. 2021, 11, 34–44. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2020, 18, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Sarvaria, A.; Madrigal, A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Da Cunha, A.; Michelin, M.A.; Murta, E.F. Pattern response of dendritic cells in the tumor microenvironment and breast cancer. World J. Clin. Oncol. 2014, 5, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2019, 9, 3176. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Chen, Y.; Tan, S.; Wu, S.; Huang, Y.; Fu, S.; Luo, F.; He, J. The Research Progress of Antiangiogenic Therapy, Immune Therapy and Tumor Microenvironment. Front. Immunol. 2022, 13, 802846. [Google Scholar] [CrossRef]
- Chen, W.; Shen, L.; Jiang, J.; Zhang, L.; Zhang, Z.; Pan, J.; Ni, C.; Chen, Z. Antiangiogenic therapy reverses the immunosuppressive breast cancer microenvironment. Biomark. Res. 2021, 9, 59. [Google Scholar] [CrossRef]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Ma, J.; Huang, L.; Hu, D.; Zeng, S.; Han, Y.; Shen, H. The role of the tumor microbe microenvironment in the tumor immune microenvironment: Bystander, activator, or inhibitor? J. Exp. Clin. Cancer Res. 2021, 40, 327. [Google Scholar] [CrossRef]
- McDonald, D.; Birmingham, A.; Knight, R. Context and the human microbiome. Microbiome 2015, 3, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, H.A.; Shigle, T.L.; Hammoudi, N.; Link, J.T.; Samaniego, F.; Kaseb, A.; Mallet, V.; Link, J.T. The oncologic burden of hepatitis C virus infection: A clinical perspective: Hepatitis C and Cancer. CA Cancer J. Clin. 2017, 67, 411–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.; Yap, L.-F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Lee, J.A.; Yoo, S.-Y.; Oh, H.J.; Jeong, S.; Cho, N.-Y.; Kang, G.H.; Kim, J.H. Differential immune microenvironmental features of microsatellite-unstable colorectal cancers according to Fusobacterium nucleatum status. Cancer Immunol. Immunother. 2020, 70, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Parhi, L.; Alon-Maimon, T.; Sol, A.; Nejman, D.; Shhadeh, A.; Fainsod-Levi, T.; Yajuk, O.; Isaacson, B.; Abed, J.; Maalouf, N.; et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef] [Green Version]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ayyoub, M.; Routy, B.; Kroemer, G. Microbiome and Anticancer Immunosurveillance. Cell 2016, 165, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Liwinski, T.; Zheng, D.; Elinav, E. The microbiome and cytosolic innate immune receptors. Immunol. Rev. 2020, 297, 207–224. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Javaheri, A.; Kruse, T.; Moonens, K.; Mejías-Luque, R.; DeBraekeleer, A.; Asche, C.I.; Tegtmeyer, N.; Kalali, B.; Bach, N.C.; Sieber, S.A.; et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2017, 2, 16189. [Google Scholar] [CrossRef] [Green Version]
- Xuan, C.; Shamonki, J.M.; Chung, A.; DiNome, M.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial Dysbiosis Is Associated with Human Breast Cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, K.J.; Ingle, J.N.; Tang, X.; Chia, N.; Jeraldo, P.R.; Walther-Antonio, M.; Kandimalla, K.K.; Johnson, S.; Yao, J.Z.; Harrington, J.; et al. A comprehensive analysis of breast cancer microbiota and host gene expression. PLoS ONE 2017, 12, e0188873. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, H.R.; Movafagh, A.; Fallah, F.; Shargh, S.A.; Mansouri, N.; Pour, A.H.; Hashemi, M. Evaluation of Methylobacterium radiotolerance and Sphyngomonas yanoikoaie in Sentinel Lymph Nodes of Breast Cancer Cases. Asian Pac. J. Cancer Prev. 2016, 17, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Altemus, J.; Niazi, F.; Green, H.; Calhoun, B.C.; Sturgis, C.; Grobmyer, S.R.; Eng, C. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget 2017, 8, 88122–88138. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The Microbiota of Breast Tissue and Its Association with Breast Cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [Google Scholar] [CrossRef] [Green Version]
- Parida, S.; Sharma, D. The power of small changes: Comprehensive analyses of microbial dysbiosis in breast cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2019, 1871, 392–405. [Google Scholar] [CrossRef]
- Chan, A.A.; Bashir, M.; Rivas, M.N.; Duvall, K.; Sieling, P.A.; Pieber, T.R.; Vaishampayan, P.A.; Love, S.M.; Lee, D.J. Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors. Sci. Rep. 2016, 6, 28061. [Google Scholar] [CrossRef] [Green Version]
- Kurath-Koller, S.; Moissl-Eichinger, C.; Gorkiewicz, G.; Kraschl, R.; Kanduth, C.; Hopfer, B.; Urlesberger, B.; Resch, B. Changes of intestinal microbiota composition and diversity in very low birth weight infants related to strategies of NEC prophylaxis: Protocol for an observational multicentre pilot study. Pilot Feasibility Stud. 2017, 3, 52. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Tian, T.; Wei, Z.; Shih, N.; Feldman, M.D.; Peck, K.N.; DeMichele, A.M.; Alwine, J.C.; Robertson, E.S. Distinct Microbial Signatures Associated with Different Breast Cancer Types. Front. Microbiol. 2018, 9, 951. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.; Pierre, J.F.; Makowski, L.; Tolley, E.; Lyn-Cook, B.; Lu, L.; Vidal, G.; Starlard-Davenport, A. Distinct microbial communities that differ by race, stage, or breast-tumor subtype in breast tissues of non-Hispanic Black and non-Hispanic White women. Sci. Rep. 2019, 9, 11940. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Brown, N.J.; Holen, I. The breast tumor microenvironment: Role in cancer development, progression and response to therapy. Expert Rev. Mol. Diagn. 2018, 18, 227–243. [Google Scholar] [CrossRef]
- Aalders, K.C.; Tryfonidis, K.; Senkus, E.; Cardoso, F. Anti-angiogenic treatment in breast cancer: Facts, successes, failures and future perspectives. Cancer Treat. Rev. 2017, 53, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Gene Funct. Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Mehraj, U.; Dar, A.H.; Wani, N.A.; Mir, M.A. Tumor microenvironment promotes breast cancer chemoresistance. Cancer Chemother. Pharmacol. 2021, 87, 147–158. [Google Scholar] [CrossRef]
- Generali, D.; Bates, G.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Immunomodulation of FOXP3+ Regulatory T Cells by the Aromatase Inhibitor Letrozole in Breast Cancer Patients. Clin. Cancer Res. 2009, 15, 1046–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.S.M.; Wang, L.; A Felizola, S.J.; Ueno, T.; Toi, M.; Loo, W.; Chow, L.W.C.; Suzuki, T.; Sasano, H. Changes of tumor infiltrating lymphocyte subtypes before and after neoadjuvant endocrine therapy in estrogen receptor-positive breast cancer patients—An immunohistochemical study of cd8+ and foxp3+ using double immunostaining with correlation to the pathobiological response of the patients. Int. J. Biol. Markers 2012, 27, 295–304. [Google Scholar] [CrossRef]
- Nalbandian, G.; Paharkova-Vatchkova, V.; Mao, A.; Nale, S.; Kovats, S. The Selective Estrogen Receptor Modulators, Tamoxifen and Raloxifene, Impair Dendritic Cell Differentiation and Activation. J. Immunol. 2005, 175, 2666–2675. [Google Scholar] [CrossRef] [Green Version]
- Komi, J.; Lassila, O. Nonsteroidal anti-estrogens inhibit the functional differentiation of human monocyte-derived dendritic cells. Blood 2000, 95, 2875–2882. [Google Scholar] [CrossRef]
- Behjati, S.; Frank, M. The Effects of Tamoxifen on Immunity. Curr. Med. Chem. 2009, 16, 3076–3080. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, K.H.; Wanyan, P.; Tian, J.H. Comparison of the efficacy and safety of denosumab versus bisphosphonates in breast cancer and bone metastases treatment: A meta-analysis of randomized controlled trials. Oncol. Lett. 2014, 7, 1997–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan—Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Sun, T.; Xu, J. Tumor-related Microbiome in the Breast Microenvironment and Breast Cancer. J. Cancer 2021, 12, 4841–4848. [Google Scholar] [CrossRef]
- Kroemer, G.; Senovilla, L.; Galluzzi, L.; Andre, F.; Zitvogel, L. Natural and therapy-induced immunosurveillance in breast cancer. Nat. Med. 2015, 21, 1128–1138. [Google Scholar] [CrossRef]
- Isaacs, J.; Anders, C.; McArthur, H.; Force, J. Biomarkers of Immune Checkpoint Blockade Response in Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2021, 22, 38. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Cortés, J.; Lipatov, O.; Im, S.-A.; Gonçalves, A.; Lee, K.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; Ohtani, S.; et al. KEYNOTE-119: Phase III study of pembrolizumab (pembro) versus single-agent chemotherapy (chemo) for metastatic triple negative breast cancer (mTNBC). Ann. Oncol. 2019, 30, v859–v860. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Narloch, J.L.; Onkar, S.; Joy, M.; Broadwater, G.; Luedke, C.; Hall, A.; Kim, R.; Pogue-Geile, K.; Sammons, S.; et al. Metastatic breast cancers have reduced immune cell recruitment but harbor increased macrophages relative to their matched primary tumors. J. Immunother. Cancer 2019, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Gianni, L.; Huang, C.-S.; Egle, D.; Bermejo, B.; Zamagni, C.; Thill, M.; Anton, A.; Zambelli, S.; Bianchini, G.; Russo, S.; et al. Abstract GS3-04: Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple negative, early high-risk and locally advanced breast cancer. NeoTRIPaPDL1 Michelangelo randomized study. Cancer Res. 2020, 80, GS3-04. [Google Scholar] [CrossRef]
- Nanda, R.; Liu, M.C.; Yau, C.; Shatsky, R.; Pusztai, L.; Wallace, A.; Chien, A.J.; Forero-Torres, A.; Ellis, E.; Han, H.; et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020, 6, 676. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.-U.; Grischke, E.-M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; Van De Vijver, K.K.; De Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Shadbad, M.; Safaei, S.; Brunetti, O.; Derakhshani, A.; Lotfinejad, P.; Mokhtarzadeh, A.; Hemmat, N.; Racanelli, V.; Solimando, A.; Argentiero, A.; et al. A Systematic Review on the Therapeutic Potentiality of PD-L1-Inhibiting MicroRNAs for Triple-Negative Breast Cancer: Toward Single-Cell Sequencing-Guided Biomimetic Delivery. Genes 2021, 12, 1206. [Google Scholar] [CrossRef] [PubMed]
- Lotfinejad, P.; Kazemi, T.; Safaei, S.; Amini, M.; Baghbani, E.; Shotorbani, S.S.; Niaragh, F.J.; Derakhshani, A.; Shadbad, M.A.; Silvestris, N. PD-L1 silencing inhibits triple-negative breast cancer development and upregulates T-cell-induced pro-inflammatory cytokines. Biomed. Pharmacother. 2021, 138, 111436. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.; Tennekoon, K.H.; Karunanayake, E.H. Interaction of Gut Microbiome and Host microRNAs with the Occurrence of Colorectal and Breast Cancer and Their Impact on Patient Immunity. OncoTargets Ther. 2021, 14, 5115–5129. [Google Scholar] [CrossRef]
- Forterre, A.; Komuro, H.; Aminova, S.; Harada, M. A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies. Cancers 2020, 12, 1852. [Google Scholar] [CrossRef]
- Ma, W.; Zhu, D.-M.; Li, J.; Chen, X.; Xie, W.; Jiang, X.; Wu, L.; Wang, G.; Xiao, Y.; Liu, Z.; et al. Coating biomimetic nanoparticles with chimeric antigen receptor T cell-membrane provides high specificity for hepatocellular carcinoma photothermal therapy treatment. Theranostics 2020, 10, 1281–1295. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitorino, M.; Alpuim Costa, D.; Vicente, R.; Caleça, T.; Santos, C. Local Breast Microbiota: A “New” Player on the Block. Cancers 2022, 14, 3811. https://doi.org/10.3390/cancers14153811
Vitorino M, Alpuim Costa D, Vicente R, Caleça T, Santos C. Local Breast Microbiota: A “New” Player on the Block. Cancers. 2022; 14(15):3811. https://doi.org/10.3390/cancers14153811
Chicago/Turabian StyleVitorino, Marina, Diogo Alpuim Costa, Rodrigo Vicente, Telma Caleça, and Catarina Santos. 2022. "Local Breast Microbiota: A “New” Player on the Block" Cancers 14, no. 15: 3811. https://doi.org/10.3390/cancers14153811
APA StyleVitorino, M., Alpuim Costa, D., Vicente, R., Caleça, T., & Santos, C. (2022). Local Breast Microbiota: A “New” Player on the Block. Cancers, 14(15), 3811. https://doi.org/10.3390/cancers14153811