
3D Bioprinting: An Enabling Technology to Understand Melanoma


 ,
,  ,
, 
Simple Summary
Abstract
1. Introduction
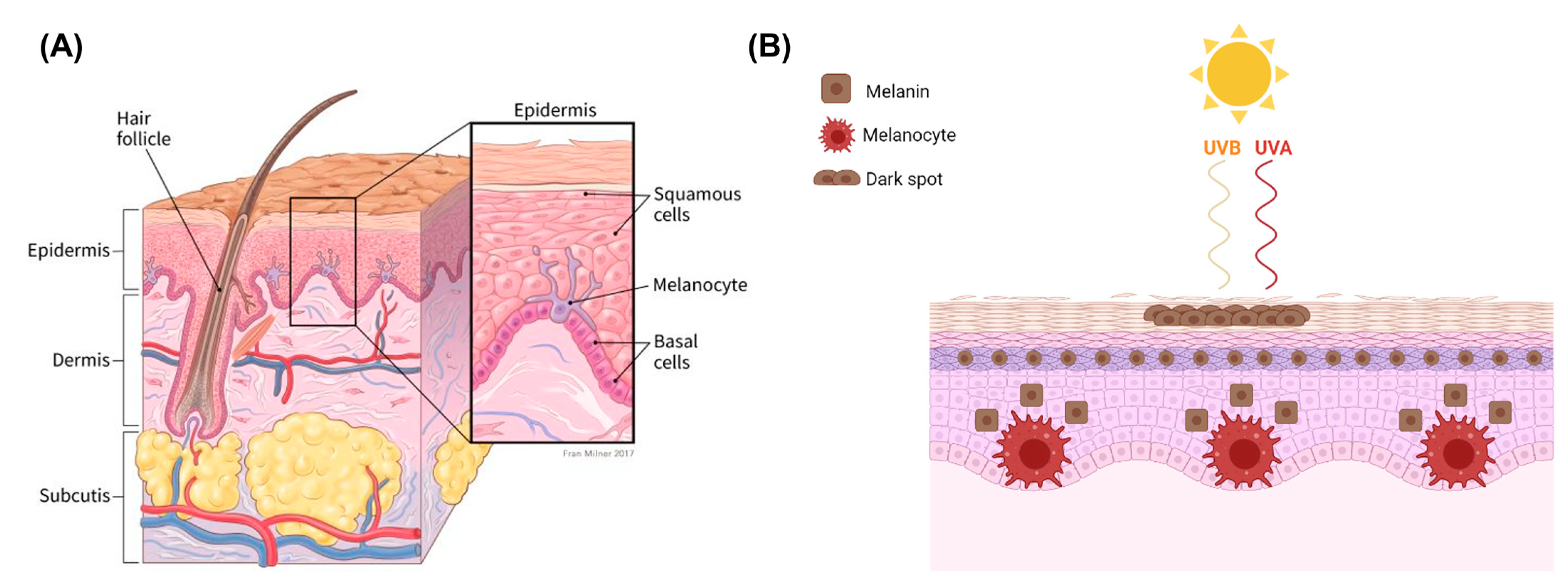
2. Melanoma
2.1. Melanocytes and Melanin
2.2. Mechanisms of Melanoma Progression and Physiology
2.3. Clinical Management
- Surgery is conducted to remove the tumour and is the standard primary treatment for patients in stages I-IIIB. Excision includes safety margins of 0.5 cm for in situ melanomas, 1 cm for tumours with a thickness of up to 2 mm, and 2 cm for tumours thicker than 2 mm [8]. Depending on the location, the surgical defect may require skin grafting. Patients with melanomas that penetrate the dermis are offered a surgical intervention to test whether the melanoma has spread to the regional lymph nodes. If the cancer has spread, the regional nodes are removed with a lymphadenectomy. To increase the rate of survival, recent efforts are developing new adjuvant therapies, specifically immunotherapy, to prevent disease progression. New trials show adjuvant immunotherapy following primary tumour removal decreases disease progression.
- Immunotherapy/biological therapy is a type of treatment that activates the immune system to amplify the immune response against cancer. In this case, the following strategies are used [65,66]:
- -
- Immune checkpoint inhibitor therapy targets immune checkpoints (proteins found on T cells and some cancer cells), which are regulators of the immune system. Checkpoints help keep immune responses from being too strong, and cancer cells can activate them in order to decrease anticancer immunity. Targeting these checkpoints releases immunity, reinstating cancer-fighting immunity. Specifically, checkpoint inhibitors are used to restore the ability of T cells to destroy cancer cells. CTLA-4, a checkpoint found on the surface of T cells, is a common target for inhibition. When attached to protein B7 on a cancer cell, CTLA-4 prevents T cells from killing cancer cells, deactivating the T cells [53]. PD-1 and PDL-1 inhibitors are also used to target the PD-1/PDL-1 interaction, which occurs between T cells and cancer cells, respectively, inhibiting T cell activity. Some of these drugs include Ipilimumab, which targets CTLA4, Pembrolizumab and nivolumab, which are PD-1 inhibitors, and Atezolizumab, which is an anti-PDL-1 [65,67]. Immune checkpoint inhibitor treatment has revolutionised melanoma care for advanced stages, in monotherapy and in combination, and is showing promising results in the adjuvant setting. There are ongoing drug development efforts at the preclinical and clinical stages investigating new targets and strategies to awaken host immunity against cancer.
- Targeted therapy uses small molecules and immunotoxins to block the growth of cancer cells by interfering with specific targeted molecules known as “molecular targets” that are involved in the growth, progression, and spread of cancer [53,68]. Despite being from the same tumour, cancer cells can be highly heterogenous. Some common drugs used in targeted therapy for melanoma are vemurafenib, dabrafenib and encorafenib, which directly attack oncogenic BRAF and the MAP Kinase pathway. In contrast to cytotoxic agents, targeted therapies do not cause as significant side effects to normal non-cancerous tissues as the drugs act directly on specific molecular cancer targets [69].
- There are multiple new avenues of research in targeted and immunotherapy. One promising strategy is the development of oncolytic virus therapy, which uses a genetically engineered version of a naturally occurring virus, injected directly into the tumours in the skin and lymph nodes, to infect and break down cancer cells without harming healthy cells. Talimogene Laherparepvec (T-vec), a modified herpes simplex virus, is a common genetically engineered oncolytic virus, able not only to suppress the growth of tumours but also to prolong overall survival [70].
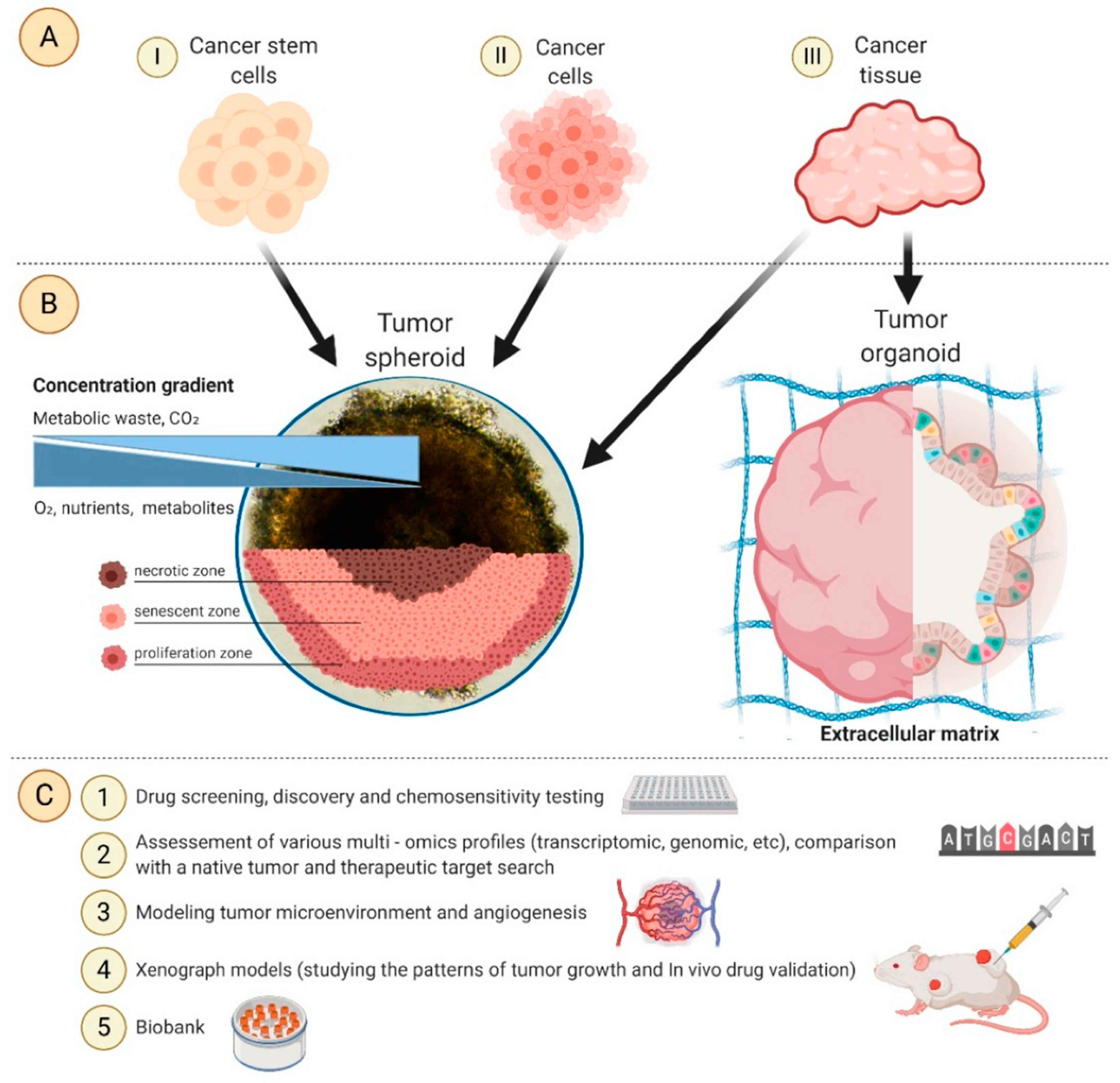
3. 3D in Vitro Models for Melanoma Modelling
3.1. Multicellular Tumour Spheroids and Organoids
3.2. Tumour-On-A-Chip
3.3. Reconstructed 3D Skin Equivalents

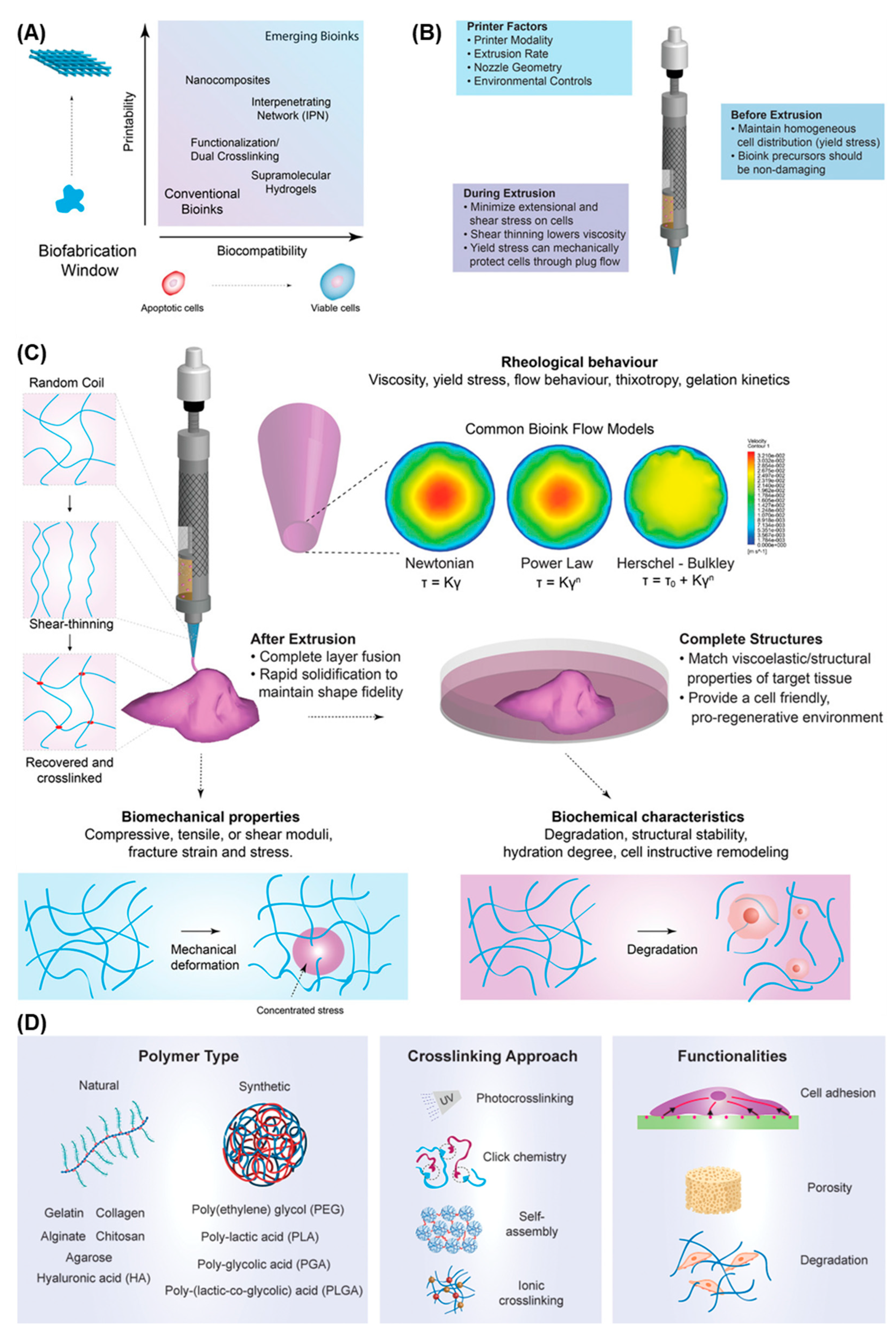
4. Bioprinting Skin and Melanoma Models
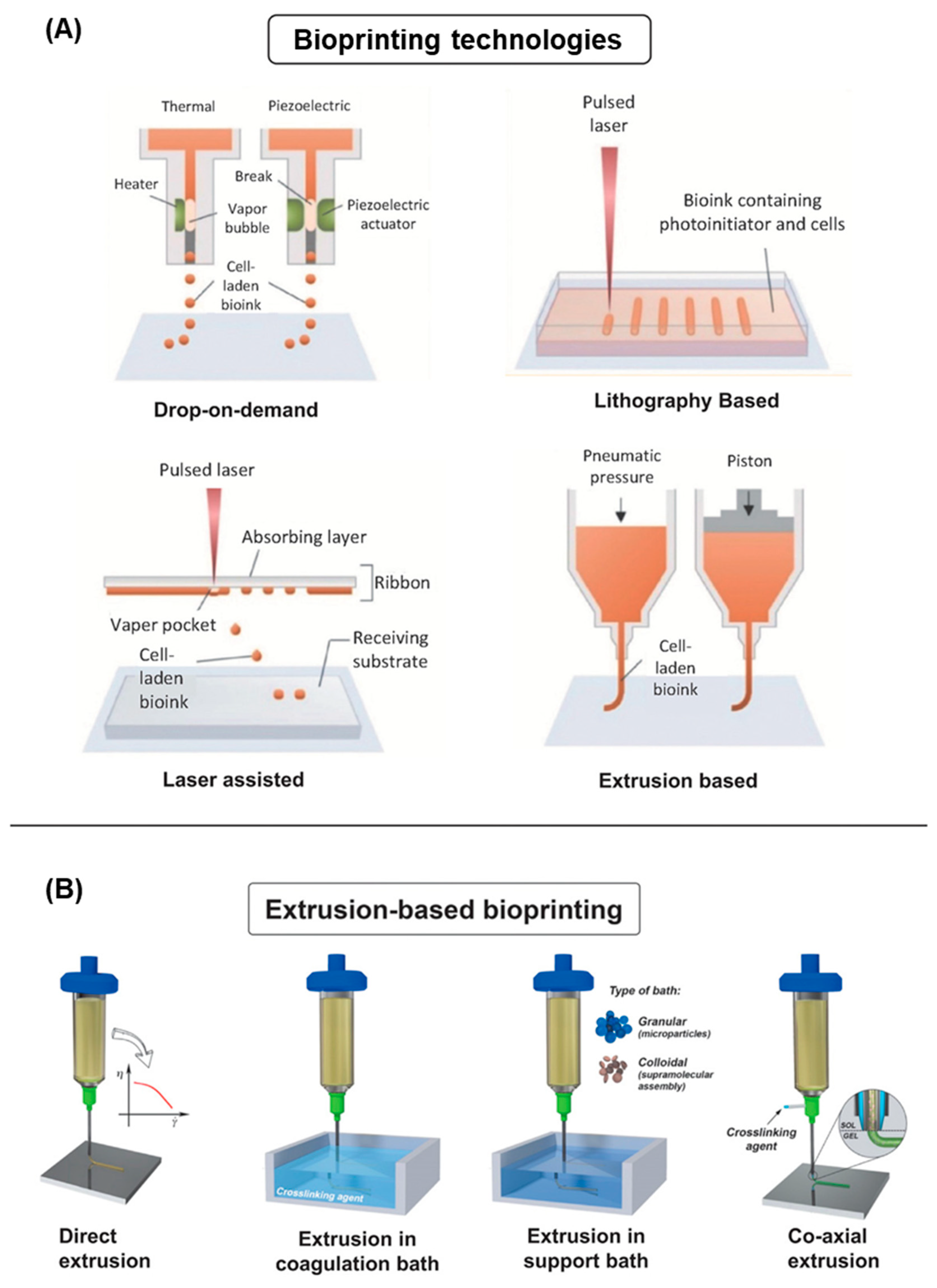
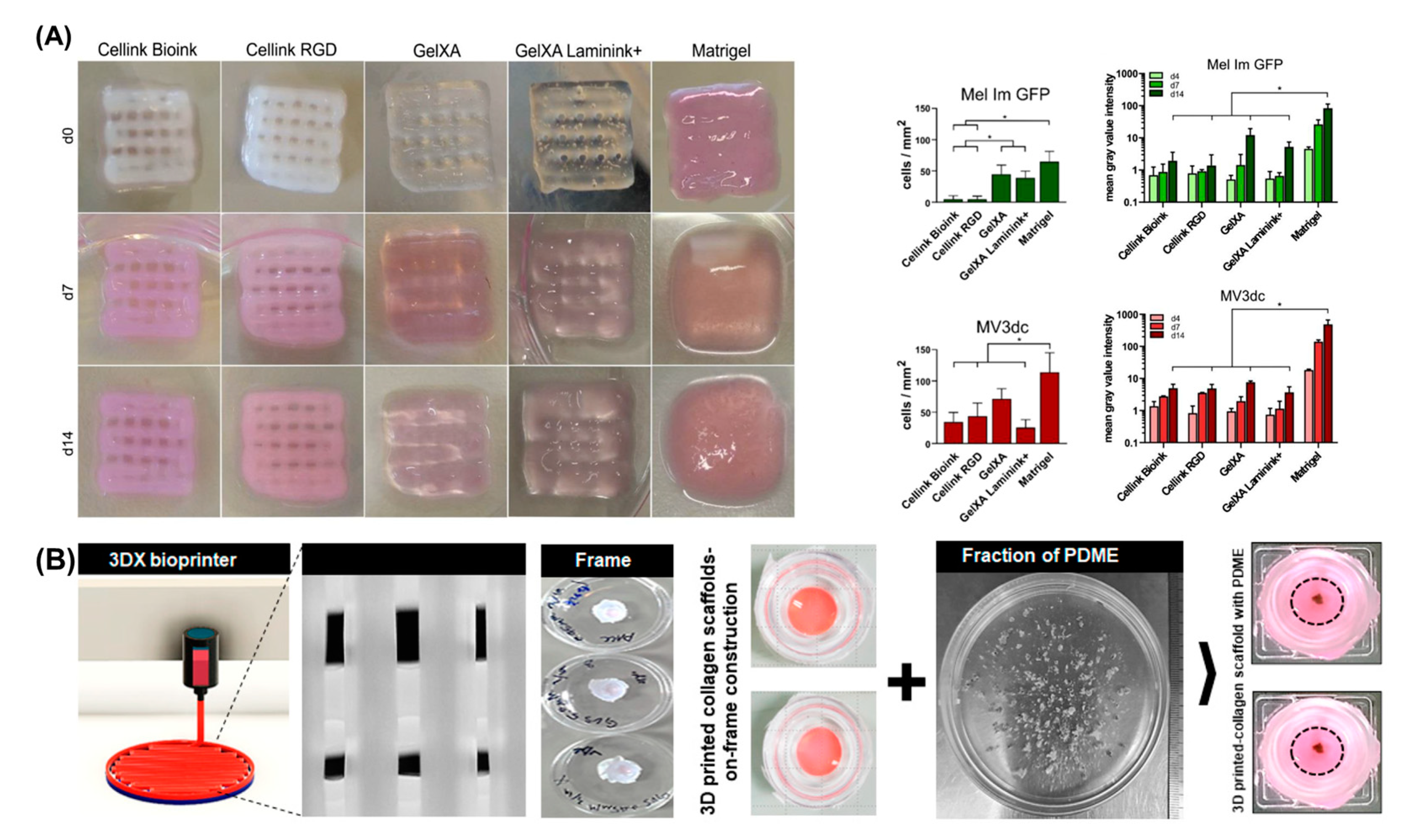
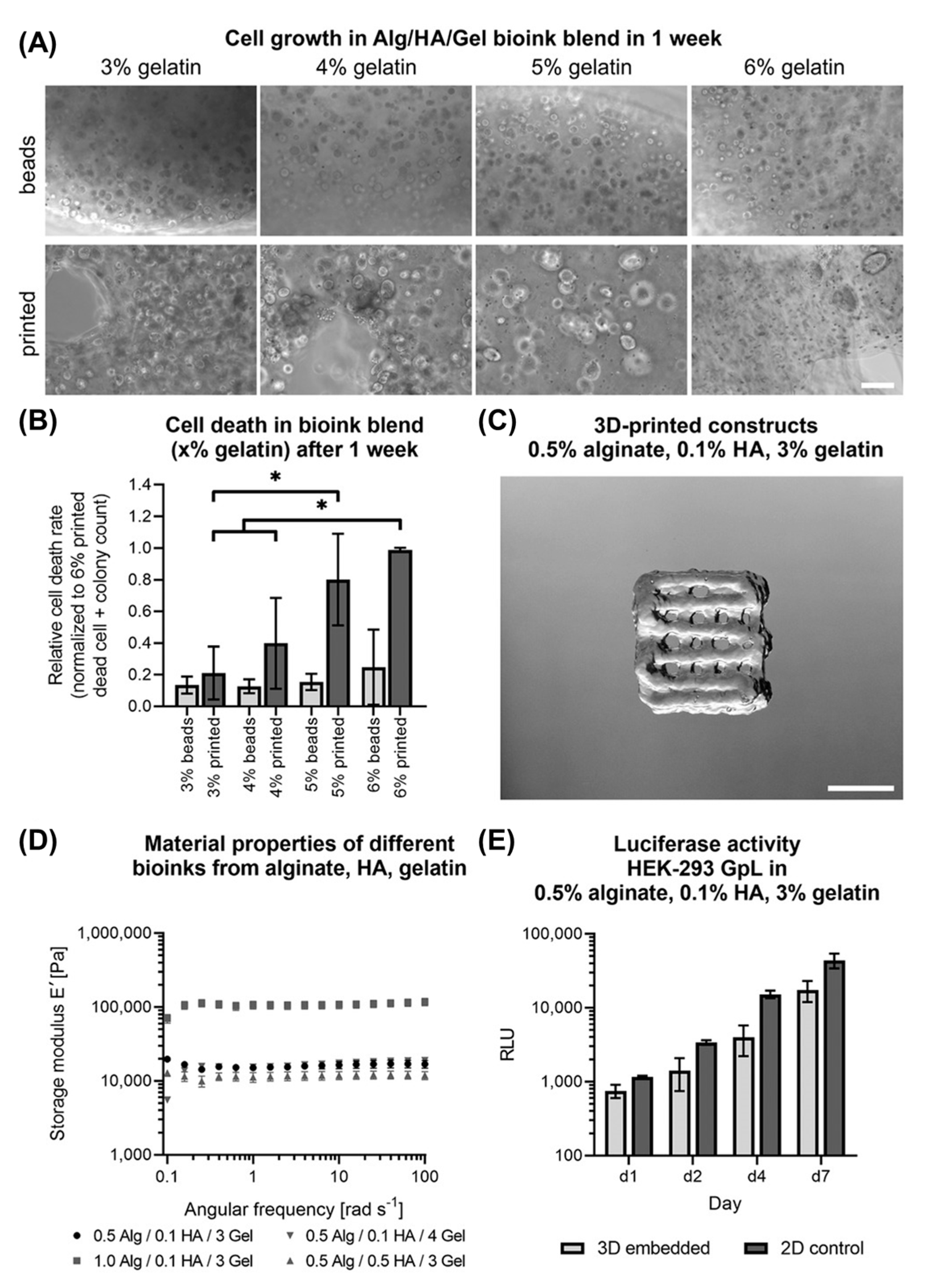
4.1. Bioinks
4.2. 3D Bioprinting Technologies
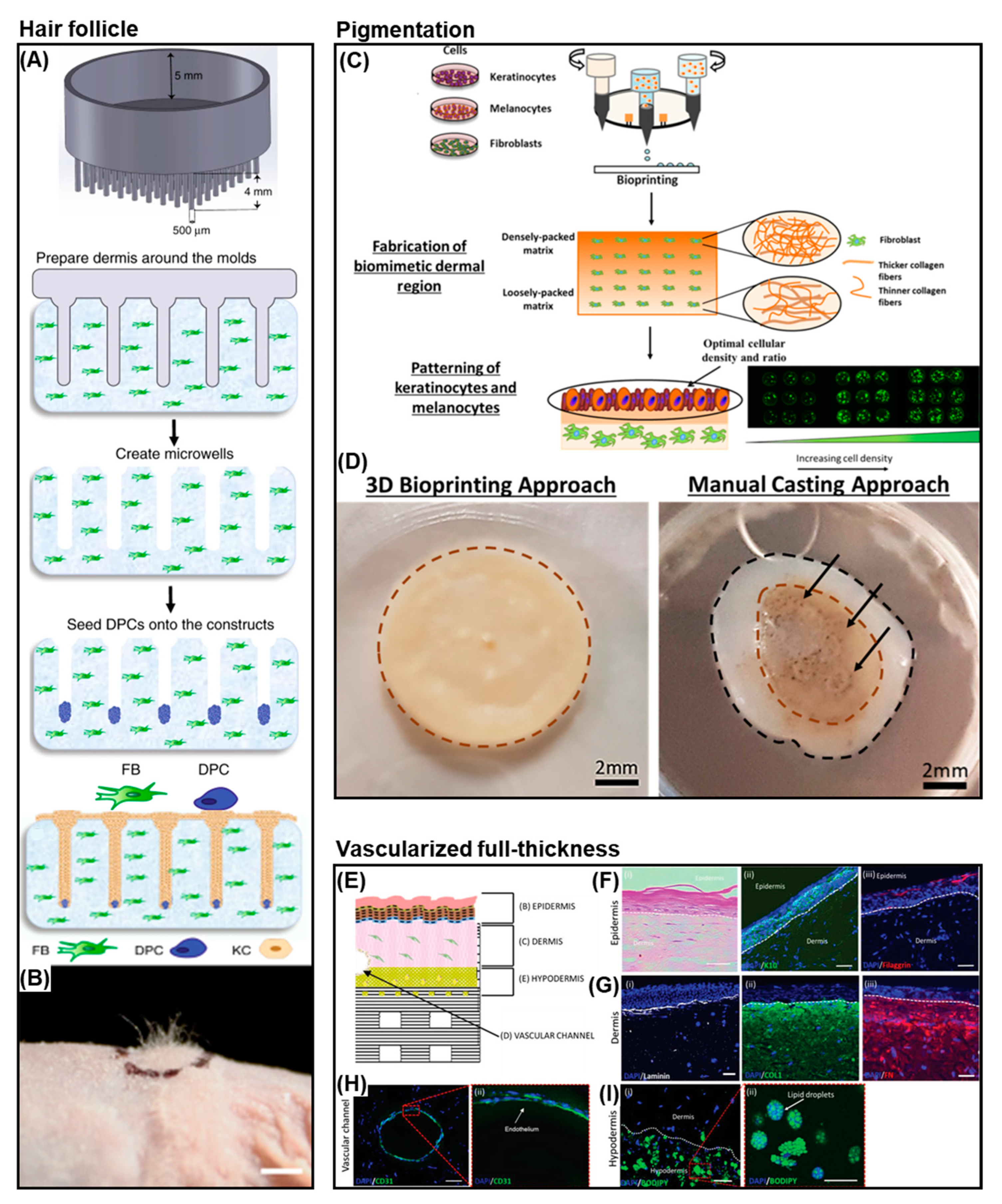
4.3. Skin Models
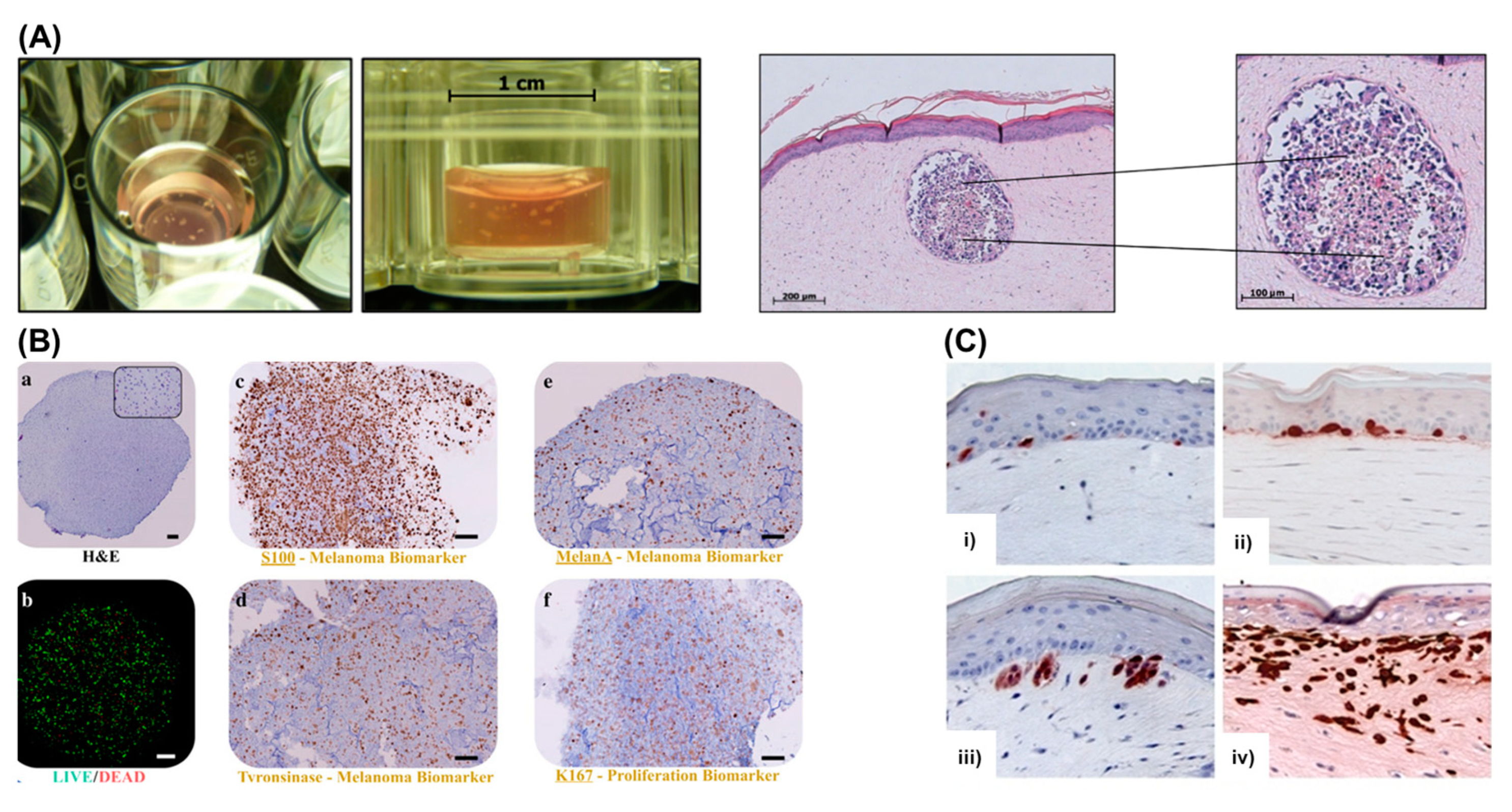
4.4. Bioprinting in Vitro Models of Melanoma
4.5. Challenges of Bioprinting Melanoma Models
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Kim, B.S.; Kwon, Y.W.; Kong, J.S.; Park, G.T.; Gao, G.; Han, W.; Kim, M.B.; Lee, H.; Kim, J.H.; Cho, D.W. 3D cell printing of in vitro stabilized skin model and in vivo pre-vascularized skin patch using tissue-specific extracellular matrix bioink: A step towards advanced skin tissue engineering. Biomaterials 2018, 168, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.; Umar Aslam Khan, M.; Shamsan Al-Arjan, W.; Izwan Abd Razak, S.; Javed, A.; Rafiq Abdul Kadir, M. Nanocomposite hydrogels for melanoma skin cancer care and treatment: In-vitro drug delivery, drug release kinetics and anti-cancer activities. Arab. J. Chem. 2021, 14, 103120. [Google Scholar] [CrossRef]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [PubMed]
- Kanavy, H.E.; Gerstenblith, M.R. Ultraviolet radiation and melanoma. Semin Cutan. Med. Surg. 2011, 30, 222–228. [Google Scholar] [CrossRef]
- Liu, Y.; Sheikh, M.S. Melanoma: Molecular Pathogenesis and Therapeutic Management. Mol. Cell Pharmacol. 2014, 6, 228. [Google Scholar]
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar] [CrossRef]
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Hoarau-Véchot, J.; Rafii, A.; Touboul, C.; Pasquier, J. Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions? Int. J. Mol. Sci. 2018, 19, 181. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Padhye, A.; D’Souza, J. Oral malignant melanoma: A silent killer? J. Indian Soc. Periodontol. 2011, 15, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Patti, R.; Cacciatori, M.; Guercio, G.; Territo, V.; Di Vita, G. Intestinal melanoma: A broad spectrum of clinical presentation. Int. J. Surg. Case Rep. 2012, 3, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- van der Weyden, L.; Brenn, T.; Patton, E.E.; Wood, G.A.; Adams, D.J. Spontaneously occurring melanoma in animals and their relevance to human melanoma. J. Pathol. 2020, 252, e5505. [Google Scholar] [CrossRef]
- Emri, G.; Paragh, G.; Tosaki, A.; Janka, E.; Kollar, S.; Hegedus, C.; Gellen, E.; Horkay, I.; Koncz, G.; Remenyik, E. Ultraviolet radiation-mediated development of cutaneous melanoma: An update. J. Photochem. Photobiol. B 2018, 185, 169–175. [Google Scholar] [CrossRef]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Abeni, D.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: I. Common and atypical naevi. Eur. J. Cancer 2005, 41, 28–44. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Munoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; Garcia, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Onco. Targets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef]
- Leong, S.P.L.; Mihm, M.C.; Murphy, G.F.; Hoon, D.S.B.; Kashani-Sabet, M.; Agarwala, S.S.; Zager, J.S.; Hauschild, A.; Sondak, V.K.; Guild, V.; et al. Progression of cutaneous melanoma: Implications for treatment. Clin. Exp. Metastasis 2012, 29, 775–796. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Seto, K.; Haneda, M.; Masago, K.; Fujita, S.; Kato, S.; Sasaki, E.; Hosoda, W.; Murakami, Y.; Kuroda, H.; Horio, Y.; et al. Negative reactions of BRAF mutation-specific immunohistochemistry to non-V600E mutations of BRAF. Pathol. Int. 2020, 70, 253–261. [Google Scholar] [CrossRef]
- Randic, T.; Kozar, I.; Margue, C.; Utikal, J.; Kreis, S. NRAS mutant melanoma: Towards better therapies. Cancer Treat. Rev. 2021, 99, 102238. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.A.; Montesion, M.; Shah, N.; Sharaf, R.; Pavlick, D.C.; Sokol, E.S.; Alexander, B.; Venstrom, J.; Elvin, J.A.; Ross, J.S.; et al. Melanoma with in-frame deletion of MAP2K1: A distinct molecular subtype of cutaneous melanoma mutually exclusive from BRAF, NRAS, and NF1 mutations. Mod. Pathol. 2020, 33, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Nagore, E.; Rachakonda, S.; Kumar, R. TERT promoter mutations in melanoma survival. Oncotarget 2019, 10, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Nambiar, R.; Rosario, S.R.; Smiraglia, D.J.; Goodrich, D.W.; Witkiewicz, A.K. Pan-cancer molecular analysis of the RB tumor suppressor pathway. Commun. Biol. 2020, 3, 158. [Google Scholar] [CrossRef]
- Mehrotra, A.; Mehta, G.; Aras, S.; Trivedi, A.; de la Serna, I.L. SWI/SNF chromatin remodeling enzymes in melanocyte differentiation and melanoma. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 151–161. [Google Scholar] [CrossRef]
- Hafner, C.; Landthaler, M.; Vogt, T. Activation of the PI3K/AKT signalling pathway in non-melanoma skin cancer is not mediated by oncogenic PIK3CA and AKT1 hotspot mutations. Exp. Dermatol. 2010, 19, e222–e227. [Google Scholar] [CrossRef]
- Cramer, S.F. Malignant melanoma in situ: Another perspective. Hum. Pathol. 1991, 22, 626. [Google Scholar] [CrossRef]
- Barden, H.; Levine, S. Histochemical observations on rodent brain melanin. Brain Res. Bull. 1983, 10, 847–851. [Google Scholar] [CrossRef]
- Theriault, L.L.; Hurley, L.S. Ultrastructure of developing melanosomes in C57 Black and pallid mice. Dev. Biol. 1970, 23, 261–275. [Google Scholar] [CrossRef]
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central Role of p53 in the Suntan Response and Pathologic Hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Guida, S.; Guida, G.; Goding, C.R. MC1R Functions, Expression, and Implications for Targeted Therapy. J. Investig. Dermatol. 2022, 142, 293–302.e291. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy Kathleen, G.; Robbins Linda, S.; Mortrud Marty, T.; Cone Roger, D. The Cloning of a Family of Genes That Encode the Melanocortin Receptors. Science 1992, 257, 1248–1251. [Google Scholar] [CrossRef]
- Manganelli, M.; Guida, S.; Ferretta, A.; Pellacani, G.; Porcelli, L.; Azzariti, A.; Guida, G. Behind the Scene: Exploiting MC1R in Skin Cancer Risk and Prevention. Genes 2021, 12, 93. [Google Scholar] [CrossRef]
- Castejon-Grinan, M.; Herraiz, C.; Olivares, C.; Jimenez-Cervantes, C.; García-Borrón, J.C. cAMP-independent non-pigmentary actions of variant melanocortin 1 receptor: AKT-mediated activation of protective responses to oxidative DNA damage. Oncogene 2018, 37, 3631–3646. [Google Scholar] [CrossRef]
- García-Borrón, J.C.; Abdel-Malek, Z.; Jiménez-Cervantes, C. MC 1R, the c AMP pathway, and the response to solar UV: Extending the horizon beyond pigmentation. Pigment. Cell Melanoma Res. 2014, 27, 699–720. [Google Scholar] [CrossRef]
- García-Borrón, J.C.; Sánchez-Laorden, B.L.; Jiménez-Cervantes, C. Melanocortin-1 receptor structure and functional regulation. Pigment. Cell Res. 2005, 18, 393–410. [Google Scholar] [CrossRef]
- Nasti, T.H.; Timares, L. MC 1R, Eumelanin and Pheomelanin: Their role in determining the susceptibility to skin cancer. Photochem. Photobiol. 2015, 91, 188–200. [Google Scholar] [CrossRef] [PubMed]
- d’Ischia, M.; Wakamatsu, K.; Cicoira, F.; Di Mauro, E.; Garcia-Borron, J.C.; Commo, S.; Galván, I.; Ghanem, G.; Kenzo, K.; Meredith, P. Melanins and melanogenesis: From pigment cells to human health and technological applications. Pigment. Cell Melanoma Res. 2015, 28, 520–544. [Google Scholar] [CrossRef] [PubMed]
- Passeron, T.; Bahadoran, P.; Bertolotto, C.; Chiaverini, C.; Buscà, R.; Valony, G.; Bille, K.; Ortonne, J.P.; Ballotti, R. Cyclic AMP promotes a peripheral distribution of melanosomes and stimulates melanophilin/Slac2-a actin association. FASEB J. 2004, 18, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Everett, M.A.; Yeargers, E.; Sayre, R.M.; Olson, R.L. Penetration of Epidermis by Ultraviolet Rays. Photochem. Photobiol. 1966, 5, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Crowley, E. UV damage, DNA repair and skin carcinogenesis. Front. Biosci. 2002, 7, d1024–d1043. [Google Scholar]
- Premi, S.; Han, L.; Mehta, S.; Knight, J.; Zhao, D.; Palmatier Meg, A.; Kornacker, K.; Brash Douglas, E. Genomic sites hypersensitive to ultraviolet radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 24196–24205. [Google Scholar] [CrossRef]
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.H.; Halaban, R.; Douki, T.; Brash, D.E. Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015, 347, 842–847. [Google Scholar] [CrossRef]
- Eddy, K.; Shah, R.; Chen, S. Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front. Oncol. 2020, 10, 626129. [Google Scholar] [CrossRef]
- Haridas, P.; McGovern, J.A.; McElwain, S.D.L.; Simpson, M.J. Quantitative comparison of the spreading and invasion of radial growth phase and metastatic melanoma cells in a three-dimensional human skin equivalent model. PeerJ 2017, 5, e3754. [Google Scholar] [CrossRef] [PubMed]
- Ciarletta, P.; Foret, L.; Ben Amar, M. The radial growth phase of malignant melanoma: Multi-phase modelling, numerical simulations and linear stability analysis. J. R. Soc. Interface 2011, 8, 345–368. [Google Scholar] [CrossRef]
- Damsky, W.E.; Bosenberg, M. Melanocytic nevi and melanoma: Unraveling a complex relationship. Oncogene 2017, 36, 5771–5792. [Google Scholar] [CrossRef] [PubMed]
- Institute, N.C.; PDQ® Adult Treatment Editorial Board. PDQ Melanoma Treatment. Bethesda, MD: National Cancer Institute. Available online: https://www.cancer.gov/types/skin/patient/melanoma-treatment-pdq (accessed on 16 September 2020).
- Nakamura, M.; Haarmann-Stemmann, T.; Krutmann, J.; Morita, A. Alternative test models for skin ageing research. Exp. Dermatol. 2018, 27, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef]
- Aunan, J.R.; Cho, W.C.; Soreide, K. The Biology of Aging and Cancer: A Brief Overview of Shared and Divergent Molecular Hallmarks. Aging Dis. 2017, 8, 628–642. [Google Scholar] [CrossRef]
- Gomes, A.P.; Ilter, D.; Low, V.; Endress, J.E.; Fernández-García, J.; Rosenzweig, A.; Schild, T.; Broekaert, D.; Ahmed, A.; Planque, M.; et al. Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature 2020, 585, 283–287. [Google Scholar] [CrossRef]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H., 3rd; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef]
- Berthod, F.; Germain, L.; Li, H.; Xu, W.; Damour, O.; Auger, F. Collagen fibril network and elastic system remodeling in a reconstructed skin transplanted on nude mice. Matrix Biol. 2001, 20, 463–473. [Google Scholar] [CrossRef]
- Kugel, C.H., 3rd; Douglass, S.M.; Webster, M.R.; Kaur, A.; Liu, Q.; Yin, X.; Weiss, S.A.; Darvishian, F.; Al-Rohil, R.N.; Ndoye, A.; et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin. Cancer Res. 2018, 24, 5347–5356. [Google Scholar] [CrossRef]
- Cancer Research UK. Worldwide Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/worldwide-cancer (accessed on 16 September 2020).
- Cancer Research UK. Melanoma Statistics UK. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/melanoma-skin-cancer (accessed on 16 September 2020).
- Foundation, T.S.C. Melanoma Statistics. Available online: https://www.skincancer.org/skin-cancer-information/skin-cancer-facts/ (accessed on 16 September 2020).
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Troy, A.B. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef]
- Bivona, T.G.; Doebele, R.C. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Kapalczynska, M.; Kolenda, T.; Przybyla, W.; Zajaczkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Blizniak, R.; Luczewski, L.; Lamperska, K. 2D and 3D cell cultures - a comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A.; Su, Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis. Model Mech. 2008, 1, 78–82. [Google Scholar] [CrossRef]
- Tannenbaum, J.; Bennett, B.T. Russell and Burch’s 3Rs then and now: The need for clarity in definition and purpose. J. Am. Assoc. Lab Anim. Sci. 2015, 54, 120–132. [Google Scholar]
- Smalley, K.S.; Lioni, M.; Herlyn, M. Life isn’t flat: Taking cancer biology to the next dimension. In Vitro Cell Dev. Biol. Anim. 2006, 42, 242–247. [Google Scholar] [CrossRef]
- Xu, X.; Farach-Carson, M.C.; Jia, X. Three-dimensional in vitro tumor models for cancer research and drug evaluation. Biotechnol. Adv. 2014, 32, 1256–1268. [Google Scholar] [CrossRef]
- Marconi, A.; Quadri, M.; Saltari, A.; Pincelli, C. Progress in melanoma modelling in vitro. Exp. Dermatol. 2018, 27, 578–586. [Google Scholar] [CrossRef]
- Müller, I.; Kulms, D. A 3D Organotypic Melanoma Spheroid Skin Model. JoVE 2018, 135, e57500. [Google Scholar] [CrossRef]
- Gilazieva, Z.; Ponomarev, A.; Rutland, C.; Rizvanov, A.; Solovyeva, V. Promising Applications of Tumor Spheroids and Organoids for Personalized Medicine. Cancers 2020, 12, 2727. [Google Scholar] [CrossRef]
- Maloney, E.; Clark, C.; Sivakumar, H.; Yoo, K.; Aleman, J.; Rajan, S.A.P.; Forsythe, S.; Mazzocchi, A.; Laxton, A.W.; Tatter, S.B.; et al. Immersion Bioprinting of Tumor Organoids in Multi-Well Plates for Increasing Chemotherapy Screening Throughput. Micromachines 2020, 11, 208. [Google Scholar] [CrossRef]
- Votanopoulos, K.I.; Forsythe, S.; Sivakumar, H.; Mazzocchi, A.; Aleman, J.; Miller, L.; Levine, E.; Triozzi, P.; Skardal, A. Model of Patient-Specific Immune-Enhanced Organoids for Immunotherapy Screening: Feasibility Study. Ann. Surg. Oncol. 2020, 27, 1956–1967. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Sadangi, S.; Lares, M.; Rehman, S.; Humayun, M.; Denecke, K.M.; Skala, M.C.; Beebe, D.J.; Setaluri, V. Microfluidic model with air-walls reveals fibroblasts and keratinocytes modulate melanoma cell phenotype, migration, and metabolism. Lab A Chip 2021, 21, 1139–1149. [Google Scholar] [CrossRef]
- Ali, N.; Hosseini, M.; Vainio, S.; Taïeb, A.; Cario-André, M.; Rezvani, H.R. Skin equivalents: Skin from reconstructions as models to study skin development and diseases. Br. J. Dermatol. 2015, 173, 391–403. [Google Scholar] [CrossRef]
- Augustine, R. Skin bioprinting: A novel approach for creating artificial skin from synthetic and natural building blocks. Prog. Biomater. 2018, 7, 77–92. [Google Scholar] [CrossRef]
- Derr, K.; Zou, J.; Luo, K.; Song, M.J.; Sittampalam, G.S.; Zhou, C.; Michael, S.; Ferrer, M.; Derr, P. Fully Three-Dimensional Bioprinted Skin Equivalent Constructs with Validated Morphology and Barrier Function. Tissue Eng. Part C Methods 2019, 25, 334–343. [Google Scholar] [CrossRef]
- Hill, D.S.; Robinson, N.D.; Caley, M.P.; Chen, M.; O’Toole, E.A.; Armstrong, J.L.; Przyborski, S.; Lovat, P.E. A Novel Fully Humanized 3D Skin Equivalent to Model Early Melanoma Invasion. Mol. Cancer Ther. 2015, 14, 2665–2673. [Google Scholar] [CrossRef]
- Kim, B.S.; Gao, G.; Kim, J.Y.; Cho, D.-W. 3D Cell Printing of Perfusable Vascularized Human Skin Equivalent Composed of Epidermis, Dermis, and Hypodermis for Better Structural Recapitulation of Native Skin. Adv. Healthc. Mater. 2019, 8, 1801019. [Google Scholar] [CrossRef]
- Lee, H.-R.; Park, J.A.; Kim, S.; Jo, Y.; Kang, D.; Jung, S. 3D microextrusion-inkjet hybrid printing of structured human skin equivalents. Bioprinting 2021, 22, e00143. [Google Scholar] [CrossRef]
- Liu, X.; Michael, S.; Bharti, K.; Ferrer, M.; Song, M.J. A biofabricated vascularized skin model of atopic dermatitis for preclinical studies. Biofabrication 2020, 12, 035002. [Google Scholar] [CrossRef]
- Pourchet, L.J.; Thepot, A.; Albouy, M.; Courtial, E.J.; Boher, A.; Blum, L.J.; Marquette, C.A. Human Skin 3D Bioprinting Using Scaffold-Free Approach. Adv. Healthc. Mater. 2017, 6, 1601101. [Google Scholar] [CrossRef]
- Rimann, M.; Bono, E.; Annaheim, H.; Bleisch, M.; Graf-Hausner, U. Standardized 3D Bioprinting of Soft Tissue Models with Human Primary Cells. J. Lab Autom. 2016, 21, 496–509. [Google Scholar] [CrossRef]
- Vorsmann, H.; Groeber, F.; Walles, H.; Busch, S.; Beissert, S.; Walczak, H.; Kulms, D. Development of a human three-dimensional organotypic skin-melanoma spheroid model for in vitro drug testing. Cell Death Dis. 2013, 4, e719. [Google Scholar] [CrossRef]
- Zhang, Z.; Michniak-Kohn, B.B. Tissue Engineered Human Skin Equivalents. Pharmaceutics 2012, 4, 26–41. [Google Scholar] [CrossRef]
- Admane, P.; Gupta, A.C.; Jois, P.; Roy, S.; Chandrasekharan Lakshmanan, C.; Kalsi, G.; Bandyopadhyay, B.; Ghosh, S. Direct 3D bioprinted full-thickness skin constructs recapitulate regulatory signaling pathways and physiology of human skin. Bioprinting 2019, 15, e00051. [Google Scholar] [CrossRef]
- Albanna, M.; Binder, K.W.; Murphy, S.V.; Kim, J.; Qasem, S.A.; Zhao, W.; Tan, J.; El-Amin, I.B.; Dice, D.D.; Marco, J.; et al. In Situ Bioprinting of Autologous Skin Cells Accelerates Wound Healing of Extensive Excisional Full-Thickness Wounds. Sci. Rep. 2019, 9, 1856. [Google Scholar] [CrossRef]
- Ashammakhi, N.; Hasan, A.; Kaarela, O.; Byambaa, B.; Sheikhi, A.; Gaharwar, A.K.; Khademhosseini, A. Advancing Frontiers in Bone Bioprinting. Adv. Healthc. Mater. 2019, 8, 1801048. [Google Scholar] [CrossRef]
- Augustine, R.; Kalva, S.N.; Ahmad, R.; Zahid, A.A.; Hasan, S.; Nayeem, A.; McClements, L.; Hasan, A. 3D Bioprinted cancer models: Revolutionizing personalized cancer therapy. Transl. Oncol. 2021, 14, 101015. [Google Scholar] [CrossRef]
- Baltazar, T.; Merola, J.; Catarino, C.; Xie, C.B.; Kirkiles-Smith, N.C.; Lee, V.; Hotta, S.; Dai, G.; Xu, X.; Ferreira, F.C.; et al. Three Dimensional Bioprinting of a Vascularized and Perfusable Skin Graft Using Human Keratinocytes, Fibroblasts, Pericytes, and Endothelial Cells. Tissue Eng. Part A 2020, 26, 227–238. [Google Scholar] [CrossRef]
- Dai, X.; Liu, L.; Ouyang, J.; Li, X.; Zhang, X.; Lan, Q.; Xu, T. Coaxial 3D bioprinting of self-assembled multicellular heterogeneous tumor fibers. Sci. Rep. 2017, 7, 1457. [Google Scholar] [CrossRef]
- Dai, X.; Ma, C.; Lan, Q.; Xu, T. 3D bioprinted glioma stem cells for brain tumor model and applications of drug susceptibility. Biofabrication 2016, 8, 045005. [Google Scholar] [CrossRef]
- Datta, P.; Ayan, B.; Ozbolat, I.T. Bioprinting for vascular and vascularized tissue biofabrication. Acta Biomater. 2017, 51, 1–20. [Google Scholar] [CrossRef]
- Datta, P.; Dey, M.; Ataie, Z.; Unutmaz, D.; Ozbolat, I.T. 3D bioprinting for reconstituting the cancer microenvironment. NPJ Precis. Oncol. 2020, 4, 18. [Google Scholar] [CrossRef]
- Koch, L.; Deiwick, A.; Schlie, S.; Michael, S.; Gruene, M.; Coger, V.; Zychlinski, D.; Schambach, A.; Reimers, K.; Vogt, P.M.; et al. Skin tissue generation by laser cell printing. Biotechnol. Bioeng. 2012, 109, 1855–1863. [Google Scholar] [CrossRef]
- Koch, L.; Gruene, M.; Unger, C.; Chichkov, B. Laser Assisted Cell Printing. Curr. Pharm. Biotechnol. 2013, 14, 91–97. [Google Scholar] [CrossRef]
- Kolesky, D.B.; Homan, K.A.; Skylar-Scott, M.A.; Lewis, J.A. Three-dimensional bioprinting of thick vascularized tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 3179. [Google Scholar] [CrossRef]
- Min, D.; Lee, W.; Bae, I.H.; Lee, T.R.; Croce, P.; Yoo, S.S. Bioprinting of biomimetic skin containing melanocytes. Exp. Dermatol. 2018, 27, 453–459. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.F.; Sousa, A.; Barrias, C.C.; Bartolo, P.J.; Granja, P.L. A single-component hydrogel bioink for bioprinting of bioengineered 3D constructs for dermal tissue engineering. Mater. Horiz. 2018, 5, 1100–1111. [Google Scholar] [CrossRef]
- Ng, W.L.; Qi, J.T.Z.; Yeong, W.Y.; Naing, M.W. Proof-of-concept: 3D bioprinting of pigmented human skin constructs. Biofabrication 2018, 10, 025005. [Google Scholar] [CrossRef]
- Sutherland, R.M. Cell and environment interactions in tumor microregions: The multicell spheroid model. Science 1988, 240, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Ryu, N.E.; Lee, S.H.; Park, H. Spheroid Culture System Methods and Applications for Mesenchymal Stem Cells. Cells 2019, 8, 1620. [Google Scholar] [CrossRef]
- Klicks, J.; Maßlo, C.; Kluth, A.; Rudolf, R.; Hafner, M. A novel spheroid-based co-culture model mimics loss of keratinocyte differentiation, melanoma cell invasion, and drug-induced selection of ABCB5-expressing cells. BMC Cancer 2019, 19, 402. [Google Scholar] [CrossRef]
- Białkowska, K.; Komorowski, P.; Bryszewska, M.; Miłowska, K. Spheroids as a Type of Three-Dimensional Cell Cultures-Examples of Methods of Preparation and the Most Important Application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef]
- Gong, X.; Lin, C.; Cheng, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of Multicellular Tumor Spheroids with Microwell-Based Agarose Scaffolds for Drug Testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef]
- Kelm, J.M.; Ehler, E.; Nielsen, L.K.; Schlatter, S.; Perriard, J.-C.; Fussenegger, M. Design of Artificial Myocardial Microtissues. Tissue Eng. 2004, 10, 201–214. [Google Scholar] [CrossRef]
- Jorgensen, A.M.; Varkey, M.; Gorkun, A.; Clouse, C.; Xu, L.; Chou, Z.; Murphy, S.V.; Molnar, J.; Lee, S.J.; Yoo, J.J.; et al. Bioprinted Skin Recapitulates Normal Collagen Remodeling in Full-Thickness Wounds. Tissue Eng. Part A 2019, 26, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Pathak, S.; Jeong, J.H. Design and manufacture of 3D cell culture plate for mass production of cell-spheroids. Sci. Rep. 2019, 9, 13976. [Google Scholar] [CrossRef] [PubMed]
- Souza, G.R.; Molina, J.R.; Raphael, R.M.; Ozawa, M.G.; Stark, D.J.; Levin, C.S.; Bronk, L.F.; Ananta, J.S.; Mandelin, J.; Georgescu, M.M.; et al. Three-dimensional tissue culture based on magnetic cell levitation. Nat. Nanotechnol. 2010, 5, 291–296. [Google Scholar] [CrossRef]
- Verjans, E.T.; Doijen, J.; Luyten, W.; Landuyt, B.; Schoofs, L. Three-dimensional cell culture models for anticancer drug screening: Worth the effort? J. Cell Physiol. 2018, 233, 2993–3003. [Google Scholar] [CrossRef]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control Release 2012, 164, 192–204. [Google Scholar] [CrossRef]
- Vadivelu, R.K.; Kamble, H.; Shiddiky, M.J.A.; Nguyen, N.-T. Microfluidic Technology for the Generation of Cell Spheroids and Their Applications. Micromachines 2017, 8, 94. [Google Scholar] [CrossRef]
- Kuriu, S.; Kadonosono, T.; Kizaka-Kondoh, S.; Ishida, T. Slicing Spheroids in Microfluidic Devices for Morphological and Immunohistochemical Analysis. Micromachines 2020, 11, 480. [Google Scholar] [CrossRef]
- Hunter, L.; Gala de Pablo, J.; Stammers, A.C.; Thomson, N.H.; Evans, S.D.; Shim, J.-u. On-chip pressure measurements and channel deformation after oil absorption. SN Appl. Sci. 2020, 2, 1501. [Google Scholar] [CrossRef]
- Huang, L.; Abdalla, A.M.E.; Xiao, L.; Yang, G. Biopolymer-Based Microcarriers for Three-Dimensional Cell Culture and Engineered Tissue Formation. Int. J. Mol. Sci. 2020, 21, 1895. [Google Scholar] [CrossRef] [PubMed]
- Antoni, D.; Burckel, H.; Josset, E.; Noel, G. Three-dimensional cell culture: A breakthrough in vivo. Int. J. Mol. Sci. 2015, 16, 5517–5527. [Google Scholar] [CrossRef] [PubMed]
- Marconi, A.; Borroni, R.G.; Truzzi, F.; Longo, C.; Pistoni, F.; Pellacani, G.; Pincelli, C. Hypoxia-Inducible Factor-1α and CD271 inversely correlate with melanoma invasiveness. Exp. Dermatol. 2015, 24, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Saltari, A.; Truzzi, F.; Quadri, M.; Lotti, R.; Palazzo, E.; Grisendi, G.; Tiso, N.; Marconi, A.; Pincelli, C. CD271 Down-Regulation Promotes Melanoma Progression and Invasion in Three-Dimensional Models and in Zebrafish. J. Investig. Dermatol. 2016, 136, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, D.; Parikh, N.; Sudol, M.; Gallick, G.E. Stimulation of the protein tyrosine kinase c-Yes but not c-Src by neurotrophins in human brain-metastatic melanoma cells. Oncogene 1998, 16, 3253–3260. [Google Scholar] [CrossRef]
- Saleh, N.A.; Rode, M.P.; Sierra, J.A.; Silva, A.H.; Miyake, J.A.; Filippin-Monteiro, F.B.; Creczynski-Pasa, T.B. Three-dimensional multicellular cell culture for anti-melanoma drug screening: Focus on tumor microenvironment. Cytotechnology 2021, 73, 35–48. [Google Scholar] [CrossRef]
- Shen, W.; Li, Y.; Li, B.; Zheng, L.; Xie, X.; Le, J.; Lu, Y.; Li, T.; Chen, F.; Jia, L. Downregulation of KCTD12 contributes to melanoma stemness by modulating CD271. Cancer Biol. Med. 2019, 16, 498–513. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, S.; Wang, J.; Zhang, Y.; Xiong, Y.; Zeng, S.X.; Lu, H. Inactivating p53 is essential for nerve growth factor receptor to promote melanoma-initiating cell-stemmed tumorigenesis. Cell Death Dis. 2020, 11, 550. [Google Scholar] [CrossRef]
- Koroknai, V.; Patel, V.; Szász, I.; Ádány, R.; Balazs, M. Gene Expression Signature of BRAF Inhibitor Resistant Melanoma Spheroids. Pathol. Oncol. Res. 2020, 26, 2557–2566. [Google Scholar] [CrossRef]
- Budden, T.; Gaudy-Marqueste, C.; Porter, A.; Kay, E.; Gurung, S.; Earnshaw, C.H.; Roeck, K.; Craig, S.; Traves, V.; Krutmann, J.; et al. Ultraviolet light-induced collagen degradation inhibits melanoma invasion. Nat. Commun. 2021, 12, 2742. [Google Scholar] [CrossRef]
- Bourland, J.; Fradette, J.; Auger, F.A. Tissue-engineered 3D melanoma model with blood and lymphatic capillaries for drug development. Sci. Rep. 2018, 8, 13191. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fang, J.; Huang, S.; Wu, X.; Xie, X.; Wang, J.; Liu, F.; Zhang, M.; Peng, Z.; Hu, N. Tumor-on-a-chip: From bioinspired design to biomedical application. Microsyst. Nanoeng. 2021, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Choudhury, D. Microfluidic bioprinting for organ-on-a-chip models. Drug Discov. Today 2019, 24, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, J.; Varadaraj, S.; Dash, S.K.; Sharma, A.; Verma, R.S. Organotypic cancer tissue models for drug screening: 3D constructs, bioprinting and microfluidic chips. Drug Discov. Today 2020, 25, 879–890. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Bolle, E.C.L.; Bartnikowski, N.; Haridas, P.; Parker, T.J.; Fraser, J.F.; Gregory, S.D.; Dargaville, T.R. Improving skin integration around long-term percutaneous devices using fibrous scaffolds in a reconstructed human skin equivalent model. J. Biomed. Mater. Res. Part B Appl. Biomater. 2020, 108, 738–749. [Google Scholar] [CrossRef]
- Mertsching, H.; Weimer, M.; Kersen, S.; Brunner, H. Human skin equivalent as an alternative to animal testing. GMS Krankenhhyg Interdiszip 2008, 3, Doc11. [Google Scholar]
- Li, L.; Fukunaga-Kalabis, M.; Herlyn, M. The Three-Dimensional Human Skin Reconstruct Model: A Tool to Study Normal Skin and Melanoma Progression. JoVE 2011, 54, e2937. [Google Scholar] [CrossRef]
- Pereira, R.F.; Bartolo, P.J. 3D bioprinting of photocrosslinkable hydrogel constructs. J. Appl. Polym. Sci. 2015, 132, 42458. [Google Scholar] [CrossRef]
- Weng, T.; Zhang, W.; Xia, Y.; Wu, P.; Yang, M.; Jin, R.; Xia, S.; Wang, J.; You, C.; Han, C.; et al. 3D bioprinting for skin tissue engineering: Current status and perspectives. J. Tissue Eng. 2021, 12, 20417314211028574. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.F.; Sousa, A.; Barrias, C.C.; Bayat, A.; Granja, P.L.; Bártolo, P.J. Advances in bioprinted cell-laden hydrogels for skin tissue engineering. Biomanuf. Rev. 2017, 2, 1–26. [Google Scholar] [CrossRef]
- Zhou, F.; Hong, Y.; Liang, R.; Zhang, X.; Liao, Y.; Jiang, D.; Zhang, J.; Sheng, Z.; Xie, C.; Peng, Z.; et al. Rapid printing of bio-inspired 3D tissue constructs for skin regeneration. Biomaterials 2020, 258, 120287. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Courseaus, J.; Khamassi, J.; Nottrodt, N.; Engelhardt, S.; Jacobsen, F.; Bierwisch, C.; Meyer, W.; Walter, T.; Weisser, J.; et al. Optimized vascular network by stereolithography for tissue engineered skin. Int. J. Bioprint. 2018, 4, 134. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.; Singh, G.; Trasatti, J.P.; Bjornsson, C.; Xu, X.; Tran, T.N.; Yoo, S.S.; Dai, G.; Karande, P. Design and fabrication of human skin by three-dimensional bioprinting. Tissue Eng. Part C Methods 2014, 20, 473–484. [Google Scholar] [CrossRef]
- Lee, W.; Debasitis, J.C.; Lee, V.K.; Lee, J.H.; Fischer, K.; Edminster, K.; Park, J.K.; Yoo, S.S. Multi-layered culture of human skin fibroblasts and keratinocytes through three-dimensional freeform fabrication. Biomaterials 2009, 30, 1587–1595. [Google Scholar] [CrossRef]
- Cadau, S.; Rival, D.; Andre-Frei, V.; Chavan, M.M.; Fayol, D.; Salducci, M.; Brisson, B.; Guillemot, F. New bioprinted skin, cosmetic in vitro model. J. Cosmet. Sci. 2017, 68, 85–90. [Google Scholar]
- Michael, S.; Sorg, H.; Peck, C.T.; Koch, L.; Deiwick, A.; Chichkov, B.; Vogt, P.M.; Reimers, K. Tissue engineered skin substitutes created by laser-assisted bioprinting form skin-like structures in the dorsal skin fold chamber in mice. PLoS ONE 2013, 8, e57741. [Google Scholar] [CrossRef]
- Cubo, N.; Garcia, M.; Del Canizo, J.F.; Velasco, D.; Jorcano, J.L. 3D bioprinting of functional human skin: Production and in vivo analysis. Biofabrication 2016, 9, 015006. [Google Scholar] [CrossRef]
- Skardal, A.; Murphy, S.V.; Crowell, K.; Mack, D.; Atala, A.; Soker, S. A tunable hydrogel system for long-term release of cell-secreted cytokines and bioprinted in situ wound cell delivery. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 1986–2000. [Google Scholar] [CrossRef]
- Shin, J.U.; Abaci, H.E.; Herron, L.; Guo, Z.; Sallee, B.; Pappalardo, A.; Jackow, J.; Wang, E.H.C.; Doucet, Y.; Christiano, A.M. Recapitulating T cell infiltration in 3D psoriatic skin models for patient-specific drug testing. Sci. Rep. 2020, 10, 4123. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.K.; Schmid, R.; Arkudas, A.; Kengelbach-Weigand, A.; Bosserhoff, A.K. Tumor Cells Develop Defined Cellular Phenotypes After 3D-Bioprinting in Different Bioinks. Cells 2019, 8, 1295. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-M.; Bang, C.; Park, M.; Shin, S.; Yun, S.; Kim, C.M.; Jeong, G.; Chung, Y.-J.; Yun, W.-S.; Lee, J.H.; et al. 3D-Printed Collagen Scaffolds Promote Maintenance of Cryopreserved Patients-Derived Melanoma Explants. Cells 2021, 10, 589. [Google Scholar] [CrossRef]
- Chimene, D.; Kaunas, R.; Gaharwar, A.K. Hydrogel Bioink Reinforcement for Additive Manufacturing: A Focused Review of Emerging Strategies. Adv. Mater. 2020, 32, 1902026. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Li, J.; Hartanto, Y.; Durham, M.; Tang, J.; Zhang, H.; Hooper, G.; Lim, K.; Woodfield, T. Advances in Extrusion 3D Bioprinting: A Focus on Multicomponent Hydrogel-Based Bioinks. Adv. Healthc. Mater. 2020, 9, e1901648. [Google Scholar] [CrossRef]
- Deo, K.A.; Singh, K.A.; Peak, C.W.; Alge, D.L.; Gaharwar, A.K. Bioprinting 101: Design, Fabrication, and Evaluation of Cell-Laden 3D Bioprinted Scaffolds. Tissue Eng. Part A 2020, 26, 318–338. [Google Scholar] [CrossRef]
- Groll, J.; Burdick, J.A.; Cho, D.W.; Derby, B.; Gelinsky, M.; Heilshorn, S.C.; Jüngst, T.; Malda, J.; Mironov, V.A.; Nakayama, K.; et al. A definition of bioinks and their distinction from biomaterial inks. Biofabrication 2018, 11, 013001. [Google Scholar] [CrossRef]
- Gungor-Ozkerim, P.S.; Inci, I.; Zhang, Y.S.; Khademhosseini, A.; Dokmeci, M.R. Bioinks for 3D bioprinting: An overview. Biomater. Sci. 2018, 6, 915–946. [Google Scholar] [CrossRef]
- Holzl, K.; Lin, S.; Tytgat, L.; Van Vlierberghe, S.; Gu, L.; Ovsianikov, A. Bioink properties before, during and after 3D bioprinting. Biofabrication 2016, 8, 032002. [Google Scholar] [CrossRef]
- Hospodiuk, M.; Moncal, K.K.; Dey, M.; Ozbolat, I.T. Extrusion-Based Biofabrication in Tissue Engineering and Regenerative Medicine. In 3D Printing and Biofabrication; Ovsianikov, A., Yoo, J., Mironov, V., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 255–281. [Google Scholar]
- Khoeini, R.; Nosrati, H.; Akbarzadeh, A.; Eftekhari, A.; Kavetskyy, T.; Khalilov, R.; Ahmadian, E.; Nasibova, A.; Datta, P.; Roshangar, L.; et al. Natural and Synthetic Bioinks for 3D Bioprinting. Adv. NanoBiomed Res. 2021, 1, 2000097. [Google Scholar] [CrossRef]
- Levato, R.; Jungst, T.; Scheuring, R.G.; Blunk, T.; Groll, J.; Malda, J. From Shape to Function: The Next Step in Bioprinting. Adv. Mater. 2020, 32, 1906423. [Google Scholar] [CrossRef]
- Morgan, F.L.C.; Moroni, L.; Baker, M.B. Dynamic Bioinks to Advance Bioprinting. Adv. Healthc. Mater. 2020, 9, e1901798. [Google Scholar] [CrossRef]
- Williams, D.; Thayer, P.; Martinez, H.; Gatenholm, E.; Khademhosseini, A. A perspective on the physical, mechanical and biological specifications of bioinks and the development of functional tissues in 3D bioprinting. Bioprinting 2018, 9, 19–36. [Google Scholar] [CrossRef]
- Daikuara, L.Y.; Chen, X.; Yue, Z.; Skropeta, D.; Wood, F.M.; Fear, M.W.; Wallace, G.G. 3D Bioprinting Constructs to Facilitate Skin Regeneration. Adv. Funct. Mater. 2022, 32, 2105080. [Google Scholar] [CrossRef]
- Kohli, N.; Sharma, V.; Brown, S.J.; García-Gareta, E. 5-Synthetic polymers for skin biomaterials. In Biomaterials for Skin Repair and Regeneration; García-Gareta, E., Ed.; Woodhead Publishing: Sawston, UK, 2019; pp. 125–149. [Google Scholar]
- ter Horst, B.; Moiemen, N.S.; Grover, L.M. Natural Polymers: Biomaterials for Skin Scaffolds in Biomaterials for Skin Repair and Regeneration; Woodhead Publishing: Sawston, UK, 2019; pp. 151–192. [Google Scholar]
- Retting, K.N.; Nguyen, D.G. Additive manufacturing in the development of 3D skin tissues. In Skin Tissue Models for Regenerative Medicine; Academic Press: Cambridge, MA, USA, 2018; pp. 377–397. [Google Scholar]
- Huang, G.; Li, F.; Zhao, X.; Ma, Y.; Li, Y.; Lin, M.; Jin, G.; Lu, T.J.; Genin, G.M.; Xu, F. Functional and Biomimetic Materials for Engineering of the Three-Dimensional Cell Microenvironment. Chem. Rev. 2017, 117, 12764–12850. [Google Scholar] [CrossRef]
- Zhang Yu, S.; Khademhosseini, A. Advances in engineering hydrogels. Science 2017, 356, eaaf3627. [Google Scholar] [CrossRef]
- Pereira, R.F.; Lourenco, B.N.; Bartolo, P.J.; Granja, P.L. Bioprinting a Multifunctional Bioink to Engineer Clickable 3D Cellular Niches with Tunable Matrix Microenvironmental Cues. Adv. Healthc. Mater. 2021, 10, e2001176. [Google Scholar] [CrossRef]
- Madduma-Bandarage, U.S.K.; Madihally, S.V. Synthetic hydrogels: Synthesis, novel trends, and applications. J. Appl. Polym. Sci. 2021, 138, 50376. [Google Scholar] [CrossRef]
- Zhu, J. Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering. Biomaterials 2010, 31, 4639–4656. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Xiao, Y.; Zhang, S.; Wang, J. Advances in crosslinking strategies of biomedical hydrogels. Biomater. Sci. 2019, 7, 843–855. [Google Scholar] [CrossRef]
- Luo, W.; Liu, H.; Wang, C.; Qin, Y.; Liu, Q.; Wang, J. Bioprinting of Human Musculoskeletal Interface. Adv. Eng. Mater. 2019, 21, 1900019. [Google Scholar] [CrossRef]
- Placone, J.K.; Engler, A.J. Recent Advances in Extrusion-Based 3D Printing for Biomedical Applications. Adv. Healthc. Mater. 2018, 7, e1701161. [Google Scholar] [CrossRef]
- Ning, L.; Chen, X. A brief review of extrusion-based tissue scaffold bio-printing. Biotechnol. J. 2017, 12, 1600671. [Google Scholar] [CrossRef]
- Chen, D.X.B. Extrusion Bioprinting Of Scaffolds for Tissue Engineering Applications; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Zhao, J.; He, N. A mini-review of embedded 3D printing: Supporting media and strategies. J. Mater. Chem. B 2020, 8, 10474–10486. [Google Scholar] [CrossRef]
- McCormack, A.; Highley, C.B.; Leslie, N.R.; Melchels, F.P.W. 3D Printing in Suspension Baths: Keeping the Promises of Bioprinting Afloat. Trends Biotechnol. 2020, 38, 584–593. [Google Scholar] [CrossRef]
- Kjar, A.; McFarland, B.; Mecham, K.; Harward, N.; Huang, Y. Engineering of tissue constructs using coaxial bioprinting. Bioact. Mater. 2021, 6, 460–471. [Google Scholar] [CrossRef]
- Gudapati, H.; Dey, M.; Ozbolat, I. A comprehensive review on droplet-based bioprinting: Past, present and future. Biomaterials 2016, 102, 20–42. [Google Scholar] [CrossRef]
- Zhou, D.; Chen, J.; Liu, B.; Zhang, X.; Li, X.; Xu, T. Bioinks for jet-based bioprinting. Bioprinting 2019, 16, e00060. [Google Scholar] [CrossRef]
- Li, X.; Liu, B.; Pei, B.; Chen, J.; Zhou, D.; Peng, J.; Zhang, X.; Jia, W.; Xu, T. Inkjet Bioprinting of Biomaterials. Chem. Rev. 2020, 120, 10793–10833. [Google Scholar] [CrossRef]
- Saunders, R.E.; Derby, B. Inkjet printing biomaterials for tissue engineering: Bioprinting. Int. Mater. Rev. 2014, 59, 430–448. [Google Scholar] [CrossRef]
- Guillemot, F.; Souquet, A.; Catros, S.; Guillotin, B. Laser-assisted cell printing: Principle, physical parameters versus cell fate and perspectives in tissue engineering. Nanomedicine 2010, 5, 507–515. [Google Scholar] [CrossRef]
- Koch, L.; Kuhn, S.; Sorg, H.; Gruene, M.; Schlie, S.; Gaebel, R.; Polchow, B.; Reimers, K.; Stoelting, S.; Ma, N.; et al. Laser Printing of Skin Cells and Human Stem Cells. Tissue Eng. Part C Methods 2010, 16, 847–854. [Google Scholar] [CrossRef]
- Guillotin, B.; Souquet, A.; Catros, S.; Duocastella, M.; Pippenger, B.; Bellance, S.; Bareille, R.; Remy, M.; Bordenave, L.; Amedee, J.; et al. Laser assisted bioprinting of engineered tissue with high cell density and microscale organization. Biomaterials 2010, 31, 7250–7256. [Google Scholar] [CrossRef]
- Antoshin, A.A.; Churbanov, S.N.; Minaev, N.V.; Zhang, D.; Zhang, Y.; Shpichka, A.I.; Timashev, P.S. LIFT-bioprinting, is it worth it? Bioprinting 2019, 15, e00052. [Google Scholar] [CrossRef]
- Dou, C.; Perez, V.; Qu, J.; Tsin, A.; Xu, B.; Li, J. A State-of-the-Art Review of Laser-Assisted Bioprinting and its Future Research Trends. ChemBioEng Rev. 2021, 8, 517–534. [Google Scholar] [CrossRef]
- Zheng, Z.; Eglin, D.; Alini, M.; Richards, G.R.; Qin, L.; Lai, Y. Visible Light-Induced 3D Bioprinting Technologies and Corresponding Bioink Materials for Tissue Engineering: A Review. Engineering 2021, 7, 966–978. [Google Scholar] [CrossRef]
- Bernal, P.N.; Delrot, P.; Loterie, D.; Li, Y.; Malda, J.; Moser, C.; Levato, R. Volumetric Bioprinting of Complex Living-Tissue Constructs within Seconds. Adv. Mater. 2019, 31, 1904209. [Google Scholar] [CrossRef]
- Kelly Brett, E.; Bhattacharya, I.; Heidari, H.; Shusteff, M.; Spadaccini Christopher, M.; Taylor Hayden, K. Volumetric additive manufacturing via tomographic reconstruction. Science 2019, 363, 1075–1079. [Google Scholar] [CrossRef]
- Loterie, D.; Delrot, P.; Moser, C. High-resolution tomographic volumetric additive manufacturing. Nat. Commun. 2020, 11, 852. [Google Scholar] [CrossRef]
- Abaci, H.E.; Coffman, A.; Doucet, Y.; Chen, J.; Jacków, J.; Wang, E.; Guo, Z.; Shin, J.U.; Jahoda, C.A.; Christiano, A.M. Tissue engineering of human hair follicles using a biomimetic developmental approach. Nat. Commun. 2018, 9, 5301. [Google Scholar] [CrossRef]
- Huang, S.; Yao, B.; Xie, J.; Fu, X. 3D bioprinted extracellular matrix mimics facilitate directed differentiation of epithelial progenitors for sweat gland regeneration. Acta Biomater. 2016, 32, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Wang, R.; Wang, Y.; Zhang, Y.; Hu, T.; Song, W.; Li, Z.; Huang, S.; Fu, X. Biochemical and structural cues of 3D-printed matrix synergistically direct MSC differentiation for functional sweat gland regeneration. Sci. Adv. 2020, 6, eaaz1094. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Enhejirigala; Yao, B.; Li, Z.; Song, W.; Li, J.; Zhu, D.; Wang, Y.; Duan, X.; Yuan, X.; et al. Using bioprinting and spheroid culture to create a skin model with sweat glands and hair follicles. Burn. Trauma 2021, 9, tkab013. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Au, P.; Tam, J.; Duda, D.G.; Fukumura, D. Engineering vascularized tissue. Nat. Biotechnol. 2005, 23, 821–823. [Google Scholar] [CrossRef]
- Vyas, C.; Pereira, R.; Huang, B.; Liu, F.; Wang, W.; Bartolo, P. Engineering the vasculature with additive manufacturing. Curr. Opin. Biomed. Eng. 2017, 2, 1–13. [Google Scholar] [CrossRef]
- Abaci, H.E.; Guo, Z.; Coffman, A.; Gillette, B.; Lee, W.H.; Sia, S.K.; Christiano, A.M. Human Skin Constructs with Spatially Controlled Vasculature Using Primary and iPSC-Derived Endothelial Cells. Adv. Healthc. Mater. 2016, 5, 1800–1807. [Google Scholar] [CrossRef]
- Yanez, M.; Rincon, J.; Dones, A.; De Maria, C.; Gonzales, R.; Boland, T. In Vivo Assessment of Printed Microvasculature in a Bilayer Skin Graft to Treat Full-Thickness Wounds. Tissue Eng. Part A 2014, 21, 224–233. [Google Scholar] [CrossRef]
- Mori, N.; Morimoto, Y.; Takeuchi, S. Skin integrated with perfusable vascular channels on a chip. Biomaterials 2017, 116, 48–56. [Google Scholar] [CrossRef]
- Jin, R.; Cui, Y.; Chen, H.; Zhang, Z.; Weng, T.; Xia, S.; Yu, M.; Zhang, W.; Shao, J.; Yang, M.; et al. Three-dimensional bioprinting of a full-thickness functional skin model using acellular dermal matrix and gelatin methacrylamide bioink. Acta Biomater. 2021, 131, 248–261. [Google Scholar] [CrossRef]
- Barros, N.R.; Kim, H.-J.; Gouidie, M.J.; Lee, K.; Bandaru, P.; Banton, E.A.; Sarikhani, E.; Sun, W.; Zhang, S.; Cho, H.-J.; et al. Biofabrication of endothelial cell, dermal fibroblast, and multilayered keratinocyte layers for skin tissue engineering. Biofabrication 2021, 13, 035030. [Google Scholar] [CrossRef]
- Pontiggia, L.; Van Hengel, I.A.J.; Klar, A.; Rütsche, D.; Nanni, M.; Scheidegger, A.; Figi, S.; Reichmann, E.; Moehrlen, U.; Biedermann, T. Bioprinting and plastic compression of large pigmented and vascularized human dermo-epidermal skin substitutes by means of a new robotic platform. J. Tissue Eng. 2022, 13, 20417314221088513. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Somasundaram, R.; Herlyn, M. Pre-clinical modeling of cutaneous melanoma. Nat. Commun. 2020, 11, 2858. [Google Scholar] [CrossRef]
- Schmid, R.; Schmidt, S.K.; Hazur, J.; Detsch, R.; Maurer, E.; Boccaccini, A.R.; Hauptstein, J.; Teßmar, J.; Blunk, T.; Schrüfer, S.; et al. Comparison of Hydrogels for the Development of Well-Defined 3D Cancer Models of Breast Cancer and Melanoma. Cancers 2020, 12, 2320. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.; Schmidt, S.K.; Detsch, R.; Horder, H.; Blunk, T.; Schrüfer, S.; Schubert, D.W.; Fischer, L.; Thievessen, I.; Heltmann-Meyer, S.; et al. A New Printable Alginate/Hyaluronic Acid/Gelatin Hydrogel Suitable for Biofabrication of In Vitro and In Vivo Metastatic Melanoma Models. Adv. Funct. Mater. 2021, 32, 2107993. [Google Scholar] [CrossRef]
- Kozlowski, M.T.; Crook, C.J.; Ku, H.T. Towards organoid culture without Matrigel. Commun. Biol. 2021, 4, 1387. [Google Scholar] [CrossRef]
- Zhang, Y.; Ellison, S.T.; Duraivel, S.; Morley, C.D.; Taylor, C.R.; Angelini, T.E. 3D printed collagen structures at low concentrations supported by jammed microgels. Bioprinting 2021, 21, e00121. [Google Scholar] [CrossRef]
- Lee, A.; Hudson, A.R.; Shiwarski, D.J.; Tashman, J.W.; Hinton, T.J.; Yerneni, S.; Bliley, J.M.; Campbell, P.G.; Feinberg, A.W. 3D bioprinting of collagen to rebuild components of the human heart. Science 2019, 365, 482–487. [Google Scholar] [CrossRef]
- Lee, J.M.; Suen, S.K.Q.; Ng, W.L.; Ma, W.C.; Yeong, W.Y. Bioprinting of Collagen: Considerations, Potentials, and Applications. Macromol. Biosci. 2021, 21, 2000280. [Google Scholar] [CrossRef]
- Isaacson, A.; Swioklo, S.; Connon, C.J. 3D bioprinting of a corneal stroma equivalent. Exp. Eye Res. 2018, 173, 188–193. [Google Scholar] [CrossRef]
- Kim, H.; Jang, J.; Park, J.; Lee, K.-P.; Lee, S.; Lee, D.-M.; Kim, K.H.; Kim, H.K.; Cho, D.-W. Shear-induced alignment of collagen fibrils using 3D cell printing for corneal stroma tissue engineering. Biofabrication 2019, 11, 035017. [Google Scholar] [CrossRef] [PubMed]
- Kilic Bektas, C.; Hasirci, V. Cell loaded 3D bioprinted GelMA hydrogels for corneal stroma engineering. Biomater. Sci. 2020, 8, 438–449. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Activated | Gene | Common Mutations | Subtype | Role during Progression | Refs. |
|---|---|---|---|---|---|
| MAPK | BRAF | V600E | Non-CSD | Initiation | [18,22,23] |
| BRAF | V600K, K601E and G469A | CSD | Initiation | [18,24] | |
| NRAS | Q61R (cutaneous melanoma), Q61K | CSD | Initiation | [18,23,25] | |
| NFI | Deletion which leads to no neurofibromin | CSD | Initiation | [18,23,26] | |
| Telomerase | TERT | Mutations in hTERT promoter leading to deregulation of cell cycle and immortalisation of cancer cells | CSD and non-CSD | Progression | [18,23,27] |
| Retinoblastoma protein | CDKN2A | Disables mutations occurring throughout the protein | CSD and non-CSD | Progression | [18,23,28] |
| Chromatin remodelling | ARID1A, ARID1B and/or ARID2 | Disables mutations occurring throughout the protein | [18,23,29] | ||
| PI3K | PTEN | Disables mutations occurring throughout the protein + deletions | Non-CSD | Advanced Progression | [18,23,30] |
| p53 | TP53 | Disables mutations occurring throughout the protein | CSD | Advanced Progression | [18] |
| TNM | Stage in Progression | Description |
|---|---|---|
| N/A | Melanocytic naevi (moles) |
|
| 0 | Melanoma in situ | |
| I | Invasive melanoma |
|
| II |  | |
| III | Advanced progression into lymph nodes | |
| IV | Metastatic melanoma |
|
| Method | Features | |
|---|---|---|
Scaffold based | Porous non-adherent 3D scaffold which physically supports cell aggregation allowing formation of spheroids with a controlled size [113] | Advantages: good tensile strength compared to other methods [10] Limitations: Simplified architecture. Can be variable across lots [114] |
Hanging drop | Drops of cell suspension are placed on the underside of a petri dish lid which hang due to surface tension. The cells then accumulate at the tip of the drop at air–liquid interface upon which they aggregate and form spheroids [115,116]. | Advantages: Can produce ~384 spheroids per trial; controllability of spheroid size; does not require specialised equipment [10,117] Limitations: Risk of droplet dehydration; time required for spheroid formation; difficult to scale up [118,119] |
Magnetic levitation | Cells are magnetised through a mixture of magnetic particles and incubated under magnetic forces to overcome gravitational forces, encouraging levitation and the formation of cellular aggregates [110,120]. | Advantages: The speed of spheroid growth is high; forms intrinsic ECM; does not require specific medium [117] Limitations: Requires magnetic beads which can be expensive and toxic to cells; produces a limited number of spheroids [119,121]. |
Spontaneous formation | Uses ultra-low attachment plates coated with an inert substance (usually agar or poly-2-hydroxyethyl methacrylate (poly-HEMA)) which inhibits cells from attaching to the surface of the wells, thereby forcing cells to amass and form spheroids. | Advantages: Easy to use; inexpensive; large scale production [10]. Limitations: Low control over the size of the spheroids; spheroids are produced through a small number of cells, therefore, setting up the ratio of two different cell types in co-cultures can be difficult [10]. |
Microfluidic platforms | Cells are placed in microchannels with a free perfusion system which allows the continuous and uniform distribution of oxygen and nutrients and the elimination of waste [117]. This system can replicate the in vivo tumour microvasculature and guarantees the permeability of gases. | Advantages: Can mimic the tumour vasculature; high throughput drug screening; continuous release of oxygen and nutrients [122,123,124,125]. Limitations: Requires specialised equipment; post culture recovery can be difficult; difficult to precisely control the flow speed [122,123,124,125] |
Matrix encapsulation | Suspended cells are surrounded by the hydrogel and placed in a calcium free solution which forms microcapsules (100 and 500 µm) in which cells aggregate to form matrix encapsulated spheroids [118]. | Advantages: Enables cell–cell and cell–ECM interaction; simple to use and inexpensive [10,118] Limitations: High chance of cell necrosis due to confinement; size heterogeneity [10,118] |
Spinner and rotating flasks | The medium is continuously agitated, inhibiting cell adhesion to the surface and leading to spheroid formation [117]. A magnetic stirrer is placed inside the spinner flask allowing the homogenous distribution of oxygen and nutrients. In continuous rotating flasks, the flask itself is rotated to allow the distribution of oxygen and nutrients. | Advantages: Large scale spheroid production; simple media changing; constant agitation provides continuous nutrients and oxygen transportation [10,118] Limitations: Cells undergo shearing under high agitation which can damage cells; slow agitation results in cell dispersion; spheroids are heterogenous ([10,118,121]) |
Microcarrier beads | Cells adhere to the natural or synthetic matrix coated beads which form spheroidal structures [126] | Advantages: Fast and inexpensive method; produced spheroids are homogenous [88,126,127]. Provides a good cell attachment surface which allows aggregation, especially for those cells which are unable to aggregate spontaneously [117]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, S.; Vyas, C.; Lim, P.; Pereira, R.F.; Virós, A.; Bártolo, P. 3D Bioprinting: An Enabling Technology to Understand Melanoma. Cancers 2022, 14, 3535. https://doi.org/10.3390/cancers14143535
Fernandes S, Vyas C, Lim P, Pereira RF, Virós A, Bártolo P. 3D Bioprinting: An Enabling Technology to Understand Melanoma. Cancers. 2022; 14(14):3535. https://doi.org/10.3390/cancers14143535
Chicago/Turabian StyleFernandes, Samantha, Cian Vyas, Peggy Lim, Rúben F. Pereira, Amaya Virós, and Paulo Bártolo. 2022. "3D Bioprinting: An Enabling Technology to Understand Melanoma" Cancers 14, no. 14: 3535. https://doi.org/10.3390/cancers14143535
APA StyleFernandes, S., Vyas, C., Lim, P., Pereira, R. F., Virós, A., & Bártolo, P. (2022). 3D Bioprinting: An Enabling Technology to Understand Melanoma. Cancers, 14(14), 3535. https://doi.org/10.3390/cancers14143535