
Is Cell-Free DNA Testing in Pancreatic Ductal Adenocarcinoma Ready for Prime Time?


Abstract
:Simple Summary
Abstract

1. Introduction
1.1. Current Management of PDA: Treatment and Disease Monitoring
1.2. Critical Needs in Pancreatic Adenocarcinoma Management
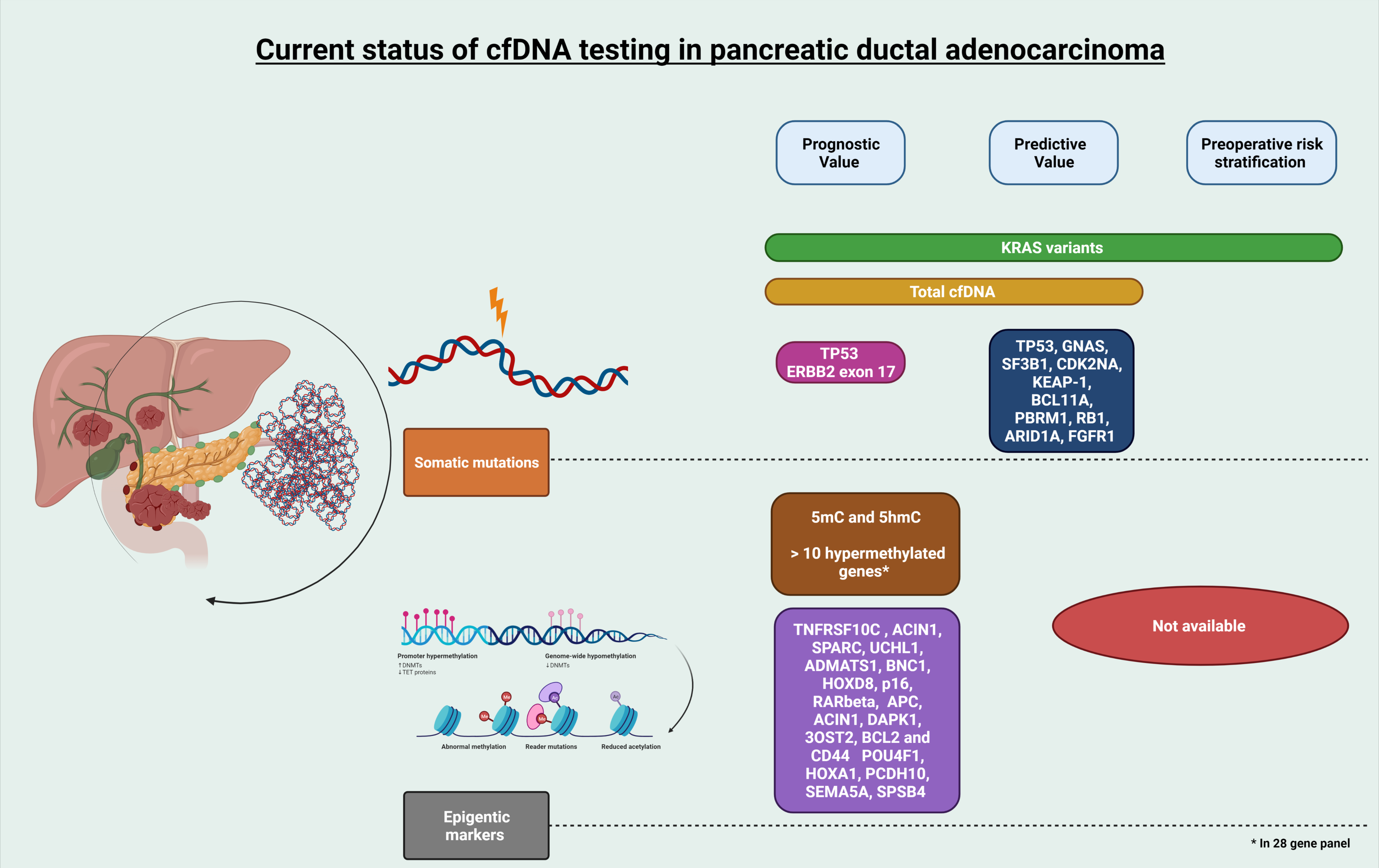
2. Detection of cfDNA in PDA
2.1. cfDNA Testing in PDA, beyond KRAS
2.2. cfDNA Epigenetic Markers
3. Prognostic Value of cfDNA in PDA
3.1. cfDNA KRAS Mutations
3.2. cfDNA KRAS with Other Mutations in PDA
3.3. cfDNA Epigenetic Markers
4. cfDNA for POR
5. Predictive Value of cfDNA Markers
cfDNA Somatic Mutations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Surveillance, Epidemiology, and End Results (SEER) Program. SEER*Stat Database: Incidence—SEER Research Data, 9 Registries, Nov 2019 Sub (1975–2017). Available online: www.seer.cancer.gov (accessed on 15 January 2022).
- Cao, F.; Wei, A.; Hu, X.; He, Y.; Zhang, J.; Xia, L.; Tu, K.; Yuan, J.; Guo, Z.; Liu, H.; et al. Integrated epigenetic biomarkers in circulating cell-free DNA as a robust classifier for pancreatic cancer. Clin. Epigenet. 2020, 12, 112. [Google Scholar] [CrossRef]
- Callery, M.P.; Chang, K.J.; Fishman, E.K.; Talamonti, M.S.; William Traverso, L.; Linehan, D.C. Pretreatment assessment of resectable and borderline resectable pancreatic cancer: Expert consensus statement. Ann. Surg. Oncol. 2009, 16, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Jones, R.P.; Psarelli, E.-E.; Jackson, R.; Ghaneh, P.; Halloran, C.M.; Palmer, D.H.; Campbell, F.; Valle, J.W.; Faluyi, O.; O’Reilly, D.A.; et al. Patterns of Recurrence After Resection of Pancreatic Ductal Adenocarcinoma. JAMA Surg. 2019, 154, 1038. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B.; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M.; et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Gay, D.Z.; Brown, K.; Mulvihill, J.D.; Boucher, K.M.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. The clinical utility of CA 19-9 in pancreatic adenocarcinoma: Diagnostic and prognostic updates. Curr. Mol. Med. 2013, 13, 340–351. [Google Scholar] [CrossRef]
- Elbanna, K.Y.; Jang, H.J.; Kim, T.K. Imaging diagnosis and staging of pancreatic ductal adenocarcinoma: A comprehensive review. Insights Imaging 2020, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Tsen, A.; Barbara, M.; Rosenkranz, L. Dilemma of elevated CA 19-9 in biliary pathology. Pancreatology 2018, 18, 862–867. [Google Scholar] [CrossRef]
- Tempero, M.A.; Uchida, E.; Takasaki, H.; Burnett, D.A.; Steplewski, Z.; Pour, P.M. Relationship of carbohydrate antigen 19-9 and Lewis antigens in pancreatic cancer. Cancer Res. 1987, 47, 5501–5503. [Google Scholar]
- Liu, C.; Deng, S.; Jin, K.; Gong, Y.; Cheng, H.; Fan, Z.; Qian, Y.; Huang, Q.; Ni, Q.; Luo, G.; et al. Lewis antigen-negative pancreatic cancer: An aggressive subgroup. Int. J. Oncol. 2020, 56, 900–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Zhang, Z.-H.; Wang, S.; Lang, J.-H. Circulating Cell-Free DNA or Circulating Tumor DNA in the Management of Ovarian and Endometrial Cancer. OncoTargets Ther. 2019, 12, 11517–11530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashworth, T.R. A Case of Cancer in Which Cells Similar to Those in the Tumours Were Seen in the Blood after Death. Med. J. Aust. 1869, 14, 146–147. [Google Scholar]
- Engell, H.C. Cancer cells in the circulating blood; a clinical study on the occurrence of cancer cells in the peripheral blood and in venous blood draining the tumour area at operation. Acta Chir. Scand. Suppl. 1955, 201, 1–70. [Google Scholar] [PubMed]
- Mandel, P.; Metaise, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Vasioukhin, V.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Stroun, M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br. J. Haematol. 1994, 86, 774–779. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Mouliere, F.; Thierry, A.R. The importance of examining the proportion of circulating DNA originating from tumor, microenvironment and normal cells in colorectal cancer patients. Expert Opin. Biol. Ther. 2012, 12 (Suppl. 1), S209–S215. [Google Scholar] [CrossRef] [PubMed]
- Gall, T.M.H.; Belete, S.; Khanderia, E.; Frampton, A.E.; Jiao, L.R. Circulating Tumor Cells and Cell-Free DNA in Pancreatic Ductal Adenocarcinoma. Am. J. Pathol. 2019, 189, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorenson, G.D.; Pribish, D.M.; Valone, F.H.; Memoli, V.A.; Bzik, D.J.; Yao, S.L. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol. Biomark. Prev. 1994, 3, 67–71. [Google Scholar]
- George, B.; Haberberger, J.; Ferguson, N.L.; Gjoerup, O.; McGregor, K.; Hendifar, A.E.; Laheru, D.A.; Weekes, C.D.; Ross, J.S.; Hemmerich, A. Correlation between comprehensive genomic profiling (CGP) utilizing tissue-based testing (T-CGP) and cell-free DNA (cfDNA) in patients (pts) with pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2021, 39, 422. [Google Scholar] [CrossRef]
- George, B.; Haberberger, J.; Ferguson, N.L.; Gjoerup, O.; McGregor, K.; Hendifar, A.E.; Laheru, D.A.; Weekes, C.D.; Ross, J.S.; Hemmerich, A. Comprehensive genomic profiling (CGP) utilizing cell-free DNA (cfDNA) in patients (pts) with pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2021, 39, 421. [Google Scholar] [CrossRef]
- Botrus, G.; Fu, Y.; Sonbol, M.B.; Drusbosky, L.; Ahn, D.H.; Borad, M.J.; Starr, J.S.; Jones, J.C.; Raman, P.; Mody, K.; et al. Serial cell-free DNA (cfDNA) sampling in advanced pancreatic ductal adenocarcinoma (PDAC) patients may predict therapeutic outcome. J. Clin. Oncol. 2021, 39, 423. [Google Scholar] [CrossRef]
- Cheng, H.; Liu, C.; Jiang, J.; Luo, G.; Lu, Y.; Jin, K.; Guo, M.; Zhang, Z.; Xu, J.; Liu, L.; et al. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int. J. Cancer 2017, 140, 2344–2350. [Google Scholar] [CrossRef]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef] [Green Version]
- Luchini, C.; Veronese, N.; Nottegar, A.; Cappelletti, V.; Daidone, M.G.; Smith, L.; Parris, C.; Brosens, L.A.A.; Caruso, M.G.; Cheng, L.; et al. Liquid Biopsy as Surrogate for Tissue for Molecular Profiling in Pancreatic Cancer: A Meta-Analysis Towards Precision Medicine. Cancers 2019, 11, 1152. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Zhang, Q.; Li, X.; Su, W.; Li, G.; Ma, T.; Gao, S.; Lou, J.; Que, R.; Zheng, L.; et al. Monitoring Tumor Burden in Response to FOLFIRINOX Chemotherapy Via Profiling Circulating Cell-Free DNA in Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 196–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.K.; Woo, S.M.; Park, B.; Yoon, K.-A.; Kim, Y.H.; Joo, J.; Park, S.J.; Kong, S.-Y. Prognostic impact of KRAS mutation in cell-free DNA in patients with pancreatic cancer. Ann. Oncol. 2017, 28, V9. [Google Scholar] [CrossRef] [Green Version]
- Kinugasa, H.; Nouso, K.; Miyahara, K.; Morimoto, Y.; Dohi, C.; Tsutsumi, K.; Kato, H.; Matsubara, T.; Okada, H.; Yamamoto, K. Detection of K-rasgene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 2015, 121, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Brychta, N.; Krahn, T.; Von Ahsen, O. Detection of KRAS Mutations in Circulating Tumor DNA by Digital PCR in Early Stages of Pancreatic Cancer. Clin. Chem. 2016, 62, 1482–1491. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [Green Version]
- Melnikov, A.A.; Scholtens, D.; Talamonti, M.S.; Bentrem, D.J.; Levenson, V.V. Methylation profile of circulating plasma DNA in patients with pancreatic cancer. J. Surg. Oncol. 2009, 99, 119–122. [Google Scholar] [CrossRef]
- Liggett, T.; Melnikov, A.; Yi, Q.L.; Replogle, C.; Brand, R.; Kaul, K.; Talamonti, M.; Abrams, R.A.; Levenson, V. Differential methylation of cell-free circulating DNA among patients with pancreatic cancer versus chronic pancreatitis. Cancer 2010, 116, 1674–1680. [Google Scholar] [CrossRef]
- Yi, J.M.; Guzzetta, A.A.; Bailey, V.J.; Downing, S.R.; Van Neste, L.; Chiappinelli, K.B.; Keeley, B.P.; Stark, A.; Herrera, A.; Wolfgang, C.; et al. Novel methylation biomarker panel for the early detection of pancreatic cancer. Clin. Cancer Res. 2013, 19, 6544–6555. [Google Scholar] [CrossRef] [Green Version]
- Eissa, M.A.L.; Lerner, L.; Abdelfatah, E.; Shankar, N.; Canner, J.K.; Hasan, N.M.; Yaghoobi, V.; Huang, B.; Kerner, Z.; Takaesu, F.; et al. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin. Epigenet. 2019, 11, 59. [Google Scholar] [CrossRef]
- Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Johansen, M.B.; Drewes, A.M.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Cell-free DNA promoter hypermethylation in plasma as a diagnostic marker for pancreatic adenocarcinoma. Clin. Epigenet. 2016, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Guler, G.D.; Ning, Y.; Ku, C.J.; Phillips, T.; McCarthy, E.; Ellison, C.K.; Bergamaschi, A.; Collin, F.; Lloyd, P.; Scott, A.; et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat. Commun. 2020, 11, 5270. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L.; Zhao, Q.; Wang, Z.; Lu, S.; Kang, Y.; Jin, G.; Tian, J. Genome-Wide Analysis of Cell-Free DNA Methylation Profiling for the Early Diagnosis of Pancreatic Cancer. Front. Genet. 2020, 11, 596078. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Sharma, A.; Chhoda, A.; Ruzgar, N.; Hasan, N.; Kwak, R.; Wolfgang, C.L.; Wang, T.H.; Kunstman, J.W.; Salem, R.R.; et al. Methylation-based Cell-free DNA Signature for Early Detection of Pancreatic Cancer. Pancreas 2021, 50, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Manoochehri, M.; Wu, Y.; Giese, N.A.; Strobel, O.; Kutschmann, S.; Haller, F.; Hoheisel, J.D.; Moskalev, E.A.; Hackert, T.; Bauer, A.S. SST gene hypermethylation acts as a pan-cancer marker for pancreatic ductal adenocarcinoma and multiple other tumors: Toward its use for blood-based diagnosis. Mol. Oncol. 2020, 14, 1252–1267. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Rashid, S.; Dash, N.R.; Gupta, S.; Saraya, A. Clinical significance of promoter methylation status of tumor suppressor genes in circulating DNA of pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2020, 146, 897–907. [Google Scholar] [CrossRef]
- Shinjo, K.; Hara, K.; Nagae, G.; Umeda, T.; Katsushima, K.; Suzuki, M.; Murofushi, Y.; Umezu, Y.; Takeuchi, I.; Takahashi, S.; et al. A novel sensitive detection method for DNA methylation in circulating free DNA of pancreatic cancer. PLoS ONE 2020, 15, e0233782. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Suehiro, Y.; Kaino, S.; Suenaga, S.; Tsuyama, T.; Matsui, H.; Higaki, S.; Fujii, I.; Suzuki, C.; Hoshida, T.; et al. Combination of CA19-9 and Blood Free-Circulating Methylated RUNX3 May Be Useful to Diagnose Stage I Pancreatic Cancer. Oncology 2021, 99, 234–239. [Google Scholar] [CrossRef]
- Kandimalla, R.; Xu, J.; Link, A.; Matsuyama, T.; Yamamura, K.; Parker, M.I.; Uetake, H.; Balaguer, F.; Borazanci, E.; Tsai, S.; et al. EpiPanGI Dx: A Cell-free DNA Methylation Fingerprint for the Early Detection of Gastrointestinal Cancers. Clin. Cancer Res. 2021, 27, 6135–6144. [Google Scholar] [CrossRef]
- Vrba, L.; Futscher, B.W.; Oshiro, M.; Watts, G.S.; Menashi, E.; Hu, C.; Hammad, H.; Pennington, D.R.; Golconda, U.; Gavini, H.; et al. Liquid biopsy, using a novel DNA methylation signature, distinguishes pancreatic adenocarcinoma from benign pancreatic disease. Clin. Epigenet. 2022, 14, 28. [Google Scholar] [CrossRef]
- Li, X.B.; Ma, J.; Liu, Z.W.; He, W.F.; Li, Z.Z.; Cui, M.; Hao, H.; Zhou, G.P.; You, L.; Wang, J.; et al. Non-invasive detection of pancreatic cancer by measuring DNA methylation of Basonuclin 1 and Septin 9 in plasma. Chin. Med. J. 2019, 132, 1504–1506. [Google Scholar] [CrossRef]
- Melson, J.; Li, Y.; Cassinotti, E.; Melnikov, A.; Boni, L.; Ai, J.; Greenspan, M.; Mobarhan, S.; Levenson, V.; Deng, Y. Commonality and differences of methylation signatures in the plasma of patients with pancreatic cancer and colorectal cancer. Int. J. Cancer 2014, 134, 2656–2662. [Google Scholar] [CrossRef]
- Park, J.W.; Baek, I.H.; Kim, Y.T. Preliminary Study Analyzing the Methylated Genes in the Plasma of Patients with Pancreatic Cancer. Scand. J. Surg. 2012, 101, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.K.; Ryu, J.K.; Yoon, W.J.; Lee, S.H.; Lee, G.Y.; Jeong, K.S.; Kim, Y.T.; Yoon, Y.B. The role of quantitative NPTX2 hypermethylation as a novel serum diagnostic marker in pancreatic cancer. Pancreas 2012, 41, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, S.D.; Stubbe, B.E.; Madsen, P.H.; Johansen, J.S.; Jensen, B.V.; Hansen, C.P.; Johansen, M.N.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Cell-free DNA promoter hypermethylation as a diagnostic marker for pancreatic ductal adenocarcinoma—An external validation study. Pancreatology 2021, 21, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Yu, Y.; Suzuki, M.; Campbell, N.; Mazdo, J.; Vasanthakumar, A.; Bhagat, T.D.; Nischal, S.; Christopeit, M.; Parekh, S.; et al. Genome-wide hydroxymethylation tested using the HELP-GT assay shows redistribution in cancer. Nucleic Acids Res. 2013, 41, e157. [Google Scholar] [CrossRef] [Green Version]
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011, 473, 398–402. [Google Scholar] [CrossRef]
- Wang, Z.; Lu, Y.; Fornage, M.; Jiao, L.; Shen, J.; Li, D.; Wei, P. Identification of novel susceptibility methylation loci for pancreatic cancer in a two-phase epigenome-wide association study. Epigenetics 2022, 1–16. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Bamlet, W.R.; Oberg, A.L.; De Andrade, M.; Matsumoto, M.E.; Tang, H.; Thibodeau, S.N.; Petersen, G.M.; Wang, L. Leukocyte DNA Methylation Signature Differentiates Pancreatic Cancer Patients from Healthy Controls. PLoS ONE 2011, 6, e18223. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yang, Y.; Kisiel, J.B.; Mahoney, D.W.; Michaud, D.S.; Guo, X.; Taylor, W.R.; Shu, X.-O.; Shu, X.; Liu, D.; et al. Integrating Genome and Methylome Data to Identify Candidate DNA Methylation Biomarkers for Pancreatic Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2021, 30, 2079–2087. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castells, A.; Puig, P.; Móra, J.; Boadas, J.; Boix, L.; Urgell, E.; Solé, M.; Capellà, G.; Lluís, F.; Fernández-Cruz, L.; et al. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: Diagnostic utility and prognostic significance. J. Clin. Oncol. 1999, 17, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Adamo, P.; Cowley, C.M.; Neal, C.P.; Mistry, V.; Page, K.; Dennison, A.R.; Isherwood, J.; Hastings, R.; Luo, J.; Moore, D.A.; et al. Profiling tumour heterogeneity through circulating tumour DNA in patients with pancreatic cancer. Oncotarget 2017, 8, 87221–87233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ako, S.; Nouso, K.; Kinugasa, H.; Dohi, C.; Matushita, H.; Mizukawa, S.; Muro, S.; Akimoto, Y.; Uchida, D.; Tomoda, T.; et al. Utility of serum DNA as a marker for KRAS mutations in pancreatic cancer tissue. Pancreatology 2017, 17, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated With Outcomes of Patients With Pancreatic Cancer. Gastroenterology 2019, 156, 108–118.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Tu, H.; Meng, Z.Q.; Chen, Z.; Wang, P.; Liu, L.M. K-ras mutational status predicts poor prognosis in unresectable pancreatic cancer. Eur. J. Surg. Oncol. 2010, 36, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Luo, G.; Jin, K.; Fan, Z.; Huang, Q.; Gong, Y.; Xu, J.; Yu, X.; Liu, C. Kras mutation correlating with circulating regulatory T cells predicts the prognosis of advanced pancreatic cancer patients. Cancer Med. 2020, 9, 2153–2159. [Google Scholar] [CrossRef]
- Del Re, M.; Vivaldi, C.; Rofi, E.; Vasile, E.; Miccoli, M.; Caparello, C.; d’Arienzo, P.D.; Fornaro, L.; Falcone, A.; Danesi, R. Early changes in plasma DNA levels of mutant KRAS as a sensitive marker of response to chemotherapy in pancreatic cancer. Sci. Rep. 2017, 7, 7931. [Google Scholar] [CrossRef]
- Diehl, A.; Hannan, L.M.; Chiorean, E.G. Prognostic value of KRAS and PI3K pathway mutations for advanced pancreatic ductal adenocarcinoma (PDAC) patients (pts). J. Clin. Oncol. 2021, 39, 424. [Google Scholar] [CrossRef]
- Kim, M.K.; Woo, S.M.; Park, B.; Yoon, K.A.; Kim, Y.H.; Joo, J.; Lee, W.J.; Han, S.S.; Park, S.J.; Kong, S.Y. Prognostic Implications of Multiplex Detection of KRAS Mutations in Cell-Free DNA from Patients with Pancreatic Ductal Adenocarcinoma. Clin. Chem. 2018, 64, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Van Laethem, J.L.; Riess, H.; Jassem, J.; Haas, M.; Martens, U.M.; Weekes, C.; Peeters, M.; Ross, P.; Bridgewater, J.; Melichar, B.; et al. Phase I/II Study of Refametinib (BAY 86-9766) in Combination with Gemcitabine in Advanced Pancreatic cancer. Target. Oncol. 2017, 12, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Ayub, M.; Rothwell, D.G.; Gulati, S.; Kilerci, B.; Hollebecque, A.; Sun Leong, H.; Smith, N.K.; Sahoo, S.; Descamps, T.; et al. Analysis of circulating cell-free DNA identifies KRAS copy number gain and mutation as a novel prognostic marker in Pancreatic cancer. Sci. Rep. 2019, 9, 11610. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Greenberg, O.; Shentzer, T.; Semenisty, V.; Epelbaum, R.; Bick, T.; Sarji, S.; Ben-Izhak, O.; Sabo, E.; Hershkovitz, D. Mutant KRAS Circulating Tumor DNA Is an Accurate Tool for Pancreatic Cancer Monitoring. Oncologist 2018, 23, 566–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pishvaian, M.J.; Joseph Bender, R.; Matrisian, L.M.; Rahib, L.; Hendifar, A.; Hoos, W.A.; Mikhail, S.; Chung, V.; Picozzi, V.; Heartwell, C.; et al. A pilot study evaluating concordance between blood-based and patient-matched tumor molecular testing within pancreatic cancer patients participating in the Know Your Tumor (KYT) initiative. Oncotarget 2017, 8, 83446–83456. [Google Scholar] [CrossRef] [Green Version]
- Semrad, T.; Barzi, A.; Lenz, H.J.; Hutchins, I.M.; Kim, E.J.; Gong, I.Y.; Tanaka, M.; Beckett, L.; Holland, W.; Burich, R.A.; et al. Pharmacodynamic separation of gemcitabine and erlotinib in locally advanced or metastatic pancreatic cancer: Therapeutic and biomarker results. Int. J. Clin. Oncol. 2015, 20, 518–524. [Google Scholar] [CrossRef] [Green Version]
- Strijker, M.; Soer, E.C.; de Pastena, M.; Creemers, A.; Balduzzi, A.; Beagan, J.J.; Busch, O.R.; van Delden, O.M.; Halfwerk, H.; van Hooft, J.E.; et al. Circulating tumor DNA quantity is related to tumor volume and both predict survival in metastatic pancreatic ductal adenocarcinoma. Int. J. Cancer 2020, 146, 1445–1456. [Google Scholar] [CrossRef]
- Sugimori, M.; Sugimori, K.; Tsuchiya, H.; Suzuki, Y.; Tsuyuki, S.; Kaneta, Y.; Hirotani, A.; Sanga, K.; Tozuka, Y.; Komiyama, S.; et al. Quantitative monitoring of circulating tumor DNA in patients with advanced pancreatic cancer undergoing chemotherapy. Cancer Sci. 2020, 111, 266–278. [Google Scholar] [CrossRef]
- Takai, E.; Totoki, Y.; Nakamura, H.; Morizane, C.; Nara, S.; Hama, N.; Suzuki, M.; Furukawa, E.; Kato, M.; Hayashi, H.; et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci. Rep. 2015, 5, 18425. [Google Scholar] [CrossRef] [Green Version]
- Toledano-Fonseca, M.; Cano, M.T.; Inga, E.; Rodríguez-Alonso, R.; Gómez-España, M.A.; Guil-Luna, S.; Mena-Osuna, R.; de la Haba-Rodríguez, J.R.; Rodríguez-Ariza, A.; Aranda, E. Circulating Cell-Free DNA-Based Liquid Biopsy Markers for the Non-Invasive Prognosis and Monitoring of Metastatic Pancreatic Cancer. Cancers 2020, 12, 1754. [Google Scholar] [CrossRef]
- Watanabe, F.; Suzuki, K.; Tamaki, S.; Abe, I.; Endo, Y.; Takayama, Y.; Ishikawa, H.; Kakizawa, N.; Saito, M.; Futsuhara, K.; et al. Longitudinal monitoring of KRAS-mutated circulating tumor DNA enables the prediction of prognosis and therapeutic responses in patients with pancreatic cancer. PLoS ONE 2019, 14, e0227366. [Google Scholar] [CrossRef]
- Tjensvoll, K.; Lapin, M.; Buhl, T.; Oltedal, S.; Steen-Ottosen Berry, K.; Gilje, B.; Søreide, J.A.; Javle, M.; Nordgård, O.; Smaaland, R. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol. Oncol. 2016, 10, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milin-Lazovic, J.; Madzarevic, P.; Rajovic, N.; Djordjevic, V.; Milic, N.; Pavlovic, S.; Veljkovic, N.; Milic, N.M.; Radenkovic, D. Meta-Analysis of Circulating Cell-Free DNA’s Role in the Prognosis of Pancreatic Cancer. Cancers 2021, 13, 3378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Ding, X.Q.; Zhu, H.; Wang, R.X.; Pan, X.R.; Tong, J.H. KRAS Mutant Allele Fraction in Circulating Cell-Free DNA Correlates With Clinical Stage in Pancreatic Cancer Patients. Front. Oncol. 2019, 9, 1295. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gupta, S.; Pandey, R.M.; Chauhan, S.S.; Saraya, A. High levels of cell-free circulating nucleic acids in pancreatic cancer are associated with vascular encasement, metastasis and poor survival. Cancer Investig. 2015, 33, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Bunduc, S.; Gede, N.; Váncsa, S.; Lillik, V.; Kiss, S.; Dembrovszky, F.; Eróss, B.; Szakács, Z.; Gheorghe, C.; Mikó, A.; et al. Prognostic role of cell-free DNA biomarkers in pancreatic adenocarcinoma: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2022, 169, 103548. [Google Scholar] [CrossRef]
- Henriksen, S.D.; Madsen, P.H.; Krarup, H.; Thorlacius-Ussing, O. DNA Hypermethylation as a Blood-Based Marker for Pancreatic Cancer: A Literature Review. Pancreas 2015, 44, 1036–1045. [Google Scholar] [CrossRef]
- Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Johansen, M.B.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Promoter hypermethylation in plasma-derived cell-free DNA as a prognostic marker for pancreatic adenocarcinoma staging. Int. J. Cancer 2017, 141, 2489–2497. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Johansen, M.B.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Cell-free DNA promoter hypermethylation in plasma as a predictive marker for survival of patients with pancreatic adenocarcinoma. Oncotarget 2017, 8, 93942–93956. [Google Scholar] [CrossRef] [Green Version]
- Dauksa, A.; Gulbinas, A.; Barauskas, G.; Pundzius, J.; Oldenburg, J.; El-Maarri, O. Whole Blood DNA Aberrant Methylation in Pancreatic Adenocarcinoma Shows Association with the Course of the Disease: A Pilot Study. PLoS ONE 2012, 7, e37509. [Google Scholar] [CrossRef] [Green Version]
- Pietrasz, D.; Wang-Renault, S.; Taieb, J.; Dahan, L.; Postel, M.; Durand-Labrunie, J.; Le Malicot, K.; Mulot, C.; Rinaldi, Y.; Phelip, J.-M.; et al. Prognostic value of circulating tumour DNA in metastatic pancreatic cancer patients: Post-hoc analyses of two clinical trials. Br. J. Cancer 2022, 126, 440–448. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Ta, R.; O’Connor, D.B.; Sulistijo, A.; Chung, B.; Conlon, K.C. The Role of Staging Laparoscopy in Resectable and Borderline Resectable Pancreatic Cancer: A Systematic Review and Meta-Analysis. Dig. Surg. 2019, 36, 251–260. [Google Scholar] [CrossRef]
- Park, S.; Jang, J.K.; Byun, J.H.; Kim, J.H.; Lee, S.S.; Kim, H.J.; Hong, S.B.; Park, S.H. CT in the prediction of margin-negative resection in pancreatic cancer following neoadjuvant treatment: A systematic review and meta-analysis. Eur. Radiol. 2021, 31, 3383–3393. [Google Scholar] [CrossRef]
- Groot, V.P.; Daamen, L.A.; He, J.; Wolfgang, C.L.; Molenaar, I.Q. Patterns of Recurrence after Surgery for Pancreatic Cancer. In Textbook of Pancreatic Cancer; Springer International Publishing: Berlin/Heidelberg, Germany, 2021; pp. 1153–1168. [Google Scholar]
- Paniccia, A.; Hosokawa, P.; Henderson, W.; Schulick, R.D.; Edil, B.H.; Mccarter, M.D.; Gajdos, C. Characteristics of 10-Year Survivors of Pancreatic Ductal Adenocarcinoma. JAMA Surg. 2015, 150, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant Chemotherapy With Gemcitabine vs Observation in Patients Undergoing Curative-Intent Resection of Pancreatic Cancer. JAMA 2007, 297, 267. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant Chemotherapy With Fluorouracil Plus Folinic Acid vs Gemcitabine Following Pancreatic Cancer Resection. JAMA 2010, 304, 1073. [Google Scholar] [CrossRef]
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy With Gemcitabine Plus Erlotinib Versus Gemcitabine Alone in Patients After R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Oba, A.; Ho, F.; Bao, Q.R.; Al-Musawi, M.H.; Schulick, R.D.; Del Chiaro, M. Neoadjuvant Treatment in Pancreatic Cancer. Front. Oncol. 2020, 10, 245. [Google Scholar] [CrossRef]
- Groot, V.P.; Mosier, S.; Javed, A.A.; Teinor, J.A.; Gemenetzis, G.; Ding, D.; Haley, L.M.; Yu, J.; Burkhart, R.A.; Hasanain, A.; et al. Circulating Tumor DNA as a Clinical Test in Resected Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 4973–4984. [Google Scholar] [CrossRef]
- Lee, J.-S.; Rhee, T.-M.; Pietrasz, D.; Bachet, J.-B.; Laurent-Puig, P.; Kong, S.-Y.; Takai, E.; Yachida, S.; Shibata, T.; Lee, J.W.; et al. Circulating tumor DNA as a prognostic indicator in resectable pancreatic ductal adenocarcinoma: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 16971. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Uemura, K.; Murakami, Y.; Kondo, N.; Nakagawa, N.; Okada, K.; Seo, S.; Hiyama, E.; Takahashi, S.; Sueda, T. Clinical Implications of Pre- and Postoperative Circulating Tumor DNA in Patients with Resected Pancreatic Ductal Adenocarcinoma. Ann. Surg. Oncol. 2021, 28, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Shi, X.; Shen, J.; Gao, S.; Wang, H.; Shen, S.; Pan, Y.; Li, B.; Xu, X.; Shao, Z.; et al. Preoperative detection of KRAS G12D mutation in ctDNA is a powerful predictor for early recurrence of resectable PDAC patients. Br. J. Cancer 2020, 122, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadano, N.; Murakami, Y.; Uemura, K.; Hashimoto, Y.; Kondo, N.; Nakagawa, N.; Sueda, T.; Hiyama, E. Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br. J. Cancer 2016, 115, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Hipp, J.; Hussung, S.; Timme-Bronsert, S.; Boerries, M.; Biesel, E.; Fichtner-Feigl, S.; Fritsch, R.; Wittel, U.A. Perioperative cell-free mutant KRAS dynamics in patients with pancreatic cancer. Br. J. Surg. 2021, 108, 239–243. [Google Scholar] [CrossRef]
- Hussung, S.; Akhoundova, D.; Hipp, J.; Follo, M.; Klar, R.F.U.; Philipp, U.; Scherer, F.; von Bubnoff, N.; Duyster, J.; Boerries, M.; et al. Longitudinal analysis of cell-free mutated KRAS and CA 19-9 predicts survival following curative resection of pancreatic cancer. BMC Cancer 2021, 21, 49. [Google Scholar] [CrossRef]
- Nakano, Y.; Kitago, M.; Matsuda, S.; Nakamura, Y.; Fujita, Y.; Imai, S.; Shinoda, M.; Yagi, H.; Abe, Y.; Hibi, T.; et al. KRAS mutations in cell-free DNA from preoperative and postoperative sera as a pancreatic cancer marker: A retrospective study. Br. J. Cancer 2018, 118, 662–669. [Google Scholar] [CrossRef] [Green Version]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Lee, B.; Lipton, L.; Cohen, J.; Tie, J.; Javed, A.A.; Li, L.; Goldstein, D.; Burge, M.; Cooray, P.; Nagrial, A.; et al. Circulating tumor DNA as a potential marker of adjuvant chemotherapy benefit following surgery for localized pancreatic cancer. Ann. Oncol. 2019, 30, 1472–1478. [Google Scholar] [CrossRef]
- Pietrasz, D.; Pécuchet, N.; Garlan, F.; Didelot, A.; Dubreuil, O.; Doat, S.; Imbert-Bismut, F.; Karoui, M.; Vaillant, J.C.; Taly, V.; et al. Plasma Circulating Tumor DNA in Pancreatic Cancer Patients Is a Prognostic Marker. Clin. Cancer Res. 2017, 23, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.G.; Kim, K.C.; Choi, S.-W.; Joo, H.J.; Baik, S.H.; Park, M.K. A Methylation profile of Circulating Cell Free DNA for the Early Detection of Gastric Cancer and the Effects after Surgical Resection. J. Clin. Exp. Oncol. 2016, 5, 1000151. [Google Scholar] [CrossRef]
- Ko, K.; Kananazawa, Y.; Yamada, T.; Kakinuma, D.; Matsuno, K.; Ando, F.; Kuriyama, S.; Matsuda, A.; Yoshida, H. Methylation status and long-fragment cell-free DNA are prognostic biomarkers for gastric cancer. Cancer Med. 2021, 10, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.H.; Symonds, E.L.; Young, G.P.; Byrne, S.; Rabbitt, P.; Roy, A.; Cornthwaite, K.; Karapetis, C.S.; Pedersen, S.K. Relationship between post-surgery detection of methylated circulating tumor DNA with risk of residual disease and recurrence-free survival. J. Cancer Res. Clin. Oncol. 2018, 144, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 79–86. [Google Scholar] [CrossRef]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Mariani, S.; Puzzoni, M.; Liscia, N.; Impera, V.; Pretta, A.; Tolu, S.; Pireddu, A.G.; Persano, M.; Musio, F.; Donisi, C.; et al. Liquid biopsy-driven anti-EGFR rechallenge in patients with metastatic colorectal cancer. J. Clin. Oncol. 2021, 39, 3577. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Kruger, S.; Heinemann, V.; Ross, C.; Diehl, F.; Nagel, D.; Ormanns, S.; Liebmann, S.; Prinz-Bravin, I.; Westphalen, C.B.; Haas, M.; et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann. Oncol. 2018, 29, 2348–2355. [Google Scholar] [CrossRef]
- Park, G.; Park, J.K.; Son, D.-S.; Shin, S.-H.; Kim, Y.J.; Jeon, H.-J.; Lee, J.; Park, W.-Y.; Lee, K.H.; Park, D. Utility of targeted deep sequencing for detecting circulating tumor DNA in pancreatic cancer patients. Sci. Rep. 2018, 8, 11631. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Sohal, D.; Hendifar, A.E.; Chung, V.; Mikhail, S.; Rahib, L.; Lyons, E.; Tibbetts, L.; et al. Outcomes in pancreatic adenocarcinoma (PDA) patients (pts) with genetic alterations in DNA damage repair (DDR) pathways: Results from the Know Your Tumor (KYT) program. J. Clin. Oncol. 2019, 37, 191. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; Kude De Almeida, F.; Baptista Simões, C.; Wiesmüller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Casolino, R.; Paiella, S.; Azzolina, D.; Beer, P.A.; Corbo, V.; Lorenzoni, G.; Gregori, D.; Golan, T.; Braconi, C.; Froeling, F.E.M.; et al. Homologous Recombination Deficiency in Pancreatic Cancer: A Systematic Review and Prevalence Meta-Analysis. J. Clin. Oncol. 2021, 39, 2617–2631. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Sehdev, A.; Gbolahan, O.; Hancock, B.A.; Stanley, M.; Shahda, S.; Wan, J.; Wu, H.H.; Radovich, M.; O’Neil, B.H. Germline and Somatic DNA Damage Repair Gene Mutations and Overall Survival in Metastatic Pancreatic Adenocarcinoma Patients Treated with FOLFIRINOX. Clin. Cancer Res. 2018, 24, 6204–6211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Palacio, S.; Mcmurry, H.S.; Ali, R.; Donenberg, T.; Silva-Smith, R.; Wideroff, G.; Sussman, D.A.; Rocha Lima, C.M.S.; Hosein, P.J. DNA damage repair deficiency as a predictive biomarker for FOLFIRINOX efficacy in metastatic pancreatic cancer. J. Gastrointest. Oncol. 2019, 10, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Terrero, G.; Datta, J.; Dennison, J.; Sussman, D.A.; Lohse, I.; Merchant, N.B.; Hosein, P.J. Ipilimumab/Nivolumab Therapy in Patients With Metastatic Pancreatic or Biliary Cancer With Homologous Recombination Deficiency Pathogenic Germline Variants. JAMA Oncol. 2022, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kagara, N.; Tanei, T.; Naoi, Y.; Shimoda, M.; Shimomura, A.; Shimazu, K.; Kim, S.J.; Noguchi, S. Correlation of Methylated Circulating Tumor DNA with Response to Neoadjuvant Chemotherapy in Breast Cancer Patients. Clin. Breast Cancer 2017, 17, 61–69.e3. [Google Scholar] [CrossRef]
- Guo, D.; Yang, L.; Yang, J.; Shi, K. Plasma cell-free DNA methylation combined with tumor mutation detection in prognostic prediction of patients with non-small cell lung cancer (NSCLC). Medicine 2020, 99, e20431. [Google Scholar] [CrossRef]
- Amatu, A.; Schirripa, M.; Tosi, F.; Lonardi, S.; Bencardino, K.; Bonazzina, E.; Palmeri, L.; Patanè, D.A.; Pizzutilo, E.G.; Mussolin, B.; et al. High Circulating Methylated DNA Is a Negative Predictive and Prognostic Marker in Metastatic Colorectal Cancer Patients Treated With Regorafenib. Front. Oncol. 2019, 9, 622. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Useros, J.; Martin-Galan, M.; Florez-Cespedes, M.; Garcia-Foncillas, J. Epigenetics of Most Aggressive Solid Tumors: Pathways, Targets and Treatments. Cancers 2021, 13, 3209. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.W.; Schwerdel, D.; Costa, I.G.; Hackert, T.; Strobel, O.; Lam, S.; Barth, T.F.; Schröppel, B.; Meining, A.; Büchler, M.W.; et al. Detection of Hot-Spot Mutations in Circulating Cell-Free DNA From Patients With Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2016, 151, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Le Calvez-Kelm, F.; Foll, M.; Wozniak, M.B.; Delhomme, T.M.; Durand, G.; Chopard, P.; Pertesi, M.; Fabianova, E.; Adamcakova, Z.; Holcatova, I.; et al. KRAS mutations in blood circulating cell-free DNA: A pancreatic cancer case-control. Oncotarget 2016, 7, 78827–78840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.W.; Schwerdel, D.; Reinacher-Schick, A.; Uhl, W.; Algul, H.; Friess, H.; Janssen, K.P.; Konig, A.; Ghadimi, M.; Gallmeier, E.; et al. A Blood-Based Multi Marker Assay Supports the Differential Diagnosis of Early-Stage Pancreatic Cancer. Theranostics 2019, 9, 1280–1287. [Google Scholar] [CrossRef]
- Liu, X.; Liu, L.; Ji, Y.; Li, C.; Wei, T.; Yang, X.; Zhang, Y.; Cai, X.; Gao, Y.; Xu, W.; et al. Enrichment of short mutant cell-free DNA fragments enhanced detection of pancreatic cancer. EBioMedicine 2019, 41, 345–356. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Study | Study Population | Technology Used | Target | Sensitivity and Specificity, Respectively |
|---|---|---|---|---|
| Cao, Wei, Hu, He, Zhang, Xia, Tu, Yuan, Guo, Liu, Xie, and Li [2] | HC vs. PDA | Methylome sequencing | 24-feature 5mC and 27-feature 5hmC model | 5mC + 5hmC—93.8% and 95.5% (AUC of 0.99) 5mC alone—87% and 82% (AUC of 0.97) 5hmC alone—78–85% and 99–100% (AUC of 0.99–0.96) |
| Melnikov et al. [38] | HC vs. PDA | Microarray | * CCND2, PLAU, SOCS1, THBS, and VHL | 76% and 59% |
| Liggett et al. [39] | HC vs. PDA vs. CP | Same microarray as Melnikov et al., 2009 | 14 promoters for PDA vs. CP 8 for HC and CP | 91.2% and 90.8% (PDA vs. CP) 82% and 78% |
| Yi et al. [40] | PDA vs. PanIN or pancreatitis | PCR | BNC1 and ADAMTS1 | Both—81% and 85% BNC1—79% and 89% ADAMTS1—48% and 92% |
| Eissa et al. [41] | PDA vs. noncancer | PCR | BNC1 and ADAMTS1 | Combined AUC of 0.95 ADAMTS1—87.2% and 95.8% (AUC = 0.91; 95%) BNC1—64.1% and 93.7% (AUC = 0.79) Both—97.3% and 91.6% (AUC = 0.95) |
| Henriksen et al. [42] | PDA vs. HC + CP + acute pancreatitis | PCR | BMP3, RASSF1A, BNC1, MESTv2, TFPI2, APDA, SFRP1, and SFRP2 With age >65 years | 76% and 83% (0.86) |
| Guler et al. [43] | PDA vs. no PDA | CHIP seq | ** Gene set of top 65% differentially hydroxymethylated genes | Training set AUC = 0.92 Validation set 1 AUC = 0.921 Validation set 2 AUC = 0.943 |
| Li et al. [44] | HC vs. PDA | MeDIP-seq | TRIM73, FAM150A, EPB41L3, SIX3, MIR663, MAPT, LOC100128977, and LOC100130148 | 93.2% and 95% |
| Ying et al. [45] | HC vs. PDA | PCR | ADAMTS1, BNC1, LRFN5, and PXDN | 100% and 90% |
| Manoochehri et al. [46] | HC vs. PDA | ddPCR | SST | 100% and 89% |
| Singh et al. [47] | HC vs. CP vs. HC | PCR | SPRC, UCHL1, NPTX2, and PENK | HC vs. CP + PDA, MI of all 4 are increased HC vs. CP, MI of UCLH1, PENK, NPTX2 increased CP vs. PDA, MI of SPARC, and NPTX2 increased CP vs. early-stage PDA, MI of SPARC increased |
| Shinjo et al. [48] | PDA vs. HC | MBD-ddPCR | ADAMTS1, HOXA1, PCDH10, SEMA5A, and SPSB4 | Panel of 5 genes with 49% and 86% 1 of 5 in 49% of PDA Panel of 5 genes + KRAS mutation in cfDNA with 68% and 86% |
| Fujimoto et al. [49] | PDA vs. benign disease and HC | PCR | RUNX3 | RUNX3 alone: 50.9% and 93.5% RUNX3 combined with CA19-9: 85.5% and 93.5% for all stages and 78% for stage I |
| Kandimalla et al. [50] | PDA vs. HC | Genome-wide DNA methylation sequencing | EpiPanGiDx | Predictive value of 85% |
| Vrba et al. [51] | PDA vs. benign cyst | PCR | 10-promoter panel in mPDA | 100% and 95% (AUC of 0.999) |
| Li et al. [52] | PDA vs. PanIN benign tumors and pancreatitis | PCR | BNC1 SEPT9 in Stage I and II | Combined—65% and 87% BCN1—50.9% and 88.7% SEPT9—36.8% and 96.2% Combined + CA 19-9 vs. CA19-9 alone—86% vs. 61.4% and 81.1% vs. 90.6% Individually they have low CT compared to HC and benign disease |
| Melson et al. [53] | PDA vs. HC | PCR # | VHL, MYF3, TMS, GPC3, and SRBC | 80% and 66% (AUC = 0.848) |
| Park et al. [54] | PDA vs. CP | PCR | NPTX2 | 80% and 76% |
| Park et al. [55] | PDA vs. HC | PCR | P16 | Higher methylation in PDA than HC (86.7 ± 29.8 vs. 33.3 ± 0.00, p = 0.016) |
| KRAS Variants | Technique | Findings |
|---|---|---|
| G12A, G12C, G12D, G12R, G12S, G12V, and G13D [64] | ddPCR | Detection of mutant KRAS is associated with poor OS (197 days vs. 60 days, HR = 2.8, p = 0.018). |
| G12D, G12V, and G12R [65] | ddPCR | KRAS mutation at G12V conferred poorer OS compared to WT (p < 0.01). No significance effect of KRAS G12D mutation. |
| G12A, G12C, G12D, G12R, G12S, G12V, and G13D [66] | ddPCR | MAF ≥ 5% of any variant was a poor predictor of PFS (HR = 2.28; 95% CI: 1.18–4.40; p = 0.014) and OS (HR = 3.46; 95% CI: 1.40–8.50; p = 0.007). MAF peak above 1% was significantly associated with radiologic progression (p = 0.0003). |
| KRAS Codon 12 mutations [67] | PCR | Detection of KRAS mutations conferred shorter OS compared to WT (3.9 months vs. 10.2 months, p < 0.001). Mutational burden could significantly correlate with TNM tumor staging (p = 0.033) and liver metastasis (p = 0.014). KRAS mutations were a negative prognostic factor for survival (HR = 7.39; 95% CI: 3.69–14.89). |
| G12V and G12D [68] | ddPCR | G12V conferred poor OS (p = 0.001). G12D conferred poor OS (p = 0.044). |
| G12D, G12V, G12R, and G13D [69] | ddPCR | Increased mutational burden conferred poor PFS (2.5 vs. 7.5 months, p = 0.03) and OS (6.5 vs. 11.5 months, p = 0.009). |
| G12D, G12V, G12R, and G12C [70] | NGS | G12R mutation conferred favorable OS compared to WT (20.4 vs. 14.5 m, HR = 0.67 (95% CI: 0.47–0.93), p = 0.0215) and PFS on first-line therapy (12.2 vs. 6.8 m, HR 0.60 (95% CI 0.40–0.85), p = 0.004). |
| G12A, G12C, G12D, G12R, G12S, G12V, and G13D [71] | ddPCR | KRAS mutations concentration >0.165 copies/L had worse OS median fractional abundance (>0.415%.) |
| G12V, G12D, and G12R in codon 12 KRAS [34] | ddPCR | mOS was significantly shorter in patients with KRAS mutant (276 days) compared with patients with WT KRAS (413 days) from cfDNA samples (p = 0.02). mOS was significantly shorter only in G12V variants compared to other KRAS mutants (219 days vs. 410 days, p = 0.006). |
| G12D, G12R, G12V, Q61H, Q61R, and A59G [72] | BEAM-PCR | Overall response rate, disease control rate, mPFS, and mOS were higher in patients without detectable KRAS mutations (48% vs. 28%, 81% vs. 69%, 8.8 vs. 5.3 months, and 18.2 vs. 6.6 months, respectively). |
| KRAS codon 12 [73] | ddPCR | Patients with WT KRAS had better OS than mutant KRAS patients (10.6 months vs. 5.6 months, p < 0.05). Patients with KRAS mutation and copy number gain had the worst prognosis with a mOS of 2.5 months (p ≤ 0.0001). |
| KRAS exon 12 [74] | PCR | Undetectable mutant KRAS conferred favorable OS (8 vs. 37.5 months from diagnosis, p < 0.004). |
| KRAS mutations: not specified [75] | DNA-based Ion-Torrent NGS assays (ClearID) | Presence of KRAS mutations in cfDNA was associated with reduced mOS (54% in mutation-positive versus 90% in mutation-negative, p < 0.05). |
| KRAS codon 12 and 13 [76] | PCR | Patients with KRAS mutations detected in cfDNA had significantly lower mPFS (1.8 vs. 4.6 months, p = 0.014) and mOS (3.0 vs. 10.5 months, p = 0.003) than those without detected plasma KRAS mutations. |
| G12A, G12C, G12D, G12V, G12R, G12S, and G13D [77] | ddPCR and NGS amplicon panel | Detectable mutant KRAS cfDNA was associated with poor OS (3.2 vs. 8.4 months, p = 0.005). |
| KRAS G12/13 mutations and KRAS Q61K [78] | ddPCR | Detectable mutant KRAS cfDNA was associated with poor PFS (308.5 vs. 168 days, p = 0.07). |
| G12D, G12R, G12V, and G13D [79] | ddPCR | Higher concentration with advanced stages (p = 0.0129). |
| RAS mutation (KRAS/NRAS codons 12, 13, 59, 61, 117, and 146) [80] | BEAM-PCR | Higher RAS MAF was associated with poor OS (142 vs. 310 days, p = 0.0261) and PFS (85 versus 175 days; p = 0.0556). |
| G12V, G12D, G12R, and Q61H [81] | ddPCR | The mOS of patients with detectable mutant KRAS cfDNA was shorter (15.8 months vs. 33.7 months; p < 0.05) |
| G12D, G12V, G12R, G13D [32] | Digital PCR and NGS | Patients with multiple liver metastasis and poor mOS had higher mutant KRAS cfDNA MAF compared to those with fewer lesions (p < 0.05). |
| Genes Studied | Comparison | Findings |
|---|---|---|
| 5mC and 5hmC pan-sequencing [2] | Identify DMPs for 5mC and DhMPs for 5hmC | 5mC: No difference between resectable vs. unresectable PDA. 5hmC: Significant different between stage I vs. II/III/IV; no difference between resectable vs. unresectable. 5mC + 5hmC: Higher in tumor size <3 cm (vs. >3 cm) and PNI; no difference between resectable vs. unresectable; no significant differences in vascular invasion or positive lymph node metastasis were found in resectable PDA patients. |
| SPARC UCLH1 PENK NPTX2 [47] | Low vs. high methylation index | SPARC: Higher in stage IV and poor survival (3 vs. 6 m); Lower in resectable (p = 0.02). NPTX2: Higher in stage IV, met dz and poor survival (3 vs. 9 m) (p = 0.04). UCLH1: Higher in stage III/IV vs. I/II (p = 0.034). |
| 28-gene panel for staging [88] | Number of methylated genes | Stage I: 7.09 (95% CI: 5.51–8.66). Stage II: 7.00 (95% CI: 5.93–8.07). Stage III: 6.77 (95% CI: 5.08–8.46). Stage IV: 10.24 (95% CI: 8.88–11.60). The number of methylated genes at stage IV was significantly higher compared to stage I/II/III PDA (p = 0.0002). |
| Specific promoters | The prediction model (SEPT9v2, SST, ALX4, CDKN2B, HIC1, MLH1, NEUROG1, and BNC1) enabled the differentiation of stage IV from stage I-III disease (AUC of 0.87 (cut point: 0.55); sensitivity of 74%, specificity of 87%)). Model (MLH1, SEPT9v2, BNC1, ALX4, CDKN2B, NEUROG1, WNT5A, and TFPI2) enabled the differentiation of stage I-II from stage III-IV disease (AUC of 0.82 (cut point: 0.66); sensitivity of 73%, specificity of 80%)). | |
| Same panel as above [89] | Number of methylated genes Risk stratification based on ASA= 3 and methylation of GSTP1, SFRP2, BNC1, SFRP1, TFPI2, and WNT5A | Patients with more than 10 hypermethylated genes had an HR of 2.03 (95% CI: 1.15–3.57). Total group (all stages of tumor) HR compared to group 1: Risk group 2: HR 2.65 (95% CI: 1.24–5.66); Risk group 3: HR of 4.34 (95% CI: 1.98–9.51); Risk group 4: HR of 21.19 (95% CI: 8.61–52.15). Stage I–II (ASA = 3, SFRP2, and MESTv2): Risk group 2: HR of 4.83 (95% CI: 2.01–11.57); Risk group 3: HR of 9.12 (95% CI: 2.18–38.25); Risk group 4: HR of 70.90 (95% CI: 12.63–397.96). Stage IV (BMP3, NPTX2, SFRP1, and MGMT): Risk group 2: HR of 5.23 (95% CI: 2.13–12.82). |
| ADAMTS1, HOXA1, PCDH10, SEMA5A, and SPSB4 ± KRAS mutations [48] | Positive vs. negative | Large tumor size and higher frequency of liver metastatic disease in cfDNA positive patients |
| p16, RARbeta, TNFRSF10C, APC, ACIN1, DAPK1, 3OST2, BCL2, and CD44 [90] | Methylation levels in CpG promoter regions | The highest tertile of methylation of ACIN1 was associated with shorter survival compared to the middle and the lowest tertile group (13 months vs. 17 months). Highest tertile of TNFRSF10C was associated with shorter survival compared to the middle and the lowest tertile group (OS, 13 months vs. 22 months). TNFRSF10C SN1 methylation was significantly associated with PNI (OR = 0.088). |
| HOXD8 and POU4F1 [91] | Detection | Median PFS and OS were 5.3 and 8.2 months in ctDNA-positive and 6.2 and 12.6 months in ctDNA-negative patients, respectively. ctDNA positivity was more often associated with young age, high CA19-9 level, and neutrophils lymphocytes ratio. ctDNA was confirmed as an independent prognostic marker for PFS (HR = 1.5, CI 95%: [1.03–2.18], p = 0.034) and OS (HR = 1.62, CI 95%: [1.05–2.5], p = 0.029). |
| Study | KRAS Variants Tested | Impact of Preop Detection | Impact of Postop Detection |
|---|---|---|---|
| Groot et al., 2019 [102] | G12V/12D/12R/Q61H | Significant | Persistent—significant |
| Lee et al., 2019 [103] | Codons 12/13/61 | Significant | |
| Yamaguchi et al., 2021 [104] | G12/12V/12R | Not significant | |
| Guo et al., 2020 [105] | G12D | Not studied | |
| Hadano et al., 2016 [106] | G12V/12D/12R | ||
| Kim et al., 2018 [71] | G12A/12C/12D/12R/12S/12V/13D | ||
| Hipp et al., 2021 [107] | G12D/12V/12R/12C | Not Significant | Significant |
| Hussung et al., 2021 [108] | Codons 12/13/61 | Significant | |
| Nakano et al., 2018 [109] | Codons 12/13 | Conversion from wild type to mutation—significant | |
| Wantanabe et al., 2019 [81] | G12V/12D/12R/Q61H | Emergence—significant | |
| Sausen et al., 2015 [110] | G12V/12D/12R/12V/12C/13D | Not studied | Significant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheel, A.; Addison, S.; Nuguru, S.P.; Manne, A. Is Cell-Free DNA Testing in Pancreatic Ductal Adenocarcinoma Ready for Prime Time? Cancers 2022, 14, 3453. https://doi.org/10.3390/cancers14143453
Sheel A, Addison S, Nuguru SP, Manne A. Is Cell-Free DNA Testing in Pancreatic Ductal Adenocarcinoma Ready for Prime Time? Cancers. 2022; 14(14):3453. https://doi.org/10.3390/cancers14143453
Chicago/Turabian StyleSheel, Ankur, Sarah Addison, Surya Pratik Nuguru, and Ashish Manne. 2022. "Is Cell-Free DNA Testing in Pancreatic Ductal Adenocarcinoma Ready for Prime Time?" Cancers 14, no. 14: 3453. https://doi.org/10.3390/cancers14143453
APA StyleSheel, A., Addison, S., Nuguru, S. P., & Manne, A. (2022). Is Cell-Free DNA Testing in Pancreatic Ductal Adenocarcinoma Ready for Prime Time? Cancers, 14(14), 3453. https://doi.org/10.3390/cancers14143453