
Alternative Treatment Options to ALK Inhibitor Monotherapy for EML4-ALK-Driven Lung Cancer

 ,
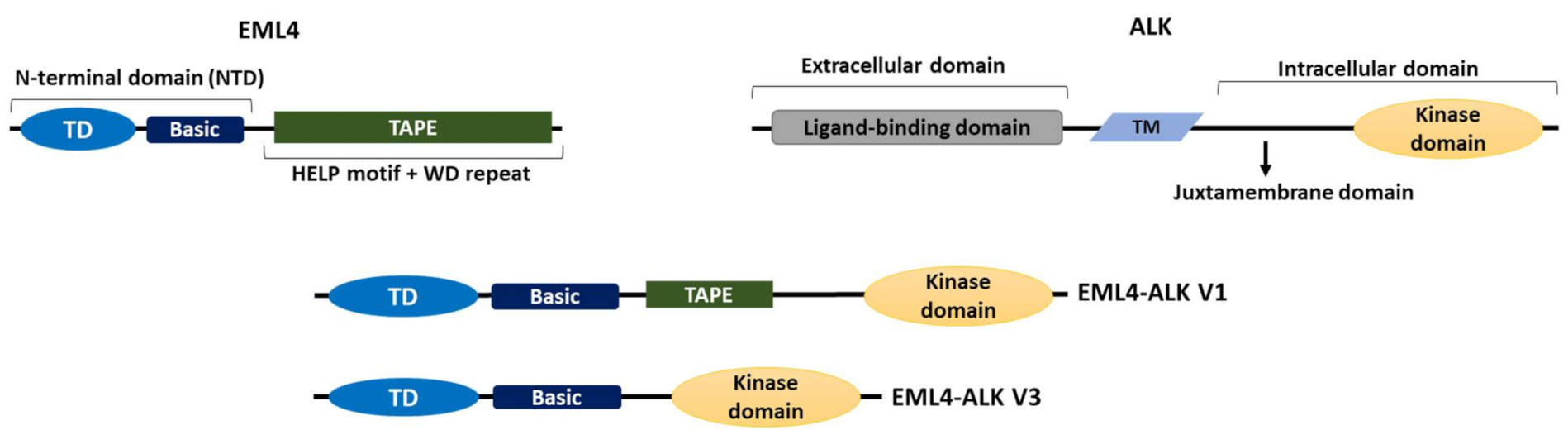
, 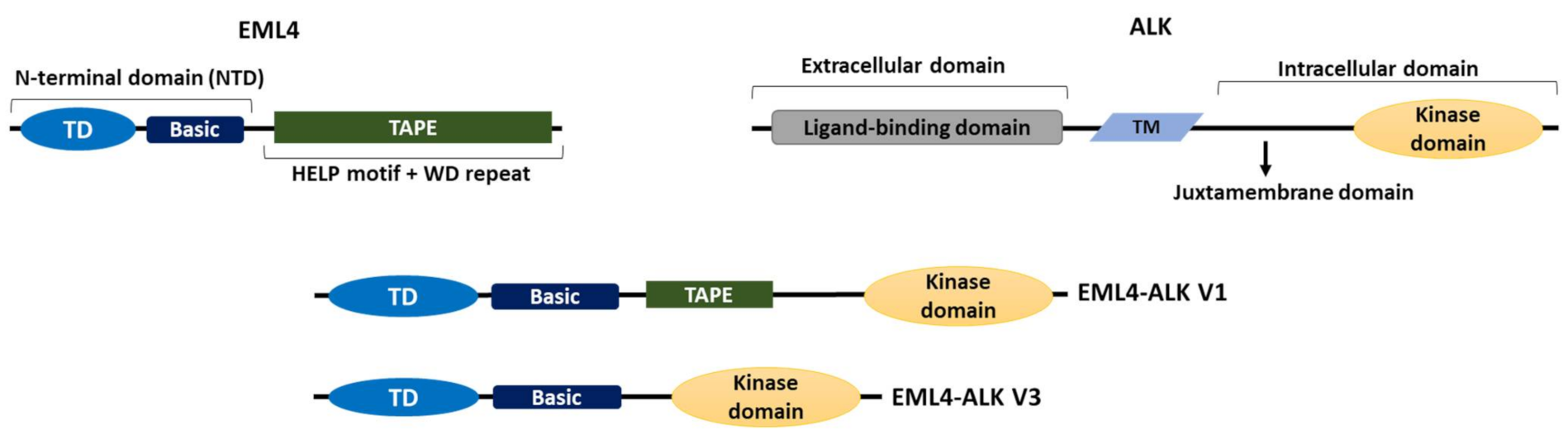{kind=link}
{kind=link}
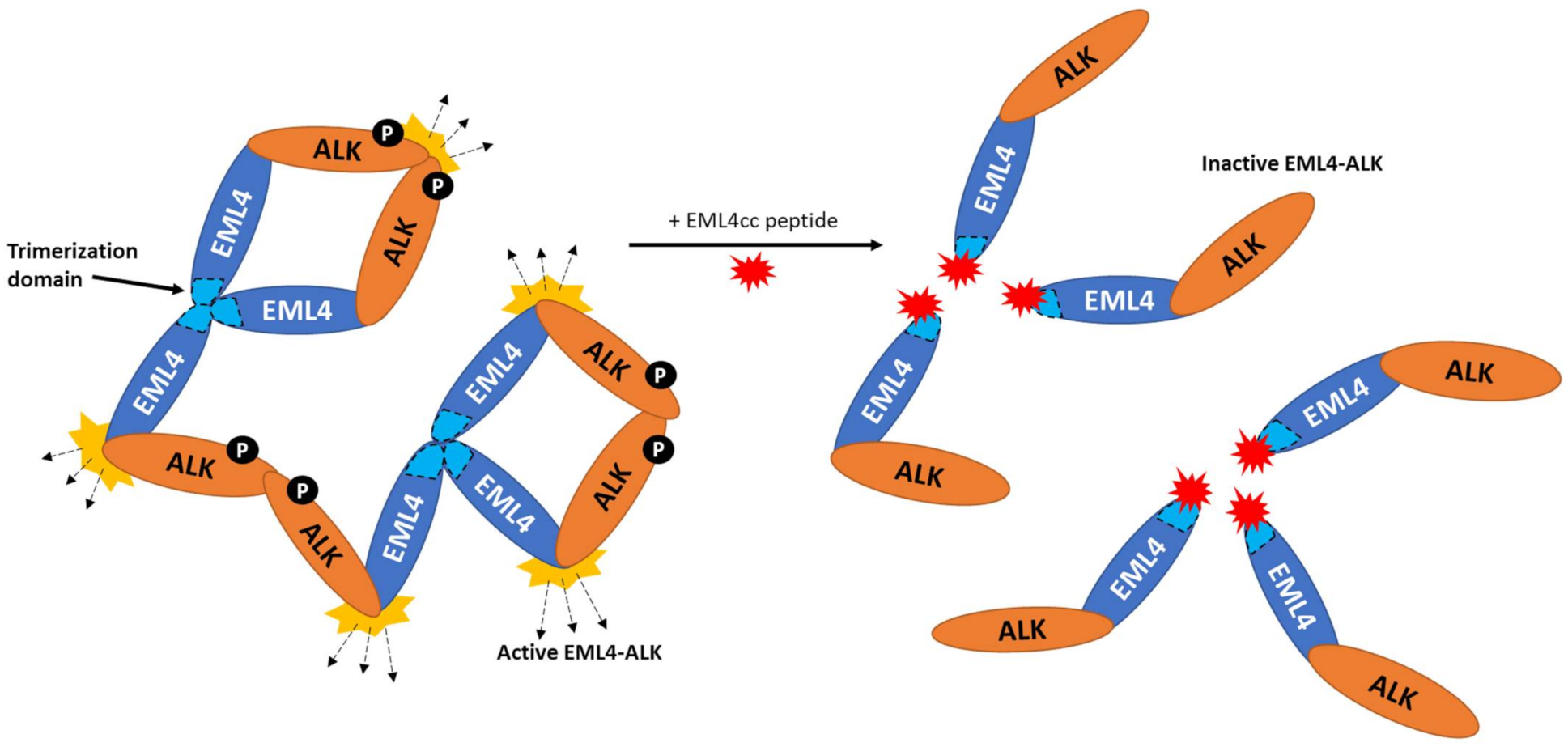{kind=link}
{kind=link}
{kind=link}
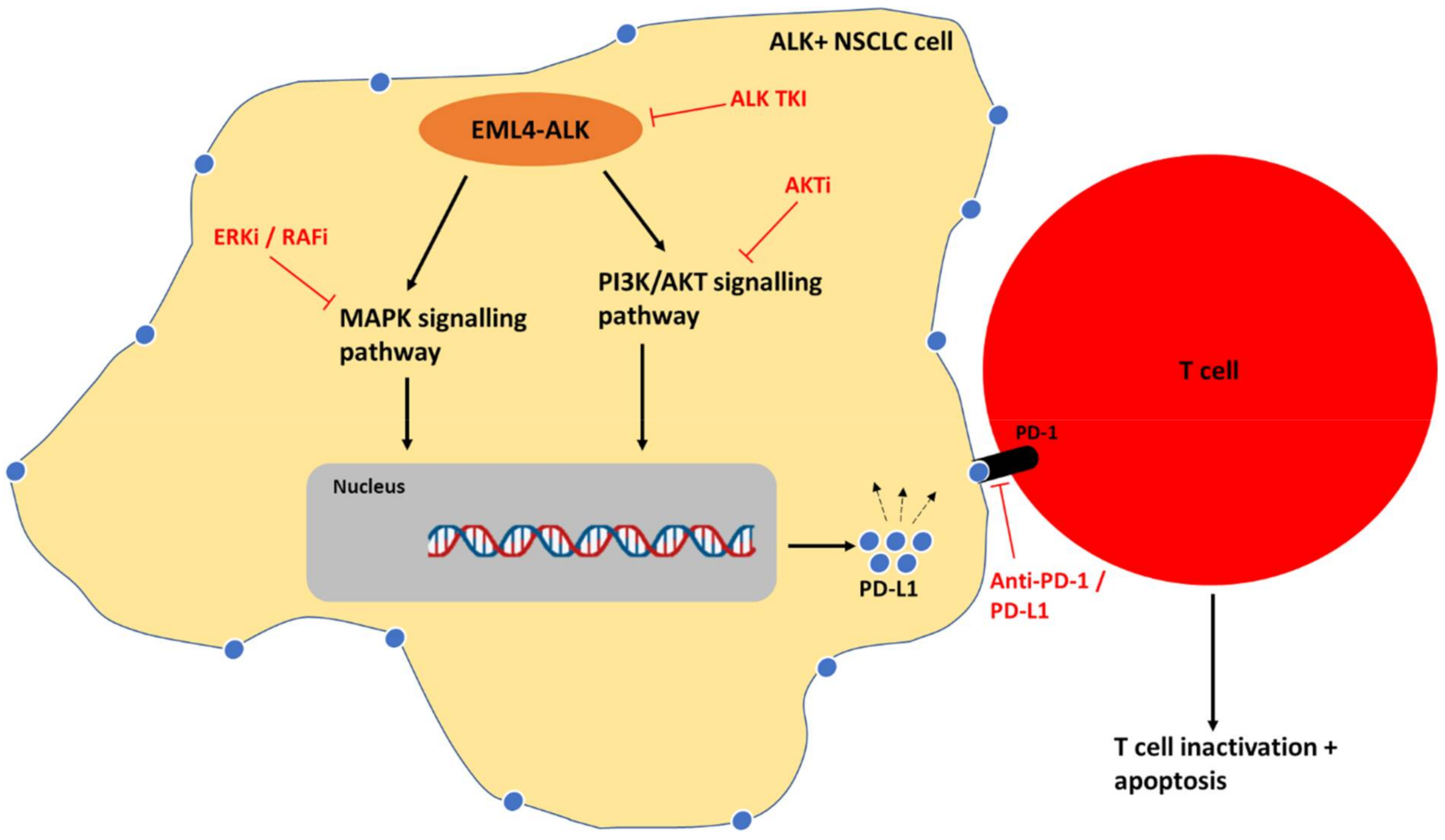{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Epidemiology and Genetics of Lung Cancer
1.2. ALK Rearrangements
1.3. The EML4-ALK Oncogenic Fusion
1.4. Targeted ALK Inhibitors and Resistance Mechanisms
1.5. Aim of Review
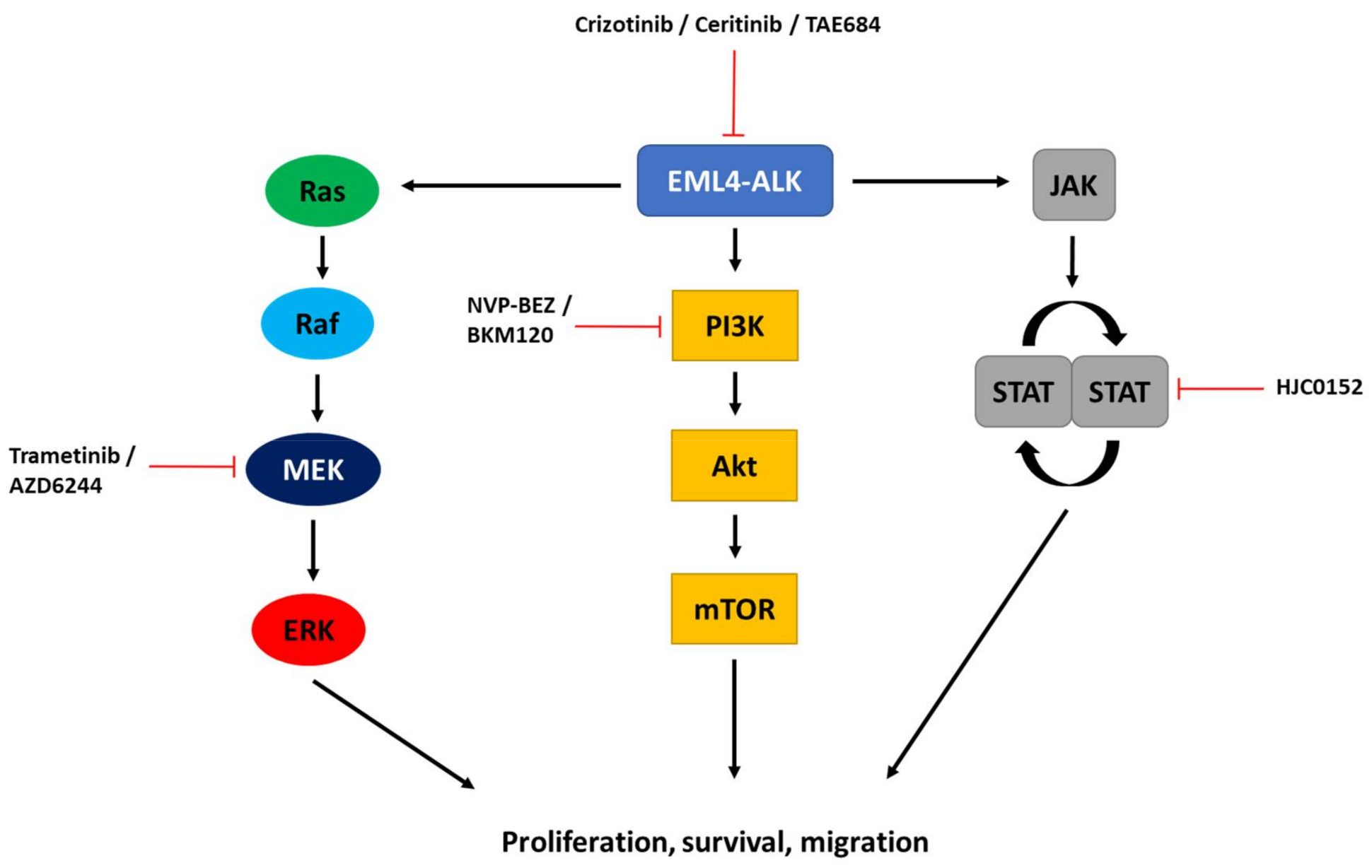
2. Targeting ALK-Dependent Signalling Pathways
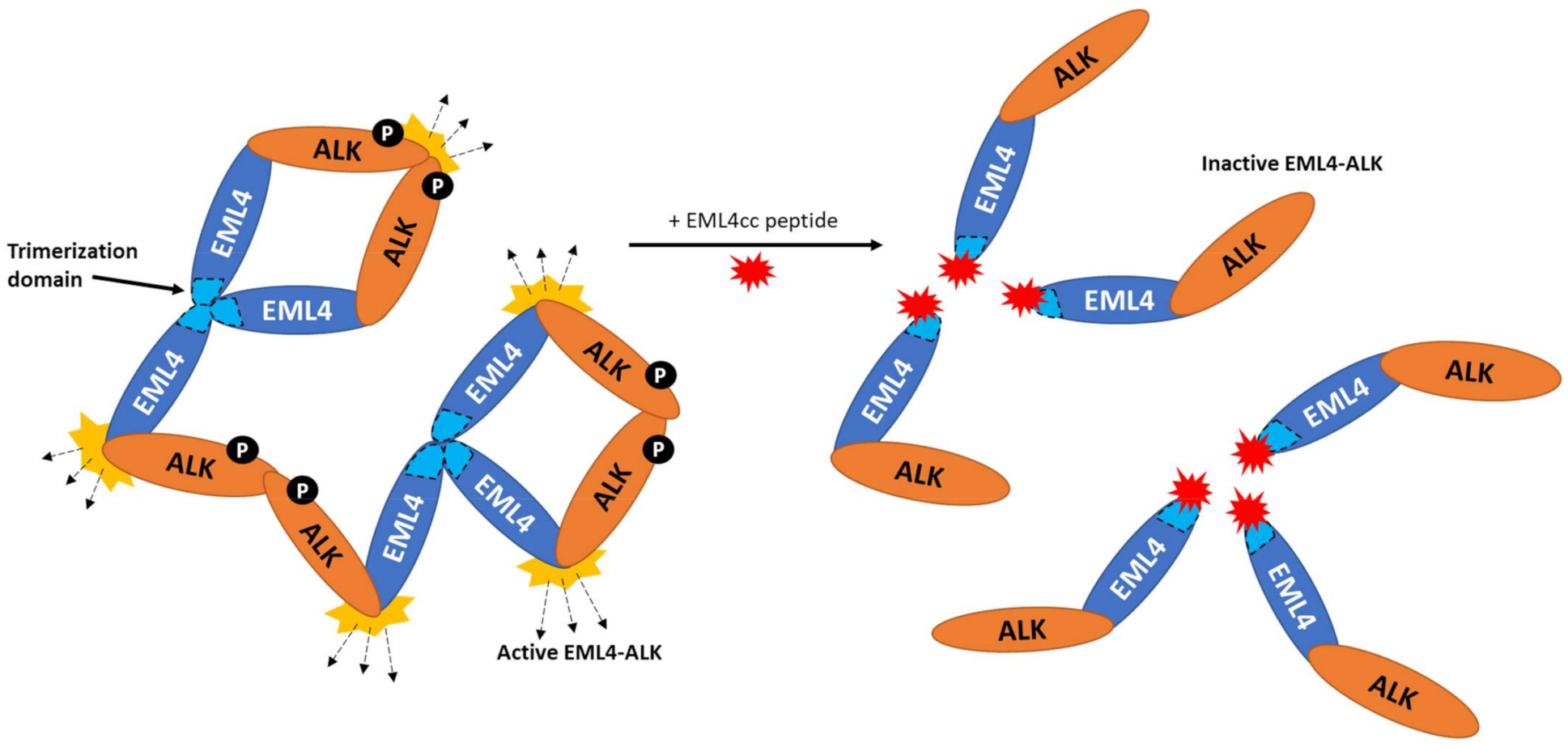
3. Targeting Pathways Independent of ALK Catalytic Activity
4. Combination of ALK Inhibitors and Chemotherapy
5. Combination of ALK Inhibitors and Radiotherapy
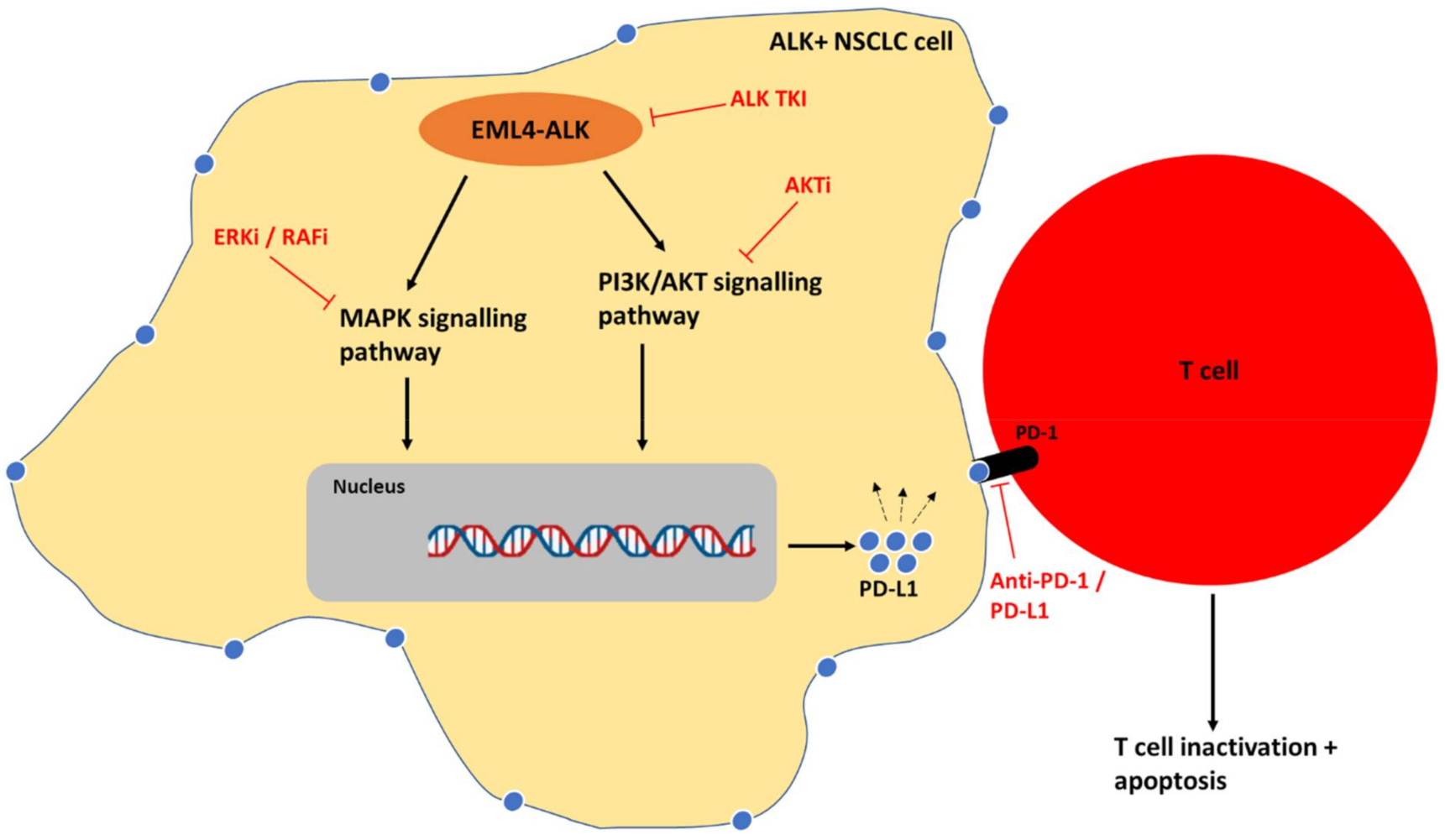
6. Combination of ALK Inhibitors and Immunotherapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Globocan. Cancer Today. 2022. Available online: http://gco.iarc.fr/today/home (accessed on 13 April 2022).
- Inamura, K. Lung Cancer: Understanding Its Molecular Pathology and the 2015 WHO Classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemal, A.; Center, M.M.; DeSantis, C.; Ward, E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1893–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thun, M.J.; Carter, B.D.; Feskanich, D.; Freedman, N.D.; Prentice, R.; Lopez, A.D.; Hartge, P.; Gapstur, S.M. 50-year trends in smoking-related mortality in the United States. N. Engl. J. Med. 2013, 368, 351–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youlden, D.R.; Cramb, S.M.; Baade, P.D. The International Epidemiology of Lung Cancer: Geographical distribution and secular trends. J. Thorac. Oncol. 2008, 3, 819–831. [Google Scholar] [CrossRef]
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global Epidemiology of Lung Cancer. Ann. Glob. Health 2019, 85, 8. [Google Scholar] [CrossRef] [Green Version]
- Lung Cancer Survival Statistics 2019. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/lung-cancer/survival (accessed on 13 April 2022).
- Black, R.C.; Khurshid, H. NSCLC: An update of driver mutations, their role in pathogenesis and clinical significance. Rhode Isl. Med. J. 2015, 98, 25. [Google Scholar]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Bayliss, R.; Choi, J.; Fennell, D.A.; Fry, A.M.; Richards, M.W. Molecular mechanisms that underpin EML4-ALK driven cancers and their response to targeted drugs. Cell. Mol. Life Sci. 2016, 73, 1209–1224. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Joubert, P.; Ansari-Pour, N.; Zhao, W.; Hoang, P.H.; Lokanga, R.; Moye, A.L.; Rosenbaum, J.; Gonzalez-Perez, A.; Martinez-Jimenez, F.; et al. Genomic and evolutionary classification of lung cancer in never smokers. Nat. Genet. 2021, 53, 1348–1359. [Google Scholar] [CrossRef]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Du, X.; Shao, Y.; Qin, H.-F.; Tai, Y.-H.; Gao, H.-J. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac. Cancer 2018, 9, 423–430. [Google Scholar] [CrossRef]
- Barreca, A.; Lasorsa, E.; Riera, L.; Machiorlatti, R.; Piva, R.; Ponzoni, M.; Kwee, I.; Bertoni, F.; Piccaluga, P.P.; Pileri, S.A.; et al. Anaplastic lymphoma kinase in human cancer. J. Mol. Endocrinol. 2011, 47, R11–R23. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Rocco, G.; Marino, F.Z.; Pirozzi, G.; Normanno, N.; Morabito, A.; Sperlongano, P.; Stiuso, P.; Luce, A.; Botti, G.; et al. Anaplastic lymphoma kinase: A glimmer of hope in lung cancer treatment? Expert Rev. Anticancer. Ther. 2013, 13, 407–420. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hutanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Yoshida, T.; Oya, Y.; Tanaka, K.; Shimizu, J.; Horio, Y.; Kuroda, H.; Sakao, Y.; Hida, T.; Yatabe, Y. Differential Crizotinib Response Duration Among ALK Fusion Variants in ALK-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3383–3389. [Google Scholar] [CrossRef] [Green Version]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.I.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Conde, E.; Rojo, F.; Gómez, J.; Enguita, A.B.; Abdulkader, I.; González, A.; Lozano, D.; Mancheno, N.; Salas, C.; Salido, M.; et al. Molecular diagnosis in non-small-cell lung cancer: Expert opinion on ALK and ROS1 testing. J. Clin. Pathol. 2022, 75, 145–153. [Google Scholar] [CrossRef]
- Cruz-Rico, G.; Avilés-Salas, A.; Segura-González, M.; Espinosa-García, A.M.; Ramírez-Tirado, L.A.; Morales-Oyarvide, V.; Rojas-Marin, C.; Cardona, A.F.; Arrieta, O. Diagnosis of EML4-ALK Translocation With FISH, Immunohistochemistry, and Real-time Polymerase Chain Reaction in Patients With Non-Small Cell Lung Cancer. Am. J. Clin. Oncol. 2017, 40, 631–638. [Google Scholar] [CrossRef]
- Wu, Y.C.; Chang, I.C.; Wang, C.L.; Chen, T.D.; Chen, Y.T.; Liu, H.P.; Chu, Y.; Chiu, Y.T.; Wu, T.H.; Chou, L.H.; et al. Comparison of IHC, FISH and RT-PCR methods for detection of ALK rearrangements in 312 non-small cell lung cancer patients in Taiwan. PLoS ONE 2013, 8, e70839. [Google Scholar] [CrossRef]
- Demidova, I.; Barinov, A.; Savelov, N.; Gagarin, I.; Grinevitch, V.; Stroiakovaski, D.; Popov, M.; Laktionov, K.; Gutorov, S.; Smolin, A.; et al. Immunohistochemistry, fluorescence in situ hybridization, and reverse transcription-polymerase chain reaction for the detection of anaplastic lymphoma kinase gene rearrangements in patients with non-small cell lung cancer: Potential advantages and methodologic pitfalls. Arch. Pathol. Lab. Med. 2014, 138, 794–802. [Google Scholar]
- Zhang, Y.G.; Jin, M.L.; Li, L.; Zhao, H.Y.; Zeng, X.; Jiang, L.; Wei, P.; Diao, X.L.; Li, X.; Cao, Q.; et al. Evaluation of ALK rearrangement in Chinese non-small cell lung cancer using FISH, immunohistochemistry, and real-time quantitative RT- PCR on paraffin-embedded tissues. PLoS ONE 2013, 8, e64821. [Google Scholar] [CrossRef] [Green Version]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Choi, Y.L.; Takeuchi, K.; Soda, M.; Inamura, K.; Togashi, Y.; Hatano, S.; Enomoto, M.; Hamada, T.; Haruta, H.; Watanabe, H.; et al. Identification of novel isoforms of the EML4-ALK transforming gene in non–small cell lung cancer. Cancer Res. 2008, 68, 4971–4976. [Google Scholar] [CrossRef] [Green Version]
- Sanders, H.R.; Li, H.-R.; Bruey, J.-M.; Scheerle, J.A.; Meloni-Ehrig, A.M.; Kelly, J.C.; Novick, C.; Albitar, M. Exon scanning by reverse transcriptase–polymerase chain reaction for detection of known and novel EML4–ALK fusion variants in non–small cell lung cancer. Cancer Genet. 2011, 204, 45–52. [Google Scholar] [CrossRef]
- Suprenant, K.A.; Dean, K.; McKee, J.; Hake, S. EMAP, an echinoderm microtubule-associated protein found in microtubule-ribosome complexes. J. Cell Sci. 1993, 104, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.W.; O’Regan, L.; Roth, D.; Montgomery, J.M.; Straube, A.; Fry, A.M.; Bayliss, R. Microtubule association of EML proteins and the EML4-ALK variant 3 oncoprotein require an N-terminal trimerization domain. Biochem. J. 2015, 467, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabir, S.R.; Yeoh, S.; Jackson, G.; Bayliss, R. EML4-ALK variants: Biological and molecular properties, and the implications for patients. Cancers 2017, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtman, S.; Rutteman, M.; De Zeeuw, C.; French, P. Echinoderm microtubule-associated protein like protein 4, a member of the echinoderm microtubule-associated protein family, stabilizes microtubules. Neuroscience 2007, 144, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Balke-Want, H.; Malchers, F.; Peifer, M.; Sos, M.L.; Koker, M.; Meder, L.; Lovely, C.M.; Heukamp, L.C.; Pao, W.; et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin. Cancer Res. 2012, 18, 4682–4690. [Google Scholar] [CrossRef] [Green Version]
- Mano, H. Non-solid oncogenes in solid tumors: EML4–ALK fusion genes in lung cancer. Cancer Sci. 2008, 99, 2349–2355. [Google Scholar] [CrossRef]
- Richards, M.W.; Law, E.W.; La’Verne, P.R.; Busacca, S.; O’Regan, L.; Fry, A.M.; Fennell, D.A.; Bayliss, R. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical β-propeller domain. Proc. Natl. Acad. Sci. USA 2014, 111, 5195–5200. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.; Richards, M.W.; Choi, J.; Fry, A.M.; Bayliss, R. Phase-separated foci of EML4-ALK facilitate signalling and depend upon an active kinase conformation. EMBO Rep. 2021, 22, e53693. [Google Scholar] [CrossRef]
- Christopoulos, P.; Endris, V.; Bozorgmehr, F.; Elsayed, M.; Kirchner, M.; Ristau, J.; Buchhalter, I.; Penzel, R.; Herth, F.J.; Heussel, C.P.; et al. EML4-ALK fusion variant V3 is a high-risk feature conferring accelerated metastatic spread, early treatment failure and worse overall survival in ALK+ non-small cell lung cancer. Int. J. Cancer 2018, 142, 2589–2598. [Google Scholar] [CrossRef] [Green Version]
- Tulpule, A.; Guan, J.; Neel, D.S.; Allegakoen, H.R.; Lin, Y.P.; Brown, D.; Chou, Y.T.; Heslin, A.; Chatterjee, N.; Perati, S.; et al. Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell 2021, 184, 2649–2664.e18. [Google Scholar] [CrossRef]
- Qin, Z.; Sun, H.; Yue, M.; Pan, X.; Chen, L.; Feng, X.; Yan, X.; Zhu, X.; Ji, H. Phase separation of EML4–ALK in firing downstream signaling and promoting lung tumorigenesis. Cell Discov. 2021, 7, 33. [Google Scholar] [CrossRef]
- Katayama, R. Therapeutic strategies and mechanisms of drug resistance in anaplastic lymphoma kinase (ALK)-rearranged lung cancer. Pharmacol. Ther. 2017, 177, 1–8. [Google Scholar] [CrossRef]
- Shaw, A.T.; Solomon, B. Targeting anaplastic lymphoma kinase in lung cancer. Clin. Cancer Res. 2011, 17, 2081–2086. [Google Scholar] [CrossRef] [Green Version]
- Camidge, D.R.; Bang, Y.-J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.I.; Kim, D.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Kim, D.-W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; Riley, G.J.; et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): Updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016, 17, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-W.; Tiseo, M.; Ahn, M.-J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.-W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: A randomized, multicenter phase II trial. J. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.I.; Perol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T.; et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision medicine takes on drug resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, B.W.; Zhai, D.; Deng, W.; Zhang, X.; Ung, J.; Nguyen, V.; Zhang, H.; Barrera, M.; Parra, A.; Cowell, J.; et al. TPX-0131, a Potent CNS-penetrant, Next-generation Inhibitor of Wild-type ALK and ALK-resistant Mutations. Mol. Cancer Ther. 2021, 20, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Kodityal, S.; Elvin, J.A.; Squillace, R.; Agarwal, N.; Miller, V.A.; Ali, S.M.; Klempner, S.J.; Ou, S.-H.I. A novel acquired ALK F1245C mutation confers resistance to crizotinib in ALK-positive NSCLC but is sensitive to ceritinib. Lung Cancer 2016, 92, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.; Milliken, J.C.; Azada, M.C.; Miller, V.A.; Ali, S.M.; Klempner, S.J. ALK F1174V mutation confers sensitivity while ALK I1171 mutation confers resistance to alectinib. The importance of serial biopsy post progression. Lung Cancer 2016, 91, 70–72. [Google Scholar] [CrossRef]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Meng, Y.; Wang, K.; Gao, M.; Du, J.; Wang, J.; Li, Z.; Zuo, D.; Wu, Y. EML4-ALK G1202R mutation induces EMT and confers resistance to ceritinib in NSCLC cells via activation of STAT3/Slug signaling. Cell. Signal. 2022, 92, 110264. [Google Scholar] [CrossRef]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non–small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [Green Version]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [Green Version]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK–positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretic, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion–positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Rihawi, K.; Alfieri, R.; Fiorentino, M.; Fontana, F.; Capizzi, E.; Cavazzoni, A.; Terracciano, M.; La Monica, S.; Ferrarini, A.; Buson, G.; et al. MYC amplification as a potential mechanism of primary resistance to crizotinib in ALK-rearranged non-small cell lung cancer: A brief report. Transl. Oncol. 2019, 12, 116–121. [Google Scholar] [CrossRef]
- Alidousty, C.; Baar, T.; Martelotto, L.G.; Heydt, C.; Wagener, S.; Fassunke, J.; Duerbaum, N.; Scheel, A.H.; Frank, S.; Holz, B.; et al. Genetic instability and recurrent MYC amplification in ALK-translocated NSCLC: A central role of TP53 mutations. J. Pathol. 2018, 246, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Isozaki, H.; Takigawa, N.; Kiura, K. Mechanisms of Acquired Resistance to ALK Inhibitors and the Rationale for Treating ALK-positive Lung Cancer. Cancers 2015, 7, 763–783. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Crizotinib resistance: Implications for therapeutic strategies. Ann. Oncol. 2016, 27, iii42–iii50. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, Y.; Zhang, H.; Shi, R.; Zhang, Z.; Liu, H.; Chen, J. EML4-ALK-mediated activation of the JAK2-STAT pathway is critical for non-small cell lung cancer transformation. BMC Pulm. Med. 2021, 21, 190. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Takezawa, K.; Sakai, K.; Azuma, K.; Kuwata, K.; Yamaguchi, H.; Hatashita, E.; Nishio, K.; Janne, P.A.; et al. Combined effect of ALK and MEK inhibitors in EML4–ALK-positive non-small-cell lung cancer cells. Br. J. Cancer 2012, 106, 763–767. [Google Scholar] [CrossRef] [Green Version]
- Kunimasa, K.; Hirotsu, Y.; Kukita, Y.; Ueda, Y.; Sato, Y.; Kimura, M.; Otsuka, T.; Hamamoto, Y.; Tamiya, M.; Inoue, T.; et al. EML4-ALK fusion variant. 3 and co-occurrent PIK3CA E542K mutation exhibiting primary resistance to three generations of ALK inhibitors. Cancer Genet. 2021, 256, 131–135. [Google Scholar] [CrossRef]
- Chen, Z.; Sasaki, T.; Tan, X.; Carretero, J.; Shimamura, T.; Li, D.; Xu, C.; Wang, Y.; Adelmant, G.O.; Capelletti, M.; et al. Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res. 2010, 70, 9827–9836. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, G.; Zhao, L.; Pan, F.; Qiang, J.; Han, S. Blocking the PI3K pathway enhances the efficacy of ALK-targeted therapy in EML4-ALK-positive nonsmall-cell lung cancer. Tumor Biol. 2014, 35, 9759–9767. [Google Scholar] [CrossRef]
- An, R.; Wang, Y.; Voeller, D.; Gower, A.; Kim, I.-K.; Zhang, Y.-W.; Giaccone, G. CRKL mediates EML4-ALK signaling and is a potential therapeutic target for ALK-rearranged lung adenocarcinoma. Oncotarget 2016, 7, 29199–29210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, H.W.; Du, J.; Boehm, J.S.; He, F.; Weir, B.A.; Wang, X.; Butaney, M.; Sequist, L.V.; Luo, B.; Engelman, J.A.; et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non–small cell lung cancers. Cancer Discov. 2011, 1, 608–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Dong Qz Fu, L.; Stoecker, M.; Wang, E.; Wang, E.H. Overexpression of CRKL correlates with poor prognosis and cell proliferation in non-small cell lung cancer. Molecular Carcinog. 2013, 52, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Tanizaki, J.; Okamoto, I.; Okabe, T.; Sakai, K.; Tanaka, K.; Hayashi, H.; Kaneda, H.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; et al. Activation of her family signaling as a mechanism of acquired resistance to alk inhibitors in eml4-alk–positive non–small cell lung cancer. Clin. Cancer Res. 2012, 18, 6219–6226. [Google Scholar] [CrossRef] [Green Version]
- Tani, T.; Yasuda, H.; Hamamoto, J.; Kuroda, A.; Arai, D.; Ishioka, K.; Ohgino, K.; Miyawaki, M.; Kawada, I.; Naoki, K.; et al. Activation of EGFR Bypass Signaling by TGFα Overexpression Induces Acquired Resistance to Alectinib in ALK-Translocated Lung Cancer Cells. Mol. Cancer Ther. 2016, 15, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Takeuchi, S.; Nakade, J.; Kita, K.; Nakagawa, T.; Nanjo, S.; Nakamuto, T.; Matsumoto, K.; Soda, M.; Mano, H.; et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin. Cancer Res. 2012, 18, 3592–3602. [Google Scholar] [CrossRef] [Green Version]
- Miyawaki, M.; Yasuda, H.; Tani, T.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Nukaga, S.; Hirano, T.; Kawada, I.; et al. Overcoming EGFR bypass signal-induced acquired resistance to ALK tyrosine kinase inhibitors in ALK-translocated lung cancer. Mol. Cancer Res. 2017, 15, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Batra, U.; Sharma, M.; Amrith, B.; Mehta, A.; Jain, P. EML4-ALK fusion as a resistance mechanism to osimertinib and its successful management with osimertinib and alectinib: Case report and review of the literature. Clin. Lung Cancer 2020, 21, e597–e600. [Google Scholar] [CrossRef]
- Du, X.; Zhang, J.; Gao, H.; Tai, Y. A novel break site of EML4-ALK report and a rare PRKAR1A-ALK report analyzed by different ALK detection platforms in non-small cell lung cancer patients. Thorac. Cancer 2021, 12, 2773–2779. [Google Scholar] [CrossRef]
- Liu, D.; Xu, X.; Wen, J.; Zhang, C.; Fan, M. Identification of a EML4-ALK exon 19 fusion variant in lung adenocarcinoma and alectinib resistance. Lung Cancer 2021, 160, 32–35. [Google Scholar] [CrossRef]
- Song, P.; Zhang, J.; Shang, C.; Zhang, L. Alectinib treatment response in lung adenocarcinoma patient with novel EML4-ALK variant. Thorac. Cancer 2018, 9, 1327–1332. [Google Scholar] [CrossRef]
- Zhu, V.W.; Nagasaka, M.; Madison, R.; Schrock, A.B.; Cui, J.; Ou, S.-H.I. A novel sequentially evolved EML4-ALK variant 3 G1202R/S1206Y double mutation in cis confers resistance to lorlatinib: A brief report and literature review. JTO Clin. Res. Rep. 2021, 2, 100116. [Google Scholar] [CrossRef]
- Nagasaka, M.; Ou, S.-H.I. Targeting Alternative Splicing as Adjunctive Treatment in EML4-ALK v3a/b+ NSCLC: Knowing Our Socratic Paradox and Learning From Spinal Muscular Atrophy. J. Thorac. Oncol. 2022, 17, 182–185. [Google Scholar] [CrossRef]
- Patel, M.; Malhotra, J.; Jabbour, S.K. Examining EML4-ALK variants in the clinical setting: The next frontier? J. Thorac. Dis. 2018, 10 (Suppl. S33), S4104. [Google Scholar] [CrossRef]
- Woo, C.; Seo, S.; Kim, S.; Jang, S.; Park, K.; Song, J.; Lee, B.; Richards, M.W.; Bayliss, R.; Lee, D.H.; et al. Differential protein stability and clinical responses ofEML4-ALK fusion variants to various ALK inhibitors in advancedALK-rearranged non-small cell lung cancer. Ann. Oncol. 2017, 28, 791–797. [Google Scholar] [CrossRef]
- Song, Z.; Lian, S.; Mak, S.; Chow, M.Z.-Y.; Xu, C.; Wang, W.; Keung, H.Y.; Lu, C.; Kebede, F.T.; Gao, Y.; et al. Deep RNA Sequencing Revealed Fusion Junctional Heterogeneity May Predict Crizotinib Treatment Efficacy in ALK-Rearranged NSCLC. J. Thorac. Oncol. 2022, 17, 264–276. [Google Scholar] [CrossRef]
- Hirai, N.; Sasaki, T.; Okumura, S.; Minami, Y.; Chiba, S.; Ohsaki, Y. Monomerization of ALK fusion proteins as a therapeutic strategy in ALK-rearranged non-small cell lung cancers. Front. Oncol. 2020, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef] [Green Version]
- Cornillie, S.P.; Bruno, B.J.; Lim, C.S.; Cheatham, I.I.I.T.E. Computational modeling of stapled peptides toward a treatment strategy for CML and broader implications in the design of lengthy peptide therapeutics. J. Phys. Chem. B 2018, 122, 3864–3875. [Google Scholar] [CrossRef]
- Iwasaki, T.; Tokuda, Y.; Kotake, A.; Okada, H.; Takeda, S.; Kawano, T.; Nakayama, Y. Cellular uptake and in vivo distribution of polyhistidine peptides. J. Control. Release 2015, 210, 115–124. [Google Scholar] [CrossRef]
- Zhao, Z.; Verma, V.; Zhang, M. Anaplastic lymphoma kinase: Role in cancer and therapy perspective. Cancer Biol. Ther. 2015, 16, 1691–1701. [Google Scholar] [CrossRef] [Green Version]
- Sang, J.; Acquaviva, J.; Friedland, J.C.; Smith, D.L.; Sequeira, M.; Zhang, C.; Jiang, Q.; Xue, L.; Lovely, C.M.; Jimenez, J.-P.; et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non–small cell lung cancer. Cancer Discov. 2013, 3, 430–443. [Google Scholar] [CrossRef] [Green Version]
- Gower, A.; Hsu, W.-H.; Hsu, S.-T.; Wang, Y.; Giaccone, G. EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer. Mol. Oncol. 2016, 10, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Rong, B.; Yang, S. Molecular mechanism and targeted therapy of Hsp90 involved in lung cancer: New discoveries and developments. Int. J. Oncol. 2018, 52, 321–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, L.E.L.; Dingemans, A.-M.C. Heat shock protein antagonists in early stage clinical trials for NSCLC. Expert Opin. Investig. Drugs 2017, 26, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long noncoding RNA and cancer: A new paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuka, M.; Tanemura, M.; Akamatsu, H. Long noncoding RNAs regulate malignant phenotypes in colorectal cancer. Biotarget 2018, 2, e4. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, J.; Xie, N.; Huang, H.; Xu, S.; Cai, J.; Qi, S. lincROR influences the stemness and crizotinib resistance in EML-ALK(+) non-small-cell lung cancer cells. Oncol. Targets Ther. 2018, 11, 3649–3657. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, Q.; Ke, Z.; Liu, Y.; Guo, H.; Fang, S.; Lu, K. LINC01001 Promotes Progression of Crizotinib-Resistant NSCLC by Modulating IGF2BP2/MYC Axis. Front. Pharmacol. 2021, 12, 759267. [Google Scholar] [CrossRef]
- Paliouras, A.R.; Buzzetti, M.; Shi, L.; Donaldson, I.J.; Magee, P.; Sahoo, S.; Leong, H.-S.; Fassan, M.; Carter, M.; Di Leva, G.; et al. Vulnerability of drug-resistant EML4-ALK rearranged lung cancer to transcriptional inhibition. EMBO Mol. Med. 2020, 12, e11099. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Adib, R.; Montgomery, J.M.; Atherton, J.; O’Regan, L.; Richards, M.W.; Straatman, K.R.; Roth, D.; Straube, A.; Bayliss, R.; Moores, C.A.; et al. Mitotic phosphorylation by NEK6 and NEK7 reduces the microtubule affinity of EML4 to promote chromosome congression. Sci. Signal. 2019, 12, eaaw2939. [Google Scholar] [CrossRef]
- O’Regan, L.; Barone, G.; Adib, R.; Woo, C.G.; Jeong, H.J.; Richardson, E.L.; Richards, M.W.; Muller, P.A.J.; Collis, S.J.; Fennell, D.A.; et al. EML4–ALK V3 oncogenic fusion proteins promote microtubule stabilization and accelerated migration through NEK9 and NEK7. J. Cell Sci. 2020, 133, jcs241505. [Google Scholar] [CrossRef]
- Soria, J.-C.; Tan, D.S.; Chiari, R.; Wu, Y.-L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.-J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Shaw, A.; Varghese, A.; Solomon, B.; Costa, D.; Novello, S.; Mino-Kenudson, M.; Awad, M.M.; Engelman, J.A.; Riely, G.J.; Monica, V.; et al. Pemetrexed-based chemotherapy in patients with advanced, ALK-positive non-small cell lung cancer. Ann. Oncol. 2013, 24, 59–66. [Google Scholar] [CrossRef]
- Lin, J.J.; Schoenfeld, A.J.; Zhu, V.W.; Yeap, B.Y.; Chin, E.; Rooney, M.; Plodkowski, A.J.; Digumarthy, S.R.; Dagogo-Jack, I.; Gainor, J.F.; et al. Efficacy of platinum/pemetrexed combination chemotherapy in ALK-positive NSCLC refractory to second-generation ALK inhibitors. J. Thorac. Oncol. 2020, 15, 258–265. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Rui, Y.; Tang, L.; Achilefu, S.; Gu, Y. Dual target gene therapy to EML4-ALK NSCLC by a gold nanoshell-based system. Theranostics 2018, 8, 2621–2633. [Google Scholar] [CrossRef]
- Below, J.; Das, J.M. Vincristine. StatPearls. Treasure Island (FL): StatPearls Publishing. January 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537122/ (accessed on 20 June 2022).
- Sampson, J.; Ju, H.-M.; Song, J.-Y.; Fry, A.M.; Bayliss, R.; Choi, J. A Polytherapy Strategy Using Vincristine and ALK Inhibitors to Sensitise EML4-ALK-Positive NSCLC. Cancers 2022, 14, 779. [Google Scholar] [CrossRef]
- Lucken, K.; O’Regan, L.; Choi, J.; Sampson, J.; Pashley, S.L.; Bayliss, R.; Khan, S.; Fry, A.M. EML4-ALK variant 3 promotes mitotic errors and spindle assembly checkpoint deficiency leading to increased microtubule poison sensitivityEML4-ALK V3 sensitizes NSCLC cells to microtubule poisons. Mol. Cancer Res. 2022, 6, 854–866. [Google Scholar] [CrossRef]
- Giaj-Levra, N.; Borghetti, P.; Bruni, A.; Ciammella, P.; Cuccia, F.; Fozza, A.; Franceschini, D.; Scotti, V.; Vagge, S.; Alongi, F. Current radiotherapy techniques in NSCLC: Challenges and potential solutions. Expert Rev. Anticancer. Ther. 2020, 20, 387–402. [Google Scholar] [CrossRef]
- Dai, Y.; Wei, Q.; Schwager, C.; Moustafa, M.; Zhou, C.; Lipson, K.E.; Weichert, W.; Debus, J.; Abdollahi, A. Synergistic effects of crizotinib and radiotherapy in experimental EML4–ALK fusion positive lung cancer. Radiother. Oncol. 2015, 114, 173–181. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudirakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.-P.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.I.; Kwak, E.L.; Siwak-Tapp, C.; Dy, J.; Bergethon, K.; Clark, J.W.; Camidge, D.R.; Solomon, B.J.; Maki, R.G.; Bang, Y.-J.; et al. Activity of Crizotinib (PF02341066), a Dual Mesenchymal-Epithelial Transition (MET) and Anaplastic Lymphoma Kinase (ALK) Inhibitor, in a Non-small Cell Lung Cancer Patient with De Novo MET Amplification. J. Thorac. Oncol. 2011, 6, 942–946. [Google Scholar] [CrossRef] [Green Version]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. JNCI J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Wei, Q.; Schwager, C.; Hanne, J.; Zhou, C.; Herfarth, K.; Rieken, S.; Lipson, K.E.; Debus, J.; Abdollahi, A. Oncogene addiction and radiation oncology: Effect of radiotherapy with photons and carbon ions in ALK-EML4 translocated NSCLC. Radiat. Oncol. 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Liang, L.; Mao, M.; Wu, L.; Chen, T.; Lyu, J.; Wang, Q.; Li. T. Efficacy and Drug Resistance Analysis of ALK Inhibitors in Combination with Stereotactic Body Radiation Therapy for Treating Lung Squamous Carcinoma Patient Harboring EML4-ALK Rearrangement. Oncol. Targets Ther. 2021, 14, 5385. [Google Scholar] [CrossRef]
- Antoni, D.; Burckel, H.; Noel, G. Effects of ALK inhibitors (PF-02341066 and PF-06463922) with radiotherapy in EML4–ALK fusion positive (H2228 and 185IG) and negative (A549) experimental lung cancer cell lines. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, e507–e508. [Google Scholar] [CrossRef]
- Sun, Y.; Nowak, K.A.; Zaorsky, N.G.; Winchester, C.-L.; Dalal, K.; Giacalone, N.J.; Liu, N.; Werner-Wasik, M.; Wasik, M.A.; Dicker, A.P.; et al. ALK inhibitor PF02341066 (crizotinib) increases sensitivity to radiation in non–small cell lung cancer expressing EML4-ALK. Mol. Cancer Ther. 2013, 12, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Liu, D.; Wen, J.; Chen, J.; Fan, M. A case report of exceptional clinical response to chemoradiotherapy and tyrosine kinase inhibitors in a patient with EML4-ALK fusion variant 1 non-small cell lung cancer. Transl. Lung Cancer Res. 2020, 9, 2500–2507. [Google Scholar] [CrossRef]
- Fleschutz, K.; Walter, L.; Leistner, R.; Heinzerling, L. ALK Inhibitors Do Not Increase Sensitivity to Radiation in EML4-ALK Non-small Cell Lung Cancer. Anticancer. Res. 2020, 40, 4937–4946. [Google Scholar] [CrossRef]
- Anagnostou, V.K.; Brahmer, J.R. Cancer immunotherapy: A future paradigm shift in the treatment of non–small cell lung cancer. Clin. Cancer Res. 2015, 21, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Chen, N.; Fang, W.; Zhan, J.; Liu, Q.; Kang, S.; He, X.; Liu, L.; Zhou, T.; Huang, J.; et al. Upregulation of PD-L1 by EML4-ALK fusion protein mediates the immune escape in ALK positive NSCLC: Implication for optional anti-PD-1/PD-L1 immune therapy for ALK-TKIs sensitive and resistant NSCLC patients. Oncoimmunology 2016, 5, e1094598. [Google Scholar] [CrossRef]
- Pyo, K.-H.; Lim, S.M.; Park, C.-W.; Jo, H.-N.; Kim, J.H.; Yun, M.-R.; Kim, D.; Xin, C.-F.; Lee, W.; Gheorghiu, B.; et al. Comprehensive analyses of immunodynamics and immunoreactivity in response to treatment in ALK-positive non-small-cell lung cancer. J. Immunother. Cancer 2020, 8, e000970. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Reynolds, C.; Waterhouse, D.; Garon, E.B.; Chandler, J.; Babu, S.; Thurmes, P.; Spira, A.; Jotte, R.; Zhu, J.; et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation—positive advanced non–small cell lung cancer (CheckMate 370). J. Thorac. Oncol. 2018, 13, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.; Liu, C.; Zheng, S.; Wang, X.; Feng, X.; Li, H.; Sun, N.; He, J. Molecular heterogeneity of anti-PD-1/PD-L1 immunotherapy efficacy is correlated with tumor immune microenvironment in East Asian patients with non-small cell lung cancer. Cancer Biol. Med. 2020, 17, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Gao, Y.; Xiong, J.; Lu, J.; Yang, J.; Wang, X.; Cai, Y.; Li, L.; Fu, X. Tumor-infiltrating CD8+ T cells in ALK-positive lung cancer are functionally impaired despite the absence of PD-L1 on tumor cells. Lung Cancer 2020, 150, 139–144. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC class I downregulation in cancer: Underlying mechanisms and potential targets for cancer immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Mu, D.; Guo, J.; Yu, W.; Zhang, J.; Ren, X.; Han, Y. Downregulation of PD-L1 and HLA-I in non-small cell lung cancer with ALK fusion. Thorac. Cancer 2022, 13, 1153–1163. [Google Scholar] [CrossRef]
- Chiu, L.-C.; Lin, S.-M.; Lo, Y.-L.; Kuo, S.C.-H.; Yang, C.-T.; Hsu, P.-C. Immunotherapy and Vaccination in Surgically Resectable Non-Small Cell Lung Cancer (NSCLC). Vaccines 2021, 9, 689. [Google Scholar] [CrossRef]
- Codony-Servat, J.; García-Roman, S.; Molina-Vila, M.Á.; Bertran-Alamillo, J.; Viteri, S.; d’Hondt, E.; Rosell, R. Anti-epidermal growth factor vaccine antibodies increase the antitumor activity of kinase inhibitors in ALK and RET rearranged lung cancer cells. Transl. Oncol. 2021, 14, 100887. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papageorgiou, S.; Pashley, S.L.; O’Regan, L.; Khan, S.; Bayliss, R.; Fry, A.M. Alternative Treatment Options to ALK Inhibitor Monotherapy for EML4-ALK-Driven Lung Cancer. Cancers 2022, 14, 3452. https://doi.org/10.3390/cancers14143452
Papageorgiou S, Pashley SL, O’Regan L, Khan S, Bayliss R, Fry AM. Alternative Treatment Options to ALK Inhibitor Monotherapy for EML4-ALK-Driven Lung Cancer. Cancers. 2022; 14(14):3452. https://doi.org/10.3390/cancers14143452
Chicago/Turabian StylePapageorgiou, Savvas, Sarah L. Pashley, Laura O’Regan, Sam Khan, Richard Bayliss, and Andrew M. Fry. 2022. "Alternative Treatment Options to ALK Inhibitor Monotherapy for EML4-ALK-Driven Lung Cancer" Cancers 14, no. 14: 3452. https://doi.org/10.3390/cancers14143452
APA StylePapageorgiou, S., Pashley, S. L., O’Regan, L., Khan, S., Bayliss, R., & Fry, A. M. (2022). Alternative Treatment Options to ALK Inhibitor Monotherapy for EML4-ALK-Driven Lung Cancer. Cancers, 14(14), 3452. https://doi.org/10.3390/cancers14143452