
Stem Cell Theory of Cancer: Implications for Translational Research from Bedside to Bench

 , ,
, , {kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
“Whether or not you can observe a thing depends upon the theory you use. It is the theory which decides what can be observed”.Albert Einstein
2. Scientific Method
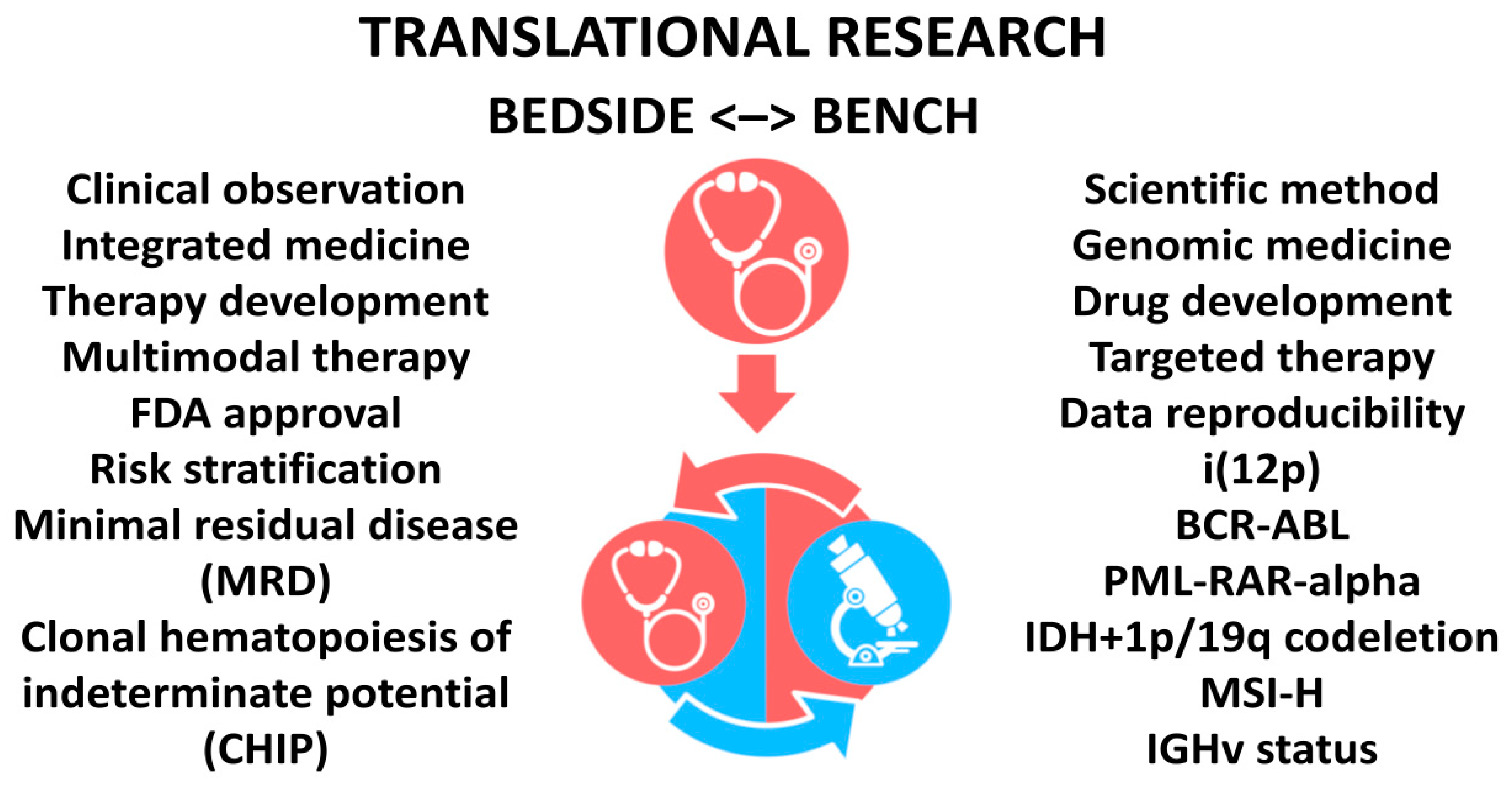
3. Translational Research
4. Heuristic Approach
4.1. Drug Development
4.2. Data Reproducibility
5. Genomic Medicine
5.1. Favorable Mutations
5.2. Innocuous Mutations
5.3. Somatic Hypermutations
6. Integrated Medicine
6.1. Clinical Risk
6.2. Recurrence Score
6.3. Risk Stratification
6.4. Minimal Residual Disease
6.5. Multimodal Therapy
7. Therapy Development
8. Limitations and Drawbacks
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fogel, D.B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp. Clin. Trials Commun. 2018, 11, 156–164. [Google Scholar] [CrossRef]
- Hingorani, A.D.; Kuan, V.; Finan, C.; Kruger, F.A.; Gaulton, A.; Chopade, S.; Sofat, R.; MacAllister, R.J.; Overington, J.P.; Hemingway, H.; et al. Improving the odds of drug development success through human genomics: Modelling study. Sci. Rep. 2019, 9, 18911. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, J.P.A. Why most published research findings are false. PLOS Med. 2005, 2, e124. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. Half of top cancer studies fail high-profile reproducibility effort. Nature 2021, 600, 368–369. [Google Scholar] [CrossRef]
- Errington, T.M.; Mathur, M.; Soderberg, C.K.; Denis, A.; Perfito, N.; Iorns, E.; Nosek, B.A. Investigating the replicability of preclinical cancer biology. eLife 2021, 10, e71601. [Google Scholar] [CrossRef]
- Tu, S.M. Origin of Cancers. In Clinical Perspectives and Implications of a Stem-Cell Theory of Cancer; Cancer Treatment and Research; Rosen, S.T., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 154, pp. 15–16. [Google Scholar]
- Tu, S.M. Story of Hydra: Portrait of Cancer as a Stem-Cell Disease; Nova: New York, NY, USA, 2019; pp. 43–53. [Google Scholar]
- Tu, S.; Bilen, M.A.; Tannir, N.M. Pillar and pitfall of cancer research. Cancer Med. 2014, 3, 1035–1037. [Google Scholar] [CrossRef]
- Huggins, C.; Stevens, R.E., Jr.; Hodges, C.V. Studies on prostatic cancer II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941, 43, 209–223. [Google Scholar]
- Li, M.C.; Whitmore, W.F., Jr.; Golbey, R.; Grabstald, H. Effects of combined drug therapy on metastatic cancer of the testis. JAMA 1960, 174, 1291–1299. [Google Scholar]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2018, 20, 273–286. [Google Scholar] [CrossRef]
- Baker, M. 1500 scientists lift the lid on reproducibility. Nature 2016, 533, 452–454. [Google Scholar] [CrossRef] [Green Version]
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 712. [Google Scholar]
- Ioannidis, J.P.A.; Kim, B.Y.S.; Trounson, A. How to design preclinical studies in nanomedicine and cell therapy to maximize the prospects of clinical translation. Nat. Biomed. Eng. 2018, 2, 797–809. [Google Scholar] [CrossRef]
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar]
- Forrest, I.S.; Chaudhary, K.; Vy, H.M.T.; Petrazzini, B.O.; Bafna, S.; Jordan, D.M.; Rocheleau, G.; Loos, R.J.F.; Nadkarni, G.N.; Cho, J.H.; et al. Population-Based Penetrance of Deleterious Clinical Variants. JAMA 2022, 327, 350–359. [Google Scholar] [CrossRef]
- Bianchini, L.; Birtwisle, L.; Saada, E.; Bazin, A.; Long, E.; Roussel, J.F.; Michiels, J.F.; Forest, F.; Dani, C.; Myklebost, O.; et al. Identification of PPAP2B as a novel recurrent translocation partner gene of HMGA2 in lipomas. Genes Chromosomes Cancer 2013, 52, 580–590. [Google Scholar]
- Robinson, D.R.; Wu, Y.-M.; Kalyana-Sundaram, S.; Cao, X.; Lonigro, R.J.; Sung, Y.S.; Chern, C.L.; Zhang, L.; Wang, R.; Su, F.; et al. Identi-fication of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat. Genet 2013, 45, 180–185. [Google Scholar]
- Seeger-Nukpezah, T.; Geynisman, D.M.; Nikonova, A.S.; Benzing, T.; Golemis, E.A. The hallmarks of cancer: Relevance to the path-ogenesis of polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 515–534. [Google Scholar]
- Darbro, B.W.; Singh, R.; Zimmerman, M.B.; Mahajan, V.; Bassuk, A.G. Autism Linked to Increased Oncogene Mutations but Decreased Cancer Rate. PLoS ONE 2016, 11, e0149041. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Leeper, H.E.; Caron, A.A.; Decker, P.A.; Jenkins, R.B.; Lachance, D.H.; Giannini, C. IDH mutation, 1p19q codeletion and ATRX loss in WHO grade II gliomas. Oncotarget 2015, 6, 30295–30305. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. SomaticSF3B1Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal archi-tecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Severson, E.A.; Riedlinger, G.M.; Connelly, C.F.; Vergilio, J.-A.; Goldfinger, M.; Ramkissoon, S.; Frampton, G.M.; Ross, J.S.; Fratella-Calabrese, A.; Gay, L.; et al. Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood 2018, 131, 2501–2505. [Google Scholar] [CrossRef] [Green Version]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated IgVH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar]
- Shubinsky, G.; Schlesinger, M. The CD38 Lymphocyte Differentiation Marker: New Insight into Its Ectoenzymatic Activity and Its Role as a Signal Transducer. Immunity 1997, 7, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Noh, S.H.; Park, S.R.; Yang, H.K.; Chung, H.C.; Chung, I.J.; Kim, S.W.; Kim, H.H.; Choi, J.H.; Kim, H.K.; Yu, W.; et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomized phase 3 trial. Lancet Oncol. 2014, 15, 1389–1396. [Google Scholar]
- Smyth, E.C.; Wotherspoon, A.; Peckitt, C.; Gonzalez, D.; Hulkki-Wilson, S.; Eltahir, Z.; Fassan, M.; Rugge, M.; Valeri, N.; Okines, A.; et al. Mismatch repair deficiency, microsatellite instability, and survival: An exploratory analysis of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial. JAMA Oncol. 2017, 3, 1197–1203. [Google Scholar]
- Pietrantonio, F.; Miceli, R.; Raimondi, A.; Kim, Y.W.; Kang, W.K.; Langley, R.E.; Choi, Y.Y.; Kim, K.-M.; Nankivell, M.G.; Morano, F.; et al. Individual Patient Data Meta-Analysis of the Value of Microsatellite Instability As a Biomarker in Gastric Cancer. J. Clin. Oncol. 2019, 37, 3392–3400. [Google Scholar] [CrossRef]
- Gryfe, R.; Kim, H.; Hsieh, E.T.; Aronson, M.D.; Holowaty, E.J.; Bull, S.B.; Redston, M.; Gallinger, S. Tumor Microsatellite Instability and Clinical Outcome in Young Patients with Colorectal Cancer. New Engl. J. Med. 2000, 342, 69–77. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Curtin, K.; Ma, K.N.; Schaffer, D.; Coleman, L.W.; Leppert, M.; Slattery, M.L. Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol. Biomark. Prev. 2001, 10, 917–923. [Google Scholar]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 321926. [Google Scholar]
- Hoevenaar, W.H.M.; Janssen, A.; Quirindongo, A.I.; Ma, H.; Klaasen, S.J.; Teixeira, A.; van Gerwen, B.; Lansu, N.; Morsink, F.H.M.; Offerhaus, G.J.A.; et al. Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 2020, 11, 1501. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.-M.; Lin, S.-H.; Logothetis, C.J. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol. 2002, 3, 508–513. [Google Scholar] [CrossRef]
- Tu, S.-M. Stem Cell Theory of Cancer: Implications of a Viral Etiology in Certain Malignancies. Cancers 2021, 13, 2738. [Google Scholar] [CrossRef]
- Tu, S.-M.; Zhang, M.; Wood, C.; Pisters, L. Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity. Cancers 2021, 13, 4006. [Google Scholar] [CrossRef]
- Tu, S.M.; Estecio, M.; Lin, S.H.; Millward, N.Z. Stem cell theory of cancer: Rude awakening or bad dreams from cancer dormancy? Cancers 2022, 14, 655. [Google Scholar]
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.Y.; Brain, E.; Causeret, S.; Delorenzi, M.; et al. 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. N. Engl. J. Med. 2016, 375, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, F.; van’t Veer, L.; Poncet, C.; Cardozo, J.L.; Delaloge, S.; Pierga, J.-Y.; Vuylsteke, P.; Brain, E.; Viale, G.; Kuemmel, S.; et al. MINDACT: Long-term results of the large prospective trial testing the 70-gene signature MammaPrint as guidance for adjuvant chemotherapy in breast cancer patients. J. Clin. Oncol. 2020, 38, 506. [Google Scholar] [CrossRef]
- Gianni, L.; Pienkowski, T.; Im, Y.H.; Tseng, L.M.; Liu, M.C.; Lluch, A.; Staroslawska, E.; de la Haba-Rodriguez, J.; Im, S.A.; Pedrini, J.L.; et al. Five-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): A multicentre, open-label, phase 2 randomised trial. Lancet Oncol. 2016, 17, 791–800. [Google Scholar]
- Tolaney, S.M.; Barry, W.T.; Dang, C.T.; Yardley, D.A.; Moy, B.; Marcom, P.K.; Albain, K.S.; Rugo, H.S.; Ellis, M.; Shapira, I.; et al. Adjuvant Paclitaxel and Trastuzumab for Node-Negative, HER2-Positive Breast Cancer. N. Engl. J. Med. 2015, 372, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Miles, D.; Kim, S.-B.; Im, Y.-H.; Im, S.-A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Tu, S.M.; Campbell, M.; Shah, A.; Logothetis, C.J. Application of a successful germ cell tumor paradigm to the challenges of common adult solid cancers. J. Cell Sci. Ther. 2021, 12, 301. [Google Scholar]
- Tu, S.-M.; Pisters, L. Curing Cancer: Lessons from a Prototype. Cancers 2021, 13, 660. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Brüggemann, M.; Kotrova, M. Minimal residual disease in adult ALL: Technical aspects and implications for correct clinical interpretation. Blood Adv. 2017, 1, 2456–2466. [Google Scholar] [CrossRef]
- Tu, S.; Bilen, M.A.; Hess, K.R.; Broaddus, R.R.; Kopetz, S.; Wei, C.; Pagliaro, L.C.; Karam, J.A.; Ward, J.; Wood, C.G.; et al. Intratumoral heterogeneity: Role of differentiation in a potentially lethal phenotype of testicular cancer. Cancer 2016, 122, 1836–1843. [Google Scholar] [CrossRef]
- Umbreit, E.C.; Siddiqui, B.A.; Hwang, M.J.; Joon, A.Y.; Maity, T.; Westerman, M.E.; Merriman, K.W.; Alhasson, H.; Uthup, J.; Guo, T.; et al. Origin of Subsequent Malignant Neoplasms in Patients with History of Testicular Germ Cell Tumor. Cancers 2020, 12, 3755. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, S.-M.; Singh, S.R.; Arnaoutakis, K.; Malapati, S.; Bhatti, S.A.; Joon, A.Y.; Atiq, O.T.; Pisters, L.L. Stem Cell Theory of Cancer: Implications for Translational Research from Bedside to Bench. Cancers 2022, 14, 3345. https://doi.org/10.3390/cancers14143345
Tu S-M, Singh SR, Arnaoutakis K, Malapati S, Bhatti SA, Joon AY, Atiq OT, Pisters LL. Stem Cell Theory of Cancer: Implications for Translational Research from Bedside to Bench. Cancers. 2022; 14(14):3345. https://doi.org/10.3390/cancers14143345
Chicago/Turabian StyleTu, Shi-Ming, Sunny R. Singh, Konstantinos Arnaoutakis, Sindhu Malapati, Sajjad A. Bhatti, Aron Y. Joon, Omar T. Atiq, and Louis L. Pisters. 2022. "Stem Cell Theory of Cancer: Implications for Translational Research from Bedside to Bench" Cancers 14, no. 14: 3345. https://doi.org/10.3390/cancers14143345
APA StyleTu, S.-M., Singh, S. R., Arnaoutakis, K., Malapati, S., Bhatti, S. A., Joon, A. Y., Atiq, O. T., & Pisters, L. L. (2022). Stem Cell Theory of Cancer: Implications for Translational Research from Bedside to Bench. Cancers, 14(14), 3345. https://doi.org/10.3390/cancers14143345