
Mechanisms of Resistance and Implications for Treatment Strategies in Chronic Myeloid Leukaemia

 ,
,
Abstract
Simple Summary
Abstract
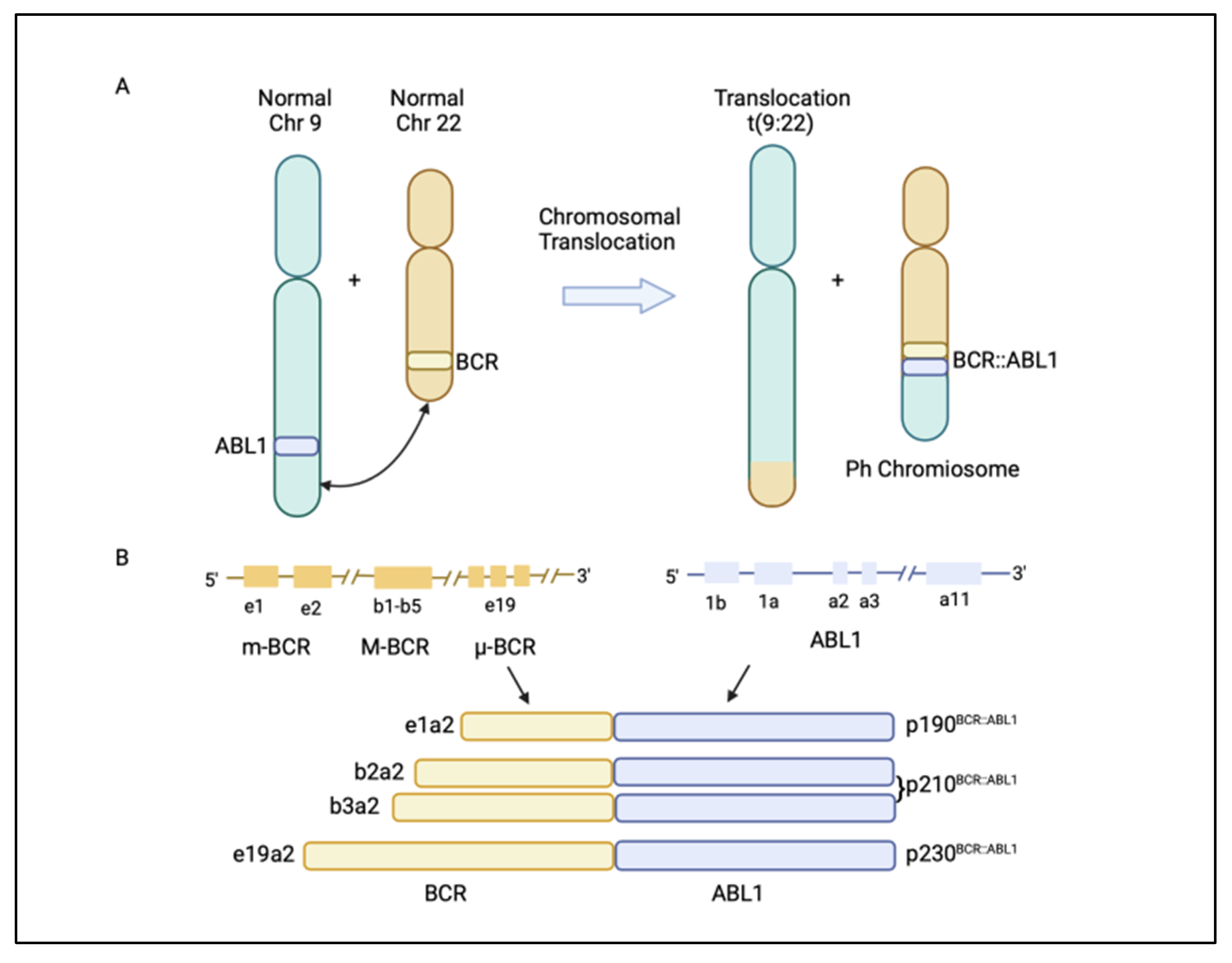
1. Introduction

2. BCR::ABL1-Dependent Mechanisms of Resistance
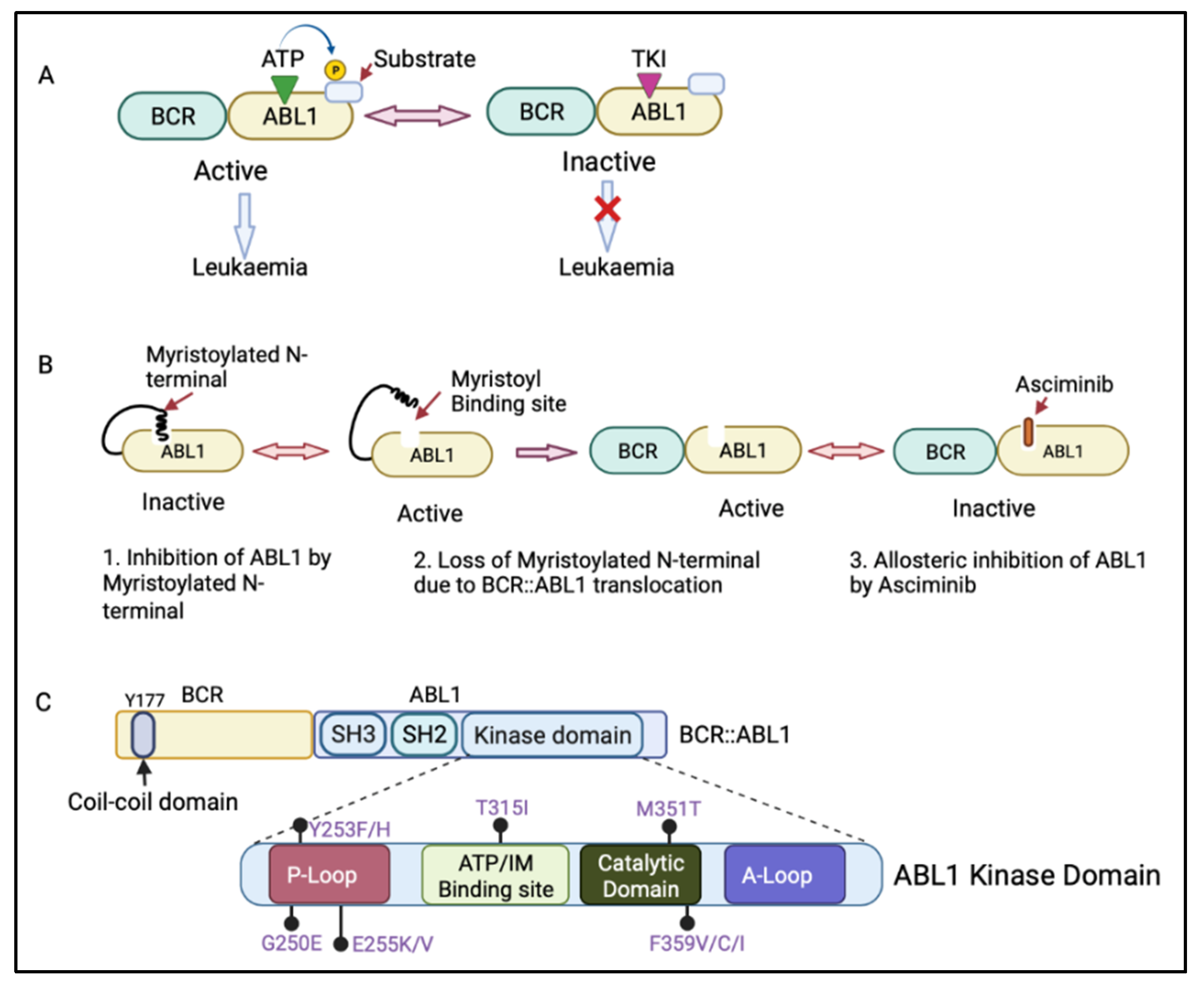
2.1. Kinase Domain Mutations

{kind=link}
{kind=link}
{kind=link}
| Kinase Domain Mutations | TKI Sensitivity |
|---|---|
| T315I, Y253F/H, E255K/V, Q252H, M244V, L248V, G250E, F317L, M351T, M355D, F359V, and H396R/P/A | Reduced sensitivity to imatinib |
| T315I, L248V, Y253H, E255K/V, and F359V/I/C | Reduced sensitivity to nilotinib |
| T315I/A, V299L, and F317L/V/I/C | Reduced sensitivity to dasatinib |
| T315I, E255V/K, V299L, G250E, E255K/V, and F317L/V/I/C | Reduced sensitivity to bosutinib |
| T315M/L | Reduced sensitivity to ponatinib |
2.2. Myristoyl Domain Mutations
2.3. BCR::ABL1 Overexpression
2.4. Altered Expression of Drug Transporters
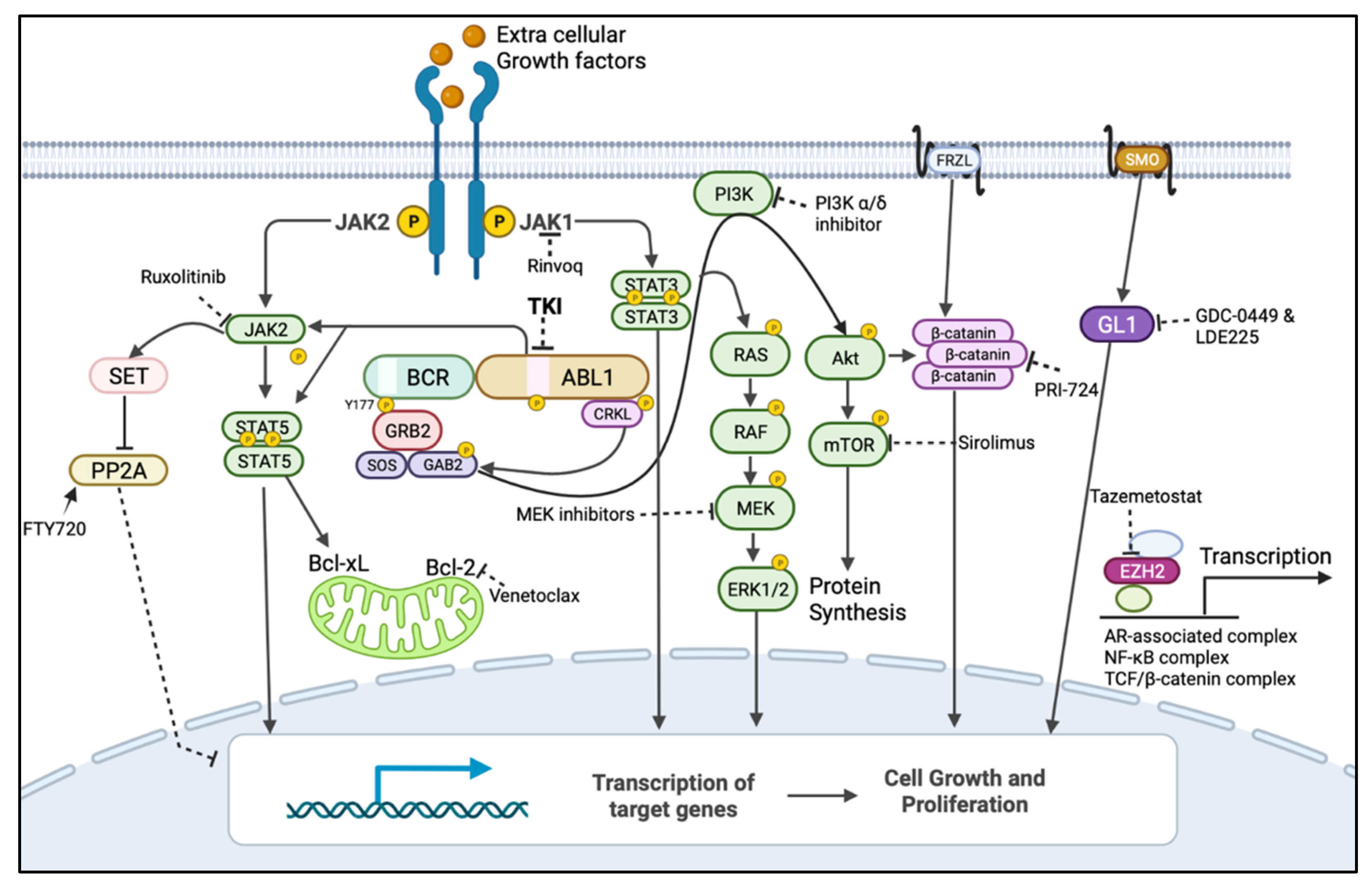
3. BCR::ABL1-Independent Mechanisms of Resistance/Persistence
3.1. Alternative Activation of MAPK Pathway
3.2. Alternative Activation of JAK/STAT Pathway
3.3. Alternative Activation of PI3K/AKT Pathway and Dysregulation of Autophagy
3.4. Activation of Wnt/β-Catenin Signalling
3.5. Protein Phosphatase 2A (PP2A) Level
3.6. Epigenetic Alterations
3.7. Inflammatory TNF-α and TGF-β Pathways
3.8. Sonic Hedgehog Pathway Activation
3.9. Dysregulation of Apoptotic Proteins’ Expression
4. Targeted Therapies against BCR::ABL1-Independent Resistant Cells
| Target Pathway | Inhibitor/s | Stage of Development | Approved/Treated Disease | Ref. |
|---|---|---|---|---|
| RAS/RAF/MEK/ERK |
| FDA approved | BRAF(V600E) melanoma | [113] |
| FDA approved | |||
| JAK/STAT |
| FDA approved |
| [114,115] |
| FDA approved |
| ||
| FDA approved |
| ||
| PI3K/AKT/mTOR |
| FDA approved |
| [116,117] |
| FDA approved |
| ||
| FDA approved |
| ||
| Wnt/β-catenin | CBP/β-catenin antagonist: PRI-724 | Phase 2 Clinical Trial (NCT01606579) | Acute myeloid leukaemia and chronic myeloid leukaemia | [118] |
| Tumour suppressor: PP2A | SET: FTY720 (Fingolimod) | FDA Approved | Multiple myeloma and mantle cell lymphoma | [119,120] |
| Epigenetic modulator: EZH2 | Tazemetostat | FDA Approved | Advanced or metastatic epithelioid sarcoma | [121] |
| Immune system | IFN-α | FDA Approved | Hairy cell leukaemia, CML, follicular non-Hodgkin lymphoma, melanoma, and AIDS-related Kaposi’s sarcoma | [34] |
| Hedgehog pathway | Vismodegib (GDC-0449) and Sonidegib (LDE225) | FDA Approved | Basal cell carcinoma and acute myeloid leukaemia | [122] |
| Intrinsic apoptotic pathway | Venetoclax | FDA Approved | Chronic lymphocytic leukaemia and acute myeloid leukaemia | [123] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bennett, J.H. Case of hypertrophy of the spleen and liver, in which death took place from suppuration of the blood. Edinburgh Med. Sug. J. 1845, 64, 413–423. [Google Scholar]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Propp, S.; Lizzi, F.A. Brief Report: Philadelphia Chromosome in Acute Lymphocytic Leukemia. Blood 1970, 36, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Flis, S.; Chojnacki, T. Chronic myelogenous leukemia, a still unsolved problem: Pitfalls and new therapeutic possibilities. Drug Des. Dev. Ther. 2019, 13, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.W.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jabbour, E.; Issa, G.; Sasaki, K.; Ravandi, F.; Maiti, A.; Daver, N.; Kadia, T.; DiNardo, C.D.; Konopleva, M.; et al. Impact of frontline treatment approach on outcomes of myeloid blast phase CML. J. Hematol. Oncol. 2021, 14, 94. [Google Scholar] [CrossRef]
- Sessions, J. Chronic myeloid leukemia in 2007. Am. J. Health-Syst. Pharm. 2007, 64, S4–S9. [Google Scholar] [CrossRef]
- McWhirter, J.R.; Wang, J.Y. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of Philadelphia chromosome-positive human leukemias. EMBO J. 1993, 12, 1533–1546. [Google Scholar] [CrossRef]
- Peiris, M.N.; Li, F.; Donoghue, D.J. BCR: A promiscuous fusion partner in hematopoietic disorders. Oncotarget 2019, 10, 2738–2754. [Google Scholar] [CrossRef]
- Pendergast, A.M.; Quilliam, L.A.; Cripe, L.D.; Bassing, C.H.; Dai, Z.; Li, N.; Batzer, A.; Rabun, K.M.; Der, C.J.; Schlessinger, J. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell 1993, 75, 175–185. [Google Scholar] [CrossRef]
- Hantschel, O.; Nagar, B.; Guettler, S.; Kretzschmar, J.; Dorey, K.; Kuriyan, J.; Superti-Furga, G. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 2003, 112, 845–857. [Google Scholar] [CrossRef]
- Vinhas, R.; Lourenco, A.; Santos, S.; Lemos, M.; Ribeiro, P.; de Sousa, A.B.; Baptista, P.V.; Fernandes, A.R. A novel BCR-ABL1 mutation in a patient with Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia. Oncol. Targets Ther. 2018, 11, 8589–8598. [Google Scholar] [CrossRef]
- Shawver, L.K.; Slamon, D.; Ullrich, A. Smart drugs: Tyrosine kinase inhibitors in cancer therapy. Cancer Cell 2002, 1, 117–123. [Google Scholar] [CrossRef]
- Radich, J.P.; Deininger, M.; Abboud, C.N.; Altman, J.K.; Berman, E.; Bhatia, R.; Bhatnagar, B.; Curtin, P.; DeAngelo, D.J.; Gotlib, J.; et al. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 1108–1135. [Google Scholar] [CrossRef]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.-T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef]
- FDA. FDA Approves Asciminib for Philadelphia Chromosome-Positive Chronic Myeloid Leukemia. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-asciminib-philadelphia-chromosome-positive-chronic-myeloid-leukemia (accessed on 9 December 2021).
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef]
- Branford, S.; Fletcher, L.; Cross, N.C.; Muller, M.C.; Hochhaus, A.; Kim, D.W.; Radich, J.P.; Saglio, G.; Pane, F.; Kamel-Reid, S.; et al. Desirable performance characteristics for BCR-ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response and comparison of response rates between clinical trials. Blood 2008, 112, 3330–3338. [Google Scholar] [CrossRef]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Breccia, M.; Molica, M.; Colafigli, G.; Massaro, F.; Quattrocchi, L.; Latagliata, R.; Mancini, M.; Diverio, D.; Guarini, A.; Alimena, G.; et al. Prognostic factors associated with a stable MR4.5 achievement in chronic myeloid leukemia patients treated with imatinib. Oncotarget 2018, 9, 7534–7540. [Google Scholar] [CrossRef]
- Wang, R.; Cong, Y.; Li, C.; Zhang, C.; Lin, H. Predictive value of early molecular response for deep molecular response in chronic phase of chronic myeloid leukemia. Medicine 2019, 98, e15222. [Google Scholar] [CrossRef]
- Hughes, T.; Boquimpani, C.; Takahashi, N.; Benyamini, N.; Clementino, N.; Shuvaev, V.; Ailawadhi, S.; Lipton, J.; Turkina, A.; Paz, R. Durable treatment-free remission after stopping second line nilotinib in patients with chronic myeloid leukemia in chronic phase: ENESTOP 96-wk update. Haematologica 2017, 102, 75. [Google Scholar]
- Ross, D.M.; Pagani, I.S.; Shanmuganathan, N.; Kok, C.H.; Seymour, J.F.; Mills, A.K.; Filshie, R.J.; Arthur, C.K.; Dang, P.; Saunders, V.A.; et al. Long-term treatment-free remission of chronic myeloid leukemia with falling levels of residual leukemic cells. Leukemia 2018, 32, 2572–2579. [Google Scholar] [CrossRef]
- Ross, D.M.; Hughes, T.P. Treatment-free remission in patients with chronic myeloid leukaemia. Nat. Rev. Clin. Oncol. 2020, 17, 493–503. [Google Scholar] [CrossRef]
- Mahon, F.-X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Bonifacio, M.; Stagno, F.; Scaffidi, L.; Krampera, M.; Di Raimondo, F. Management of Chronic Myeloid Leukemia in Advanced Phase. Front. Oncol 2019, 9, 1132. [Google Scholar] [CrossRef]
- Soverini, S.; Bassan, R.; Lion, T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: Recent advances and remaining challenges. J. Hematol. Oncol. 2019, 12, 39. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Loscocco, F.; Visani, G.; Galimberti, S.; Curti, A.; Isidori, A. BCR-ABL Independent Mechanisms of Resistance in Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 939. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; O’Hare, T.; Deininger, M.W. Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors. Hematol. Oncol. Clin. N. Am. 2017, 31, 589–612. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, C.; Kumar, P.S.; Sarma, P. Novel mutations in the kinase domain of BCR-ABL gene causing imatinib resistance in chronic myeloid leukemia patients. Sci. Rep. 2019, 9, 2412. [Google Scholar] [CrossRef]
- Redaelli, S.; Piazza, R.; Rostagno, R.; Magistroni, V.; Perini, P.; Marega, M.; Gambacorti-Passerini, C.; Boschelli, F. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J. Clin. Oncol. 2009, 27, 469–471. [Google Scholar] [CrossRef]
- Zabriskie, M.S.; Eide, C.A.; Tantravahi, S.K.; Vellore, N.A.; Estrada, J.; Nicolini, F.E.; Khoury, H.J.; Larson, R.A.; Konopleva, M.; Cortes, J.E.; et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 2014, 26, 428–442. [Google Scholar] [CrossRef]
- Pagani, I.S.; Dang, P.; Saunders, V.A.; Braley, J.; Thieleke, A.; Branford, S.; Hughes, T.P.; Ross, D.M. Clinical utility of genomic DNA Q-PCR for the monitoring of a patient with atypical e19a2 BCR-ABL1 transcripts in chronic myeloid leukemia. Leuk. Lymphoma 2020, 61, 2527–2529. [Google Scholar] [CrossRef]
- Cortes, J.E.; Hughes, T.P.; Mauro, M.J.; Hochhaus, A.; Rea, D.; Goh, Y.T.; Janssen, J.; Steegmann, J.L.; Heinrich, M.C.; Talpaz, M.; et al. Asciminib, a First-in-Class STAMP Inhibitor, Provides Durable Molecular Response in Patients (pts) with Chronic Myeloid Leukemia (CML) Harboring the T315I Mutation: Primary Efficacy and Safety Results from a Phase 1 Trial. Blood 2020, 136, 47–50. [Google Scholar] [CrossRef]
- Garcia-Gutierrez, V.; Luna, A.; Alonso-Dominguez, J.M.; Estrada, N.; Boque, C.; Xicoy, B.; Giraldo, P.; Angona, A.; Alvarez-Larran, A.; Sanchez-Guijo, F.; et al. Safety and efficacy of asciminib treatment in chronic myeloid leukemia patients in real-life clinical practice. Blood Cancer J. 2021, 11, 16. [Google Scholar] [CrossRef]
- Soverini, S.; Abruzzese, E.; Bocchia, M.; Bonifacio, M.; Galimberti, S.; Gozzini, A.; Iurlo, A.; Luciano, L.; Pregno, P.; Rosti, G.; et al. Next-generation sequencing for BCR-ABL1 kinase domain mutation testing in patients with chronic myeloid leukemia: A position paper. J. Hematol. Oncol. 2019, 12, 131. [Google Scholar] [CrossRef]
- Zhan, J.Y.; Ma, J.; Zheng, Q.C. Molecular dynamics investigation on the Asciminib resistance mechanism of I502L and V468F mutations in BCR-ABL. J. Mol. Graph. Model. 2019, 89, 242–249. [Google Scholar] [CrossRef]
- Qiang, W.; Antelope, O.; Zabriskie, M.S.; Pomicter, A.D.; Vellore, N.A.; Szankasi, P.; Rea, D.; Cayuela, J.M.; Kelley, T.W.; Deininger, M.W.; et al. Mechanisms of resistance to the BCR-ABL1 allosteric inhibitor asciminib. Leukemia 2017, 31, 2844–2847. [Google Scholar] [CrossRef]
- Yaghmaie, M.; Yeung, C.C. Molecular Mechanisms of Resistance to Tyrosine Kinase Inhibitors. Curr. Hematol. Malig. Rep. 2019, 14, 395–404. [Google Scholar] [CrossRef]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Eadie, L.N.; Dang, P.; Saunders, V.A.; Yeung, D.T.; Osborn, M.P.; Grigg, A.P.; Hughes, T.P.; White, D.L. The clinical significance of ABCB1 overexpression in predicting outcome of CML patients undergoing first-line imatinib treatment. Leukemia 2017, 31, 75–82. [Google Scholar] [CrossRef]
- Mahon, F.X.; Belloc, F.; Lagarde, V.; Chollet, C.; Moreau-Gaudry, F.; Reiffers, J.; Goldman, J.M.; Melo, J.V. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003, 101, 2368–2373. [Google Scholar] [CrossRef]
- Eadie, L.N.; Saunders, V.A.; Branford, S.; White, D.L.; Hughes, T.P. The new allosteric inhibitor asciminib is susceptible to resistance mediated by ABCB1 and ABCG2 overexpression in vitro. Oncotarget 2018, 9, 13423. [Google Scholar] [CrossRef]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Venables, A.; Zrim, S.; Zannettino, A.; Lynch, K.; Manley, P.W.; Hughes, T. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: Higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood 2007, 110, 4064–4072. [Google Scholar] [CrossRef]
- Hiwase, D.K.; Saunders, V.; Hewett, D.; Frede, A.; Zrim, S.; Dang, P.; Eadie, L.; To, L.B.; Melo, J.; Kumar, S.; et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: Therapeutic implications. Clin. Cancer Res. 2008, 14, 3881–3888. [Google Scholar] [CrossRef]
- Lu, L.; Saunders, V.; Leclercq, T.; Hughes, T.; White, D. Ponatinib is not transported by ABCB1, ABCG2 or OCT-1 in CML cells. Leukemia 2015, 29, 1792. [Google Scholar] [CrossRef]
- Dohse, M.; Scharenberg, C.; Shukla, S.; Robey, R.W.; Volkmann, T.; Deeken, J.F.; Brendel, C.; Ambudkar, S.V.; Neubauer, A.; Bates, S.E. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab. Dispos. 2010, 38, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Hanfstein, B.; Erben, P.; Wolf, D.; Ernst, T.; Fabarius, A.; Saussele, S.; Purkayastha, D.; Woodman, R.C.; Hofmann, W.K.; et al. MDR1 expression predicts outcome of Ph+ chronic phase CML patients on second-line nilotinib therapy after imatinib failure. Leukemia 2014, 28, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Shanmuganathan, N.; Branford, S. The Hidden Pathogenesis of CML: Is BCR-ABL1 the First Event? Curr. Hematol. Malig. Rep. 2019, 14, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Eiring, A.M.; Khorashad, J.S.; Anderson, D.J.; Yu, F.; Redwine, H.M.; Mason, C.C.; Reynolds, K.R.; Clair, P.M.; Gantz, K.C.; Zhang, T.Y. β-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia 2015, 29, 2328. [Google Scholar] [CrossRef]
- Mojtahedi, H.; Yazdanpanah, N.; Rezaei, N. Chronic myeloid leukemia stem cells: Targeting therapeutic implications. Stem Cell Res. Ther. 2021, 12, 603. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Rana, A.; Datoguia, T.S.; Hamerschlak, N.; Brumatti, G. BCR-ABL1 Tyrosine kinase complex signaling transduction: Challenges to overcome resistance in chronic myeloid leukemia. Pharmaceutics 2022, 14, 215. [Google Scholar] [CrossRef]
- Ma, L.; Shan, Y.; Bai, R.; Xue, L.; Eide, C.A.; Ou, J.; Zhu, L.J.; Hutchinson, L.; Cerny, J.; Khoury, H.J.; et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci. Transl. Med. 2014, 6, 252ra121. [Google Scholar] [CrossRef]
- Suzuki, M.; Abe, A.; Imagama, S.; Nomura, Y.; Tanizaki, R.; Minami, Y.; Hayakawa, F.; Ito, Y.; Katsumi, A.; Yamamoto, K. BCR-ABL-independent and RAS/MAPK pathway-dependent form of imatinib resistance in Ph-positive acute lymphoblastic leukemia cell line with activation of EphB4. Eur. J. Haematol. 2010, 84, 229–238. [Google Scholar] [CrossRef]
- Kumar, A.; Bhattacharyya, J.; Jaganathan, B.G. Adhesion to stromal cells mediates imatinib resistance in chronic myeloid leukemia through ERK and BMP signaling pathways. Sci. Rep. 2017, 7, 9535. [Google Scholar] [CrossRef]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef]
- Brachet-Botineau, M.; Polomski, M.; Neubauer, H.A.; Juen, L.; Hedou, D.; Viaud-Massuard, M.C.; Prie, G.; Gouilleux, F. Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers 2020, 12, 240. [Google Scholar] [CrossRef]
- Eiring, A.; Kraft, I.; Page, B.; O’hare, T.; Gunning, P.; Deininger, M. STAT3 as a mediator of BCR-ABL1-independent resistance in chronic myeloid leukemia. Leuk. Suppl. 2014, 3, S5. [Google Scholar] [CrossRef]
- Kuepper, M.K.; Butow, M.; Herrmann, O.; Ziemons, J.; Chatain, N.; Maurer, A.; Kirschner, M.; Maie, T.; Costa, I.G.; Eschweiler, J.; et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia 2019, 33, 1964–1977. [Google Scholar] [CrossRef]
- Gullaksen, S.E.; Skavland, J.; Gavasso, S.; Tosevski, V.; Warzocha, K.; Dumrese, C.; Ferrant, A.; Gedde-Dahl, T.; Hellmann, A.; Janssen, J.; et al. Single cell immune profiling by mass cytometry of newly diagnosed chronic phase chronic myeloid leukemia treated with nilotinib. Haematologica 2017, 102, 1361–1367. [Google Scholar] [CrossRef][Green Version]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Holbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, V.; Gupta, S.K.; Kumari, G.; Verma, M. Combating TKI resistance in CML by inhibiting the PI3K/Akt/mTOR pathway in combination with TKIs: A review. Med. Oncol. 2021, 38, 10. [Google Scholar] [CrossRef]
- Burchert, A.; Wang, Y.; Cai, D.; Von Bubnoff, N.; Paschka, P.; Müller-Brüsselbach, S.; Ottmann, O.; Duyster, J.; Hochhaus, A.; Neubauer, A. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia 2005, 19, 1774. [Google Scholar] [CrossRef]
- Wagle, M.; Eiring, A.M.; Wongchenko, M.; Lu, S.; Guan, Y.; Wang, Y.; Lackner, M.; Amler, L.; Hampton, G.; Deininger, M.W.; et al. A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia 2016, 30, 1493–1501. [Google Scholar] [CrossRef]
- Mitchell, R.; Hopcroft, L.E.M.; Baquero, P.; Allan, E.K.; Hewit, K.; James, D.; Hamilton, G.; Mukhopadhyay, A.; O’Prey, J.; Hair, A.; et al. Targeting BCR-ABL-Independent TKI Resistance in Chronic Myeloid Leukemia by mTOR and Autophagy Inhibition. J. Natl. Cancer Inst. 2018, 110, 467–478. [Google Scholar] [CrossRef]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R. Targeting autophagy potentiates tyrosine kinase inhibitor–induced cell death in Philadelphia chromosome–positive cells, including primary CML stem cells. J. Clin. Investig. 2013, 123, 3634. [Google Scholar] [CrossRef][Green Version]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef] [PubMed]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, E.; Del Re, M.; Galimberti, S.; Restante, G.; Rofi, E.; Crucitta, S.; Barate, C.; Petrini, M.; Danesi, R.; Di Paolo, A. Concise Review: Chronic Myeloid Leukemia: Stem Cell Niche and Response to Pharmacologic Treatment. Stem Cells Transl. Med. 2018, 7, 305–314. [Google Scholar] [CrossRef]
- Leo, E.; Mancini, M.; Aluigi, M.; Luatti, S.; Castagnetti, F.; Testoni, N.; Soverini, S.; Santucci, M.A.; Martinelli, G. BCR-ABL1-associated reduction of beta catenin antagonist Chibby1 in chronic myeloid leukemia. PLoS ONE 2013, 8, e81425. [Google Scholar] [CrossRef]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of beta-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074. [Google Scholar] [CrossRef]
- Ciccone, M.; Calin, G.A.; Perrotti, D. From the Biology of PP2A to the PADs for Therapy of Hematologic Malignancies. Front. Oncol. 2015, 5, 21. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 2013, 123, 4144–4157. [Google Scholar] [CrossRef]
- Branford, S.; Kim, D.D.H.; Apperley, J.F.; Eide, C.A.; Mustjoki, S.; Ong, S.T.; Nteliopoulos, G.; Ernst, T.; Chuah, C.; Gambacorti-Passerini, C.; et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia 2019, 33, 1835–1850. [Google Scholar] [CrossRef]
- Kim, T.; Tyndel, M.S.; Kim, H.J.; Ahn, J.S.; Choi, S.H.; Park, H.J.; Kim, Y.K.; Kim, S.Y.; Lipton, J.H.; Zhang, Z.; et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood 2017, 129, 38–47. [Google Scholar] [CrossRef]
- Mitani, K.; Nagata, Y.; Sasaki, K.; Yoshida, K.; Chiba, K.; Tanaka, H.; Shiraishi, Y.; Miyano, S.; Makishima, H.; Nakamura, Y.; et al. Somatic mosaicism in chronic myeloid leukemia in remission. Blood 2016, 128, 2863–2866. [Google Scholar] [CrossRef][Green Version]
- Togasaki, E.; Takeda, J.; Yoshida, K.; Shiozawa, Y.; Takeuchi, M.; Oshima, M.; Saraya, A.; Iwama, A.; Yokote, K.; Sakaida, E.; et al. Frequent somatic mutations in epigenetic regulators in newly diagnosed chronic myeloid leukemia. Blood Cancer J. 2017, 7, e559. [Google Scholar] [CrossRef]
- Palandri, F.; Castagnetti, F.; Iacobucci, I.; Martinelli, G.; Amabile, M.; Gugliotta, G.; Poerio, A.; Testoni, N.; Breccia, M.; Bocchia, M.; et al. The response to imatinib and interferon-alpha is more rapid than the response to imatinib alone: A retrospective analysis of 495 Philadelphia-positive chronic myeloid leukemia patients in early chronic phase. Haematologica 2010, 95, 1415–1419. [Google Scholar] [CrossRef]
- Xie, H.; Peng, C.; Huang, J.; Li, B.E.; Kim, W.; Smith, E.C.; Fujiwara, Y.; Qi, J.; Cheloni, G.; Das, P.P.; et al. Chronic Myelogenous Leukemia- Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discov. 2016, 6, 1237–1247. [Google Scholar] [CrossRef]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.M.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016, 6, 1248. [Google Scholar] [CrossRef]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Anusha; Dalal, H.; Subramanian, S.; Snijesh, V.P.; Gowda, D.A.; Krishnamurthy, H.; Damodar, S.; Vyas, N. Exovesicular-Shh confers Imatinib resistance by upregulating Bcl2 expression in chronic myeloid leukemia with variant chromosomes. Cell Death Dis. 2021, 12, 259. [Google Scholar] [CrossRef] [PubMed]
- Parry, N.; Wheadon, H.; Copland, M. The application of BH3 mimetics in myeloid leukemias. Cell Death Dis. 2021, 12, 222. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Yin, C.; Zong, S. hnRNPK/Beclin1 signaling regulates autophagy to promote imatinib resistance in Philadelphia chromosome-positive acute lymphoblastic leukemia cells. Exp. Hematol. 2022, 108, 46–54. [Google Scholar] [CrossRef]
- Medina, A.; Puig, N.; Flores-Montero, J.; Jimenez, C.; Sarasquete, M.E.; Garcia-Alvarez, M.; Prieto-Conde, I.; Chillon, C.; Alcoceba, M.; Gutierrez, N.C.; et al. Comparison of next-generation sequencing (NGS) and next-generation flow (NGF) for minimal residual disease (MRD) assessment in multiple myeloma. Blood Cancer J. 2020, 10, 108. [Google Scholar] [CrossRef]
- Chan, L.N.; Murakami, M.A.; Robinson, M.E.; Caeser, R.; Sadras, T.; Lee, J.; Cosgun, K.N.; Kume, K.; Khairnar, V.; Xiao, G.; et al. Signalling input from divergent pathways subverts B cell transformation. Nature 2020, 583, 845–851. [Google Scholar] [CrossRef]
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jorgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood 2014, 124, 1492–1501. [Google Scholar] [CrossRef]
- Sweet, K.; Hazlehurst, L.; Sahakian, E.; Powers, J.; Nodzon, L.; Kayali, F.; Hyland, K.; Nelson, A.; Pinilla-Ibarz, J. A phase I clinical trial of ruxolitinib in combination with nilotinib in chronic myeloid leukemia patients with molecular evidence of disease. Leuk. Res. 2018, 74, 89–96. [Google Scholar] [CrossRef]
- Yagi, K.; Shimada, A.; Sendo, T. Pharmacological inhibition of JAK3 enhances the antitumor activity of imatinib in human chronic myeloid leukemia. Eur. J. Pharmacol. 2018, 825, 28–33. [Google Scholar] [CrossRef]
- Ozel, B.; Kipcak, S.; Biray Avci, C.; Gunduz, C.; Saydam, G.; Aktan, C.; Selvi Gunel, N. Combination of dasatinib and okadaic acid induces apoptosis and cell cycle arrest by targeting protein phosphatase PP2A in chronic myeloid leukemia cells. Med. Oncol. 2022, 39, 46. [Google Scholar] [CrossRef]
- Perrotti, D.; Agarwal, A.; Lucas, C.M.; Narla, G.; Neviani, P.; Odero, M.D.; Ruvolo, P.P.; Verrills, N.M. Comment on “PP2A inhibition sensitizes cancer stem cells to ABL tyrosine kinase inhibitors in BCR-ABL human leukemia”. Sci. Transl. Med. 2019, 11, eaau0416. [Google Scholar] [CrossRef]
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 51–63. [Google Scholar] [CrossRef]
- Horne, G.A.; Stobo, J.; Kelly, C.; Mukhopadhyay, A.; Latif, A.L.; Dixon-Hughes, J.; McMahon, L.; Cony-Makhoul, P.; Byrne, J.; Smith, G.; et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020, 34, 1775–1786. [Google Scholar] [CrossRef]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar]
- Mu, H.; Zhu, X.; Jia, H.; Zhou, L.; Liu, H. Combination Therapies in Chronic Myeloid Leukemia for Potential Treatment-Free Remission: Focus on Leukemia Stem Cells and Immune Modulation. Front. Oncol. 2021, 11, 643382. [Google Scholar] [CrossRef]
- Simonsson, B.; Gedde-Dahl, T.; Markevarn, B.; Remes, K.; Stentoft, J.; Almqvist, A.; Bjoreman, M.; Flogegard, M.; Koskenvesa, P.; Lindblom, A.; et al. Combination of pegylated IFN-alpha2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 2011, 118, 3228–3235. [Google Scholar] [CrossRef]
- Gisslinger, H.; Zagrijtschuk, O.; Buxhofer-Ausch, V.; Thaler, J.; Schloegl, E.; Gastl, G.A.; Wolf, D.; Kralovics, R.; Gisslinger, B.; Strecker, K.; et al. Ropeginterferon alfa-2b, a novel IFN.Nalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 2015, 126, 1762–1769. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Zhou, H.; Mak, D.H.; Schober, W.; Leverson, J.D.; Zhang, B.; Bhatia, R.; Huang, X. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 2016, 8, ra117–ra355. [Google Scholar] [CrossRef]
- Maiti, A.; Franquiz, M.J.; Ravandi, F.; Cortes, J.E.; Jabbour, E.J.; Sasaki, K.; Marx, K.; Daver, N.G.; Kadia, T.M.; Konopleva, M.Y.; et al. Venetoclax and BCR-ABL Tyrosine Kinase Inhibitor Combinations: Outcome in Patients with Philadelphia Chromosome-Positive Advanced Myeloid Leukemias. Acta Haematol. 2020, 143, 567–573. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Han, E.S.; Wen, W.; Dellinger, T.H.; Wu, J.; Lu, S.A.; Jove, R.; Yim, J.H. Ruxolitinib synergistically enhances the anti-tumor activity of paclitaxel in human ovarian cancer. Oncotarget 2018, 9, 24304–24319. [Google Scholar] [CrossRef]
- Mogul, A.; Corsi, K.; McAuliffe, L. Baricitinib: The Second FDA-Approved JAK Inhibitor for the Treatment of Rheumatoid Arthritis. Ann. Pharmacother. 2019, 53, 947–953. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a therapeutic target for cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapamycin for longevity: Opinion article. Aging 2019, 11, 8048–8067. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef]
- Tinsley, S.L.; Allen-Petersen, B.L. PP2A and cancer epigenetics: A therapeutic opportunity waiting to happen. NAR Cancer 2022, 4, zcac002. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Perl, A.; Leonard, D.; Sangodkar, J.; Narla, G. Therapeutic targeting of PP2A. Int. J. Biochem. Cell Biol. 2018, 96, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Gong, Y.; Zhang, T.; Huang, J.; Tan, Z.; Xue, L. Finding an easy way to harmonize: A review of advances in clinical research and combination strategies of EZH2 inhibitors. Clin. Epigenet. 2021, 13, 62. [Google Scholar] [CrossRef]
- Jamieson, C.; Martinelli, G.; Papayannidis, C.; Cortes, J.E. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov. 2020, 1, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Lasica, M.; Anderson, M.A. Review of Venetoclax.x in CLL, AML and Multiple Myeloma. J. Pers. Med. 2021, 11, 463. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poudel, G.; Tolland, M.G.; Hughes, T.P.; Pagani, I.S. Mechanisms of Resistance and Implications for Treatment Strategies in Chronic Myeloid Leukaemia. Cancers 2022, 14, 3300. https://doi.org/10.3390/cancers14143300
Poudel G, Tolland MG, Hughes TP, Pagani IS. Mechanisms of Resistance and Implications for Treatment Strategies in Chronic Myeloid Leukaemia. Cancers. 2022; 14(14):3300. https://doi.org/10.3390/cancers14143300
Chicago/Turabian StylePoudel, Govinda, Molly G. Tolland, Timothy P. Hughes, and Ilaria S. Pagani. 2022. "Mechanisms of Resistance and Implications for Treatment Strategies in Chronic Myeloid Leukaemia" Cancers 14, no. 14: 3300. https://doi.org/10.3390/cancers14143300
APA StylePoudel, G., Tolland, M. G., Hughes, T. P., & Pagani, I. S. (2022). Mechanisms of Resistance and Implications for Treatment Strategies in Chronic Myeloid Leukaemia. Cancers, 14(14), 3300. https://doi.org/10.3390/cancers14143300