
Insights into the Possible Molecular Mechanisms of Resistance to PARP Inhibitors


Abstract
Simple Summary
Abstract
1. Introduction
2. Primary Resistance
2.1. PTEN Deficiency and PI3K/AKT Pathway Activation
2.2. ATM Roles and Loss of NHEJ Pathway
2.3. ALC1 Overexpression
3. Secondary Resistance
3.1. Upregulation of ABC Transporters
3.2. Decreased PARP1 Trapping
3.3. Restoration of HR
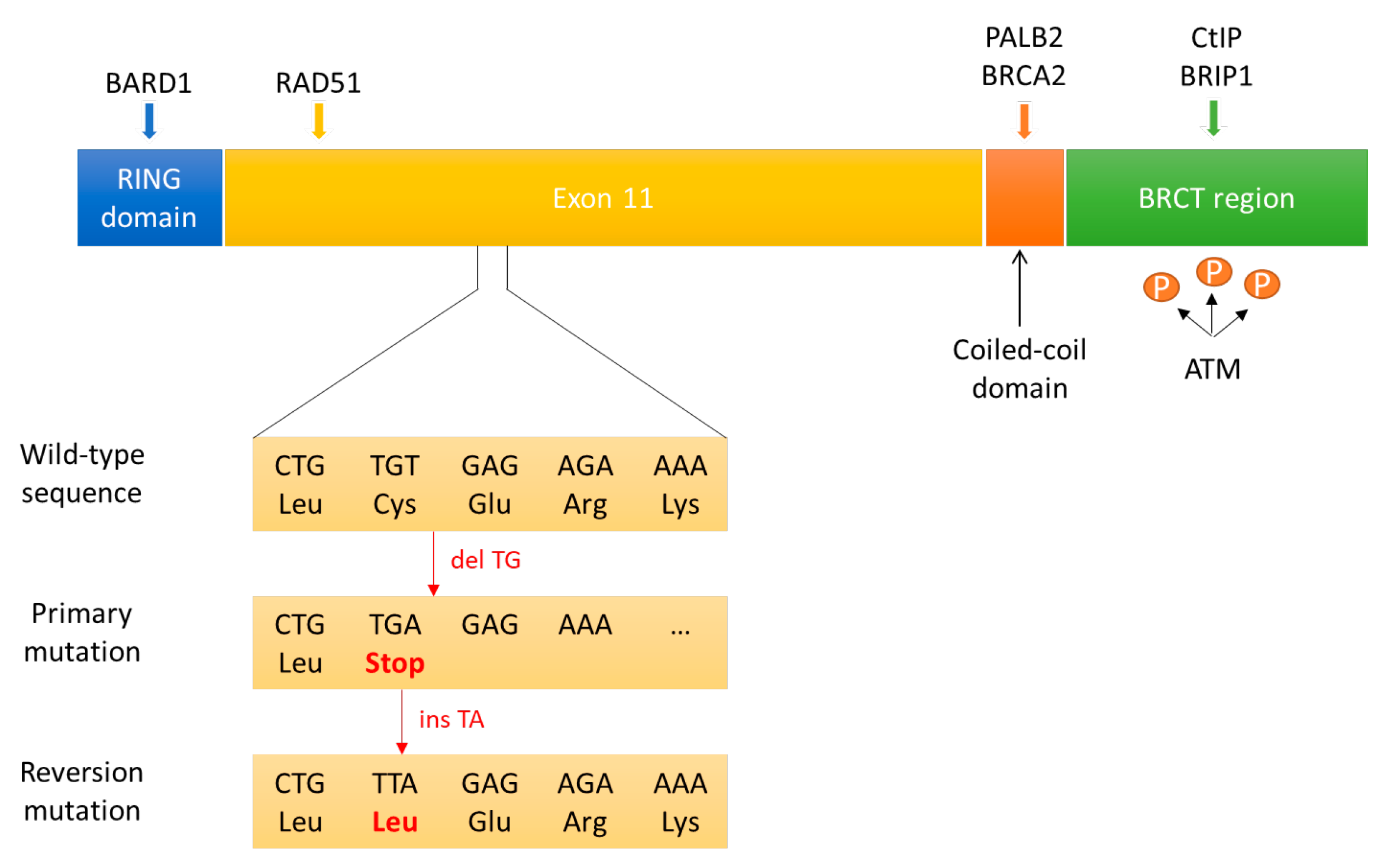
3.3.1. Reversion Mutations in BRCA1/2
3.3.2. Hypomorphic BRCA1 Allele
3.3.3. Epigenetic Reversion of BRCA1 Promoter Hypermethylation
3.3.4. Loss of End Resection Regulation
3.3.5. Overexpression of RAD51 and RAD51 Paralogs
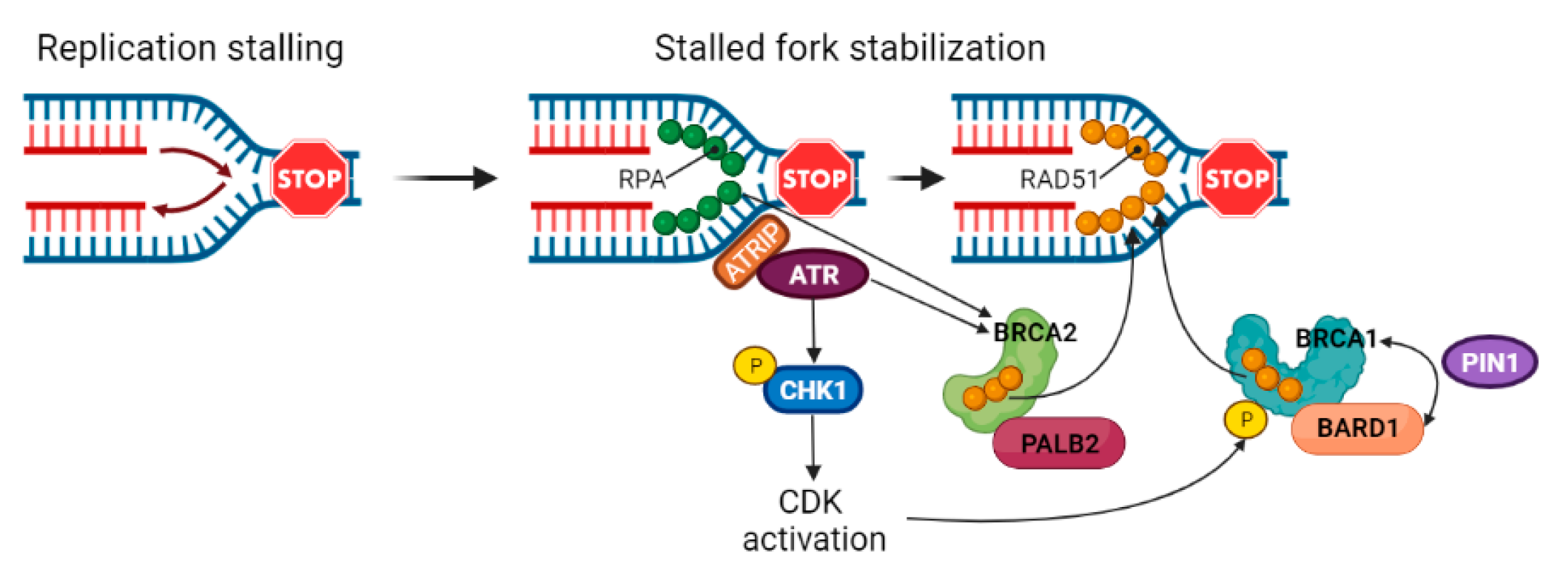
3.4. Stabilization of Stalled Forks
4. Future Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mortusewicz, O.; Amé, J.C.; Schreiber, V.; Leonhardt, H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007, 35, 7665–7675. [Google Scholar] [CrossRef] [PubMed]
- Dawicki-McKenna, J.M.; Langelier, M.F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain. Mol. Cell 2015, 60, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Wu, W.F.; Langelier, M.F.; Yang, J.C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef]
- El Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef]
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117. [Google Scholar] [CrossRef]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef]
- Haince, J.F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef]
- Zahradka, P.; Ebisuzaki, K. A shuttle mechanism for DNA-protein interactions. The regulation of poly(ADP-ribose) polymerase. Eur. J. Biochem. 1982, 127, 579–585. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with laparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Rehman, F.L.; Bajrami, I.; Brough, R.; Wallberg, F.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Chen, L.; Campbell, J.; et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of laparib toxicity. PLoS ONE 2013, 8, e61520. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Bothra, S.J. PARP inhibitors in hereditary breast and ovarian cancer and other cancers: A review. Adv. Genet. 2021, 108, 35–80. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, N.; Ryder, S.; Forbes, C.; Ross, J.; Quek, R.G. A systematic review of the international prevalence of BRCA mutation in breast cancer. Clin. Epidemiol. 2019, 11, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 mutations among 1342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011, 121, 353–357. [Google Scholar] [CrossRef]
- Rosen, M.N.; Goodwin, R.A.; Vickers, M.M. BRCA mutated pancreatic cancer: A change is coming. World J. Gastroenterol. 2021, 27, 1943–1958. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Hwang, J.S.; Wang, A.; Bae, I. Inhibition of constitutively activated phosphoinositide 3-kinase/AKT pathway enhances anti- tumor activity of chemotherapeutic agents in breast cancer susceptibility gene 1-defective breast cancer cells. Mol. Carcinog. 2013, 52, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN Deficiency in Endometrioid Endometrial Adenocarcinomas Predicts Sensitivity to PARP Inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef] [PubMed]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef]
- McCormick, A.; Donoghue, P.; Dixon, M.; O’Sullivan, R.; O’Donnell, R.L.; Murray, J.; Kaufmann, A.; Curtin, N.J.; Edmondson, R.J. Ovarian Cancers Harbor Defects in Nonhomologous End Joining Resulting in Resistance to Rucaparib. Clin. Cancer Res. 2017, 23, 2050–2060. [Google Scholar] [CrossRef]
- Juhász, S.; Smith, R.; Schauer, T.; Spekhardt, D.; Mamar, H.; Zentout, S.; Chapuis, C.; Huet, S.; Timinszky, G. The chromatin remodeler ALC1 underlies resistance to PARP inhibitor treatment. Sci. Adv. 2020, 6, eabb8626. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Sol, W.; Kersbergen, A.; Schlicker, A.; Guyader, C.; Xu, G.; Wessels, L.; Borst, P.; Jonkers, J.; Rottenberg, S. BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res. 2015, 75, 732–741. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Drost, R.; Dhillon, K.K.; van der Gulden, H.; van der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bernhardy, A.J.; Nacson, J.; Krais, J.J.; Tan, Y.F.; Nicolas, E.; Radke, M.R.; Handorf, E.; Llop-Guevara, A.; Balmaña, J.; et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun. 2019, 10, 5661. [Google Scholar] [CrossRef] [PubMed]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef] [PubMed]
- Ter Brugge, P.; Kristel, P.; van der Burg, E.; Boon, U.; de Maaker, M.; Lips, E.; Mulder, L.; de Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw148, Erratum in J. Natl. Cancer Inst. 2020, 112, 1075. [Google Scholar] [CrossRef]
- Veeck, J.; Ropero, S.; Setien, F.; Gonzalez-Suarez, E.; Osorio, A.; Benitez, J.; Herman, J.G.; Esteller, M. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J. Clin. Oncol. 2010, 28, e563–e564, author reply e565-6. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Zhang, J.R.; Li, S.D.; He, Y.Y.; Yang, Y.X.; Liu, X.L.; Wan, X.P. Aberrant methylation of breast and ovarian cancer susceptibility gene 1 in chemosensitive human ovarian cancer cells does not involve the phosphatidylinositol 30-kinase-Akt pathway. Cancer Sci. 2010, 101, 1618–1623. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11, 819. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237.e6. [Google Scholar] [CrossRef]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of replication fork stability in BRCA1-and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef]
- Dungrawala, H.; Bhat, K.P.; Le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol. Cell 2017, 67, 374–386.e5. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A. The PKB/AKT pathway in cancer. Curr. Pharm. Des. 2010, 16, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. 2011, 11, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.C.; Lyons, T.R.; Young, C.D.; Hansen, K.C.; Anderson, S.M.; Holt, J.T. AKT regulates BRCA1 stability in response to hormone signaling. Mol. Cell. Endocrinol. 2010, 319, 129–142. [Google Scholar] [CrossRef][Green Version]
- Kang, H.J.; Yi, Y.W.; Kim, H.J.; Hong, Y.B.; Seong, Y.S.; Bae, I. BRCA1 negatively regulates IGF-1 expression through an estrogen-responsive element-like site. Cell Death Dis. 2012, 3, e336. [Google Scholar] [CrossRef]
- Xiang, T.; Ohashi, A.; Huang, Y.; Pandita, T.K.; Ludwig, T.; Powell, S.N.; Yang, Q. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008, 68, 10040–10044. [Google Scholar] [CrossRef]
- Saal, L.H.; Gruvberger-Saal, S.K.; Persson, C.; Lövgren, K.; Jumppanen, M.; Staaf, J.; Jönsson, G.; Pires, M.M.; Maurer, M.; Holm, K.; et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat. Genet. 2008, 40, 102–107. [Google Scholar] [CrossRef]
- Foulkes, W.D. BRCA1—Sowing the seeds crooked in the furrow. Nat. Genet. 2008, 40, 8–9. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef]
- McEllin, B.; Camacho, C.V.; Mukherjee, B.; Hahm, B.; Tomimatsu, N.; Bachoo, R.M.; Burma, S. PTEN loss compromises homologous recombination repair in astrocytes: Implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010, 70, 5457–5464. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Minami, A.; Nakanishi, A.; Ogura, Y.; Kitagishi, Y.; Matsuda, S. Connection between Tumor Suppressor BRCA1 and PTEN in Damaged DNA Repair. Front. Oncol. 2014, 4, 318. [Google Scholar] [CrossRef] [PubMed]
- Chock, K.L.; Allison, J.M.; Shimizu, Y.; El Shamy, W.M. BRCA1-IRIS overexpression promotes cisplatin resistance in ovarian cancer cells. Cancer Res. 2010, 70, 8782–8791. [Google Scholar] [CrossRef][Green Version]
- Burstein, H.J. Novel agents and future directions for refractory breast cancer. Semin. Oncol. 2011, 38, S17–S24. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; im, J.S.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell. Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4. [Google Scholar] [CrossRef]
- Buis, J.; Stoneham, T.; Spehalski, E.; Ferguson, D.O. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat. Struct. Mol. Biol. 2012, 19, 246–252. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Ducy, M.; Sesma-Sanz, L.; Guitton-Sert, L.; Lashgari, A.; Gao, Y.; Brahiti, N.; Rodrigue, A.; Margaillan, G.; Caron, M.C.; Côté, J.; et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 2019, 44, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural basis of homologous recombination. Cell. Mol. Life Sci. 2020, 77, 3–18. [Google Scholar] [CrossRef]
- Cortesi, L.; Piombino, C.; Toss, A. Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? J. Pers. Med. 2021, 11, 245. [Google Scholar] [CrossRef]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef]
- Jowsey, P.; Morrice, N.A.; Hastie, C.J.; McLauchlan, H.; Toth, R.; Rouse, J. Characterisation of the sites of DNA damage-induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair 2007, 6, 1536–1544. [Google Scholar] [CrossRef]
- Zhang, F.; Gong, Z. Regulation of DNA double-strand break repair pathway choice: A new focus on 53BP1. J. Zhejiang Univ. Sci. B 2021, 22, 38–46. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Daley, J.M.; Sung, P. 53BP1, BRCA1 and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef]
- Feng, L.; Li, N.; Li, Y.; Wang, J.; Gao, M.; Wang, W.; Chen, J. Cell cycle-dependent inhibition of 53BP1 signaling by BRCA1. Cell Discov. 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chanut, P.; Britton, S.; Coates, J.; Jackson, S.P.; Calsou, P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016, 7, 12889. [Google Scholar] [CrossRef] [PubMed]
- Ahel, D.; Hořejší, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef]
- Ma, N.F.; Hu, L.; Fung, J.M.; Xie, D.; Zheng, B.J.; Chen, L.; Tang, D.J.; Fu, L.; Wu, Z.; Chen, M.; et al. Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 2008, 47, 503–510. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Gupta., P.; Ashby, C.R., Jr.; Chen, Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef]
- Shen, D.W.; Fojo, A.; Chin, J.E.; Roninson, I.B.; Richert, N.; Pastan, I.; Gottesman, M.M. Human multidrug-resistant cell lines: Increased mdr1 expression can precede gene amplification. Science 1986, 232, 643–645. [Google Scholar] [CrossRef]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef]
- Parekh, H.; Wiesen, K.; Simpkins, H. Acquisition of taxol resistance via P-glycoprotein- and non-P-glycoprotein-mediated mechanisms in human ovarian carcinoma cells. Biochem. Pharmacol. 1997, 53, 461–470. [Google Scholar] [CrossRef]
- Wang, Y.C.; Juric, D.; Francisco, B.; Yu, R.X.; Duran, G.E.; Chen, G.K.; Chen, X.; Sikic, B.I. Regional activation of chromosomal arm 7q with and without gene amplification in taxane-selected human ovarian cancer cell lines. Genes Chromosomes Cancer 2006, 45, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Wils, P.; Phung-Ba, V.; Warnery, A.; Lechardeur, D.; Raeissi, S.; Hidalgo, I.J.; Scherman, D. Polarized transport of docetaxel and vinblastine mediated by P-glycoprotein in human intestinal epithelial cell monolayers. Biochem. Pharmacol. 1994, 48, 1528–1530. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Pascal, J.M.; Ellenberger, T. The rise and fall of poly(ADP-ribose): An enzymatic perspective. DNA Repair 2015, 32, 10–16. [Google Scholar] [CrossRef]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6. [Google Scholar] [CrossRef]
- Williams, R.S.; Glover, J.N. Structural consequences of a cancer-causing BRCA1-BRCT missense mutation. J. Biol. Chem. 2003, 278, 2630–2635. [Google Scholar] [CrossRef]
- Williams, R.S.; Chasman, D.I.; Hau, D.D.; Hui, B.; Lau, A.Y.; Glover, J.N. Detection of protein folding defects caused by BRCA1-BRCT truncation and missense mutations. J. Biol. Chem. 2003, 278, 53007–53016. [Google Scholar] [CrossRef]
- Lee, M.S.; Green, R.; Marsillac, S.M.; Coquelle, N.; Williams, R.S.; Yeung, T.; Foo, D.; Hau, D.D.; Hui, B.; Monteiro, A.N.; et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010, 70, 4880–4890. [Google Scholar] [CrossRef]
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Chen, J.; Plug, A. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997, 88, 265–275. [Google Scholar] [CrossRef]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef]
- Mattarocci, S.; Hafner, L.; Lezaja, A.; Shyian, M.; Shore, D. Rif1: A Conserved Regulator of DNA Replication and Repair Hijacked by Telomeres in Yeasts. Front. Genet. 2016, 7, 45. [Google Scholar] [CrossRef]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef]
- Lo, K.W.; Kan, H.M.; Chan, L.N.; Xu, W.G.; Wang, K.P.; Wu, Z.; Sheng, M.; Zhang, M. The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J. Biol. Chem. 2005, 280, 8172–8179. [Google Scholar] [CrossRef]
- Becker, J.R.; Cuella-Martin, R.; Barazas, M.; Liu, R.; Oliveira, C.; Oliver, A.W.; Bilham, K.; Holt, A.B.; Blackford, A.N.; Heierhorst, J.; et al. The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat. Commun. 2018, 9, 5406. [Google Scholar] [CrossRef]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Preventing replication fork collapse to maintain genome integrity. DNA Repair 2015, 32, 149–157. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Lin, C.C.; Dai, H.; Mo, W.; Li, Y.; Stephan, C.; Davies, P.; Lu, Z.; Mills, G.B.; Lee, J.S.; et al. Defective replication stress response is inherently linked to the cancer stem cell phenotype. Cell Rep. 2018, 23, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Lemacon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef]
- Binz, S.K.; Sheehan, A.M.; Wold, M.S. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair 2004, 3, 1015–1024. [Google Scholar] [CrossRef]
- Murphy, A.K.; Fitzgerald, M.; Ro, T.; Kim, J.H.; Rabinowitsch, A.I.; Chowdhury, D.; Schildkraut, C.L.; Borowiec, J.A. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J. Cell Biol. 2014, 206, 493–507. [Google Scholar] [CrossRef]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kowalczykowski, S.C. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51- mediated displacement of RPA and presynaptic complex formation. J. Biol. Chem. 2002, 277, 31663–31672. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.; et al. Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Techer, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef]
- Kim, T.M.; Son, M.Y.; Dodds, S.; Hu, L.; Luo, G.; Hasty, P. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 2015, 43, 893–903. [Google Scholar] [CrossRef]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 maintains fork stability in BRCA1/2-deficient tumors and promotes alternative end-joining DNA repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef]
- Ying, S.; Chen, Z.; Medhurst, A.L.; Neal, J.A.; Bao, Z.; Mortusewicz, O.; McGouran, J.; Song, X.; Shen, H.; Hamdy, F.C.; et al. DNA-PKcs and PARP1 Bind to Unresected Stalled DNA Replication Forks Where They Recruit XRCC1 to Mediate Repair. Cancer Res. 2016, 76, 1078–1088. [Google Scholar] [CrossRef]
- Ding, X.; Chaudhuri, A.R.; Callen, E.; Pang, Y.; Biswas, K.; Klarmann, K.D.; Martin, B.K.; Burkett, S.; Cleveland, L.; Stauffer, S.; et al. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 2016, 7, 12425. [Google Scholar] [CrossRef]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmañà, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.L.; Wijnhoven, P.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Sun, A.; Cao, W.; Eliason, A.; Mendez, K.M.; Getzler, A.J.; Tsuda, S.; Diao, H.; Mukori, C.; Bruno, N.E.; et al. Physiological expression and function of the MDR1 transporter in cytotoxic T lymphocytes. J. Exp. Med. 2020, 217, e20191388. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resistance Mechanism | Evidence | References |
|---|---|---|
| Primary resistance | ||
| PI3K/AKT pathway activation | Cell lines | Yi et al. [31] |
| Wild-type PTEN | Cell lines | Dedes et al. [32] |
| Loss of NHEJ | Cell lines | Balmus et al. [33], Patel et al. [34], Mc Cormick et al. [35] |
| ALC1 overexpression | Cell lines | Juhász et al. [36] |
| Secondary resistance | ||
| Upregulation of ABC transporters | Mouse models, cell lines | Jaspers et al. [37], Vaidyanathan et al. [38] |
| Decreased PARP1 trapping | Mouse models, cell lines | Pettitt et al. [39], Gogola et al. [40] |
| Restoration of HR | ||
| -BRCA1/2 reversion mutations | Tumour DNA and ctDNA from cancer patients | Tobalina et al. [41] |
| -Hypomorphic BRCA1 allele | Cell lines, mouse models, PDXs | Drost et al. [42], Wang et al. [43], Cruz et al. [44], Wang et al. [45], Castroviejo-Bermejo et al. [46] |
| -Loss of BRCA1 promoter methylation | Cell lines, PDXs | Ter Brugge et al. [47], Veeck et al. [48], Wang et al. [49] |
| -Loss of end resection regulation (53BP1, RIF1, REV7, Sheldin complex or DYNLL1 depletion) | Cell lines | Belotserkovskaya et al. [50], Xu et al. [51], Noordermeer et al. [52], Gupta et al. [53], He et al. [54] |
| -RAD51 overexpression | Ovarian cancer samples, cell lines | Kondrashova et al. [55], Liu et al. [56], Marzio et al. [57] |
| Stabilization of stalled fork (FANCD2 overexpression, RADX depletion, SMARCAL1 inactivation,) | Cell lines | Michl et al. [58], Chaudhuri et al. [59], Taglialatela et al. [60], Dungrawala et al. [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piombino, C.; Cortesi, L. Insights into the Possible Molecular Mechanisms of Resistance to PARP Inhibitors. Cancers 2022, 14, 2804. https://doi.org/10.3390/cancers14112804
Piombino C, Cortesi L. Insights into the Possible Molecular Mechanisms of Resistance to PARP Inhibitors. Cancers. 2022; 14(11):2804. https://doi.org/10.3390/cancers14112804
Chicago/Turabian StylePiombino, Claudia, and Laura Cortesi. 2022. "Insights into the Possible Molecular Mechanisms of Resistance to PARP Inhibitors" Cancers 14, no. 11: 2804. https://doi.org/10.3390/cancers14112804
APA StylePiombino, C., & Cortesi, L. (2022). Insights into the Possible Molecular Mechanisms of Resistance to PARP Inhibitors. Cancers, 14(11), 2804. https://doi.org/10.3390/cancers14112804