
Antigen-Specific T Cell Immunotherapy Targeting Claudin18.2 in Gastric Cancer

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients, Specimens and Ethical Statement
2.2. HLA Typing
2.3. Epitope Prediction and Peptide Synthesis
2.4. T Cell Response Analysis
2.5. Cytokine Cytometric Bead Array Analysis
2.6. Cytotoxicity Assay
2.7. Generation of Dendritic Cells (DCs) and Peptide Reactive T Cells
2.8. Claudin18.2 Expression
2.9. Statistics SPSS Was Used for All Statistical Analyses
3. Results
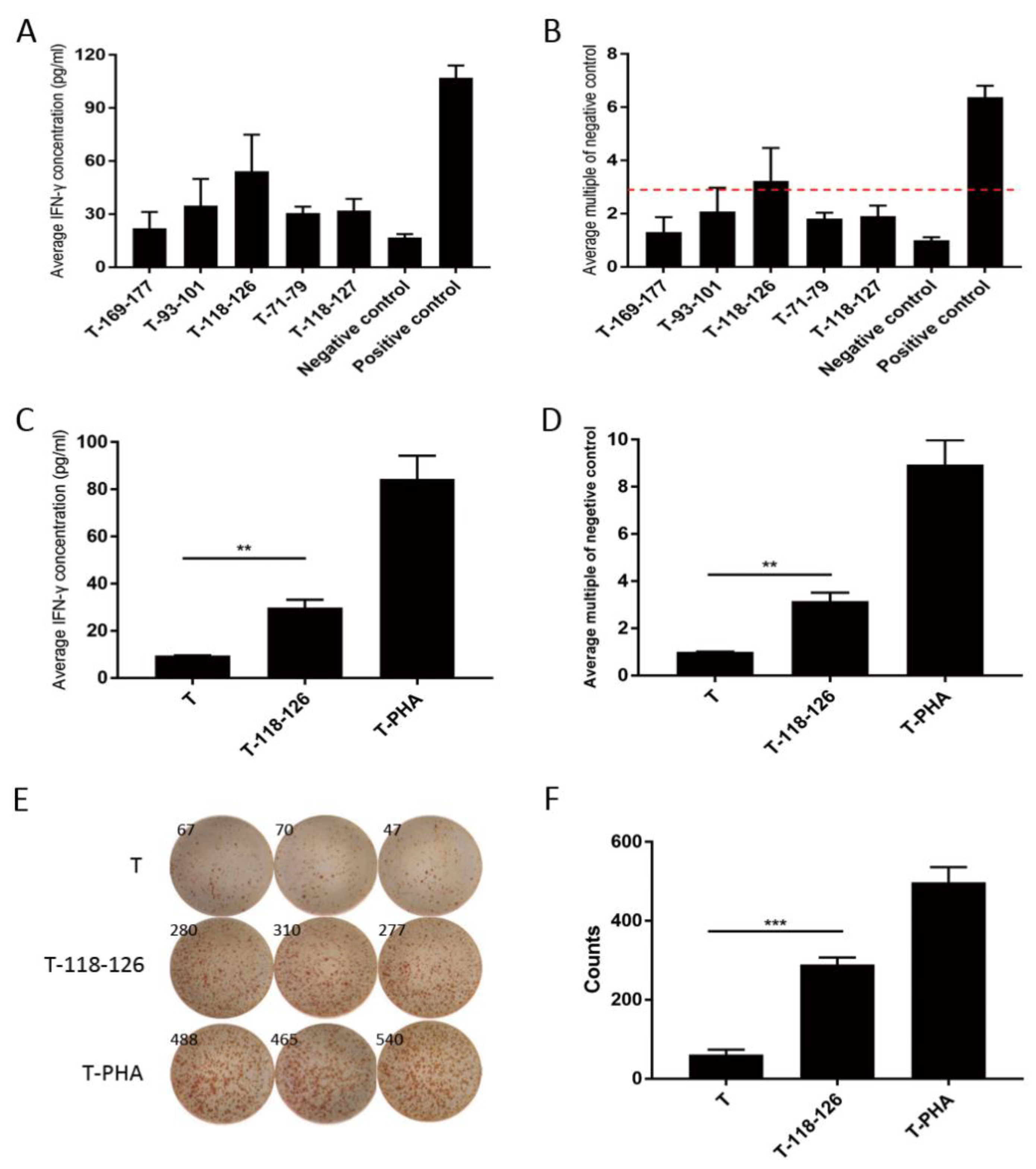
3.1. Claudin18.2 Peptides Specifically Induced Immune Activation in GC Patients
3.2. Claudin18.2 Peptide Stimulated T Cells Showed Promising Anti-Tumor Ability In Vitro
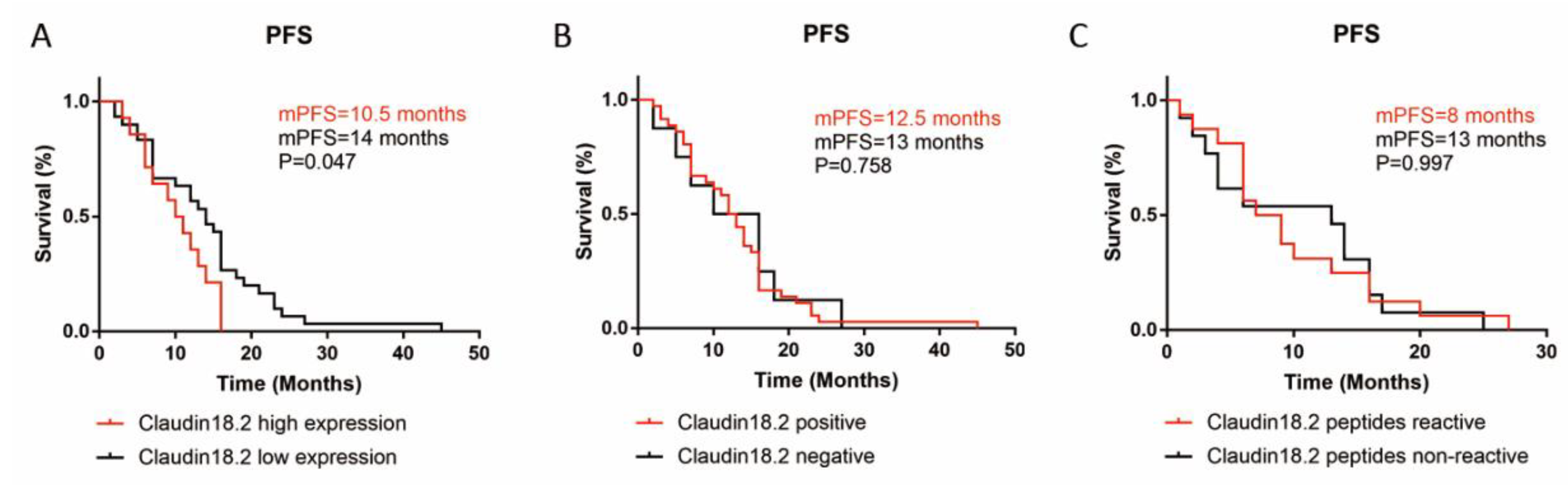
3.3. Claudin18.2 Highly Expressed in GC
3.4. Claudin18.2 Peptide Reactivity Was Higher in Claudin18.2 High Expressing GC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Jim, M.A.; Pinheiro, P.S.; Carreira, H.; Espey, D.K.; Wiggins, C.L.; Weir, H.K. Stomach cancer survival in the United States by race and stage (2001–2009): Findings from the CONCORD-2 study. Cancer 2017, 123, 4994–5013. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-H.; Shen, L.; Li, J.; Zhou, Z.-W.; Liang, H.; Zhang, X.-T.; Tang, L.; Xin, Y.; Jin, J.; Zhang, Y.-J.; et al. The Chinese Society of Clinical Oncology (CSCO): Clinical guidelines for the diagnosis and treatment of gastric cancer. Cancer Commun. 2019, 39, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, D.; Okines, A.F.; Ashley, S. Capecitabine and Oxaliplatin for Advanced Esophagogastric Cancer. N. Engl. J. Med. 2010, 362, 858–859. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.-C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.-Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, J.; Xiong, J.; Wu, C.; Bai, Y.; Liu, W.; Tong, J.; Liu, Y.; Xu, R.; et al. Randomized, Double-Blind, Placebo-Controlled Phase III Trial of Apatinib in Patients with Chemotherapy-Refractory Advanced or Metastatic Adenocarcinoma of the Stomach or Gastroesophageal Junction. J. Clin. Oncol. 2016, 34, 1448–1454. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Shitara, K.; Hara, H.; Yoshikawa, T.; Fujitani, K.; Nishina, T.; Hosokawa, A.; Asakawa, T.; Kawakami, S.; Muro, K. Pertuzumab plus trastuzumab and chemotherapy for Japanese patients with HER2-positive metastatic gastric or gastroesophageal junction cancer: A subgroup analysis of the JACOB trial. Int. J. Clin. Oncol. 2020, 25, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Ricci, A.; Rizzo, A.; Llimpe, F.R.; Di Fabio, F.; De Biase, D.; Rihawi, K. Novel HER2-Directed Treatments in Advanced Gastric Carcinoma: AnotHER Paradigm Shift? Cancers 2021, 13, 1664. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-K.; Boku, N.; Satoh, T.; Ryu, M.-H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.-S.; Muro, K.; Kang, W.K.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.-P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Shoji, H.; Nagashima, K.; Yamamoto, S.; Ishikawa, M.; Imazeki, H.; Aoki, M.; Miyamoto, T.; Hirano, H.; Honma, Y.; et al. Correlation between immune-related adverse events and prognosis in patients with gastric cancer treated with nivolumab. BMC Cancer 2019, 19, 974. [Google Scholar] [CrossRef] [PubMed]
- Rihawi, K.; Ricci, A.D.; Rizzo, A.; Brocchi, S.; Marasco, G.; Pastore, L.V.; Llimpe, F.L.R.; Golfieri, R.; Renzulli, M. Tumor-Associated Macrophages and Inflammatory Microenvironment in Gastric Cancer: Novel Translational Implications. Int. J. Mol. Sci. 2021, 22, 3805. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Koslowski, M.; Dhaene, K.; Usener, D.; Brandenburg, G.; Seitz, G.; Huber, C.; Türeci, Ö. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin. Cancer Res. 2008, 14, 7624–7634. [Google Scholar] [CrossRef] [Green Version]
- Rohde, C.; Yamaguchi, R.; Mukhina, S.; Sahin, U.; Itoh, K.; Türeci, Ö. Comparison of Claudin 18.2 expression in primary tumors and lymph node metastases in Japanese patients with gastric adenocarcinoma. Jpn. J. Clin. Oncol. 2019, 49, 870–876. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Toom, S.; Huang, Y. Anti-claudin 18.2 antibody as new targeted therapy for advanced gastric cancer. J. Hematol. Oncol. 2017, 10, 105. [Google Scholar] [CrossRef]
- Türeci, Ö.; Sahin, U.; Schulze-Bergkamen, H.; Zvirbule, Z.; Lordick, F.; Koeberle, D.; Thuss-Patience, P.; Ettrich, T.; Arnold, D.; Bassermann, F.; et al. A multicentre, phase IIa study of zolbetuximab as a single agent in patients with recurrent or refractory advanced adenocarcinoma of the stomach or lower oesophagus: The MONO study. Ann. Oncol. 2019, 30, 1487–1495. [Google Scholar] [CrossRef] [Green Version]
- Türeci, Ö.; Mitnacht-Kraus, R.; Wöll, S.; Yamada, T.; Sahin, U. Characterization of zolbetuximab in pancreatic cancer models. Oncoimmunology 2019, 8, e1523096. [Google Scholar] [CrossRef] [Green Version]
- Schuler, M.; Al-Batran, S.-E.; Zvirbule, Z.; Manikhas, G.; Lordick, F.; Rusyn, A.; Vinnyk, Y.; Vynnychenko, I.; Fadeeva, N.; Nechaeva, M.; et al. Final results of the FAST study, an international, multicenter, randomized, phase II trial of epirubicin, oxaliplatin, and capecitabine (EOX) with or without the anti-CLDN18.2 antibody IMAB362 as first-line therapy in patients with advanced CLDN18.2+ gastric and gastroesophageal junction (GEJ) adenocarcinoma. Ann. Oncol. 2016, 27, vi208. [Google Scholar]
- Jiang, H.; Shi, Z.; Wang, P.; Wang, C.; Yang, L.; Du, G.; Zhang, H.; Shi, B.; Jia, J.; Li, Q.; et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J. Natl. Cancer Inst. 2019, 111, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Wang, B.; Li, Z.; Li, J.; Wang, H.; Chen, L.; Jiang, H.; Wu, M.; Xiao, J.; Peng, X.; et al. Phase I trial of Claudin 18.2-specific chimeric antigen receptor T cells for advanced gastric and pancreatic adenocarcinoma. J. Clin. Oncol. 2019, 37, 2509. [Google Scholar] [CrossRef]
- Xu, B.; Liu, F.; Liu, Q.; Shi, T.; Wang, Z.; Wu, N.; Xu, X.; Li, L.; Fan, X.; Yu, L.; et al. Highly expressed Claudin18.2 as a potential therapeutic target in advanced gastric signet-ring cell carcinoma (SRCC). J. Gastrointest. Oncol. 2020, 11, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Hida, N.; Maeda, Y.; Katagiri, K.; Takasu, H.; Harada, M.; Itoh, K. A simple culture protocol to detect peptide-specific cytotoxic T lymphocyte precursors in the circulation. Cancer Immunol. Immunother. 2002, 51, 219–228. [Google Scholar]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.-I.; Restifo, N.P.; Yang, J.C. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Nelde, A.; Walz, J.S.; Kowalewski, D.J.; Schuster, H.; Wolz, O.-O.; Peper, J.K.; Cardona Gloria, Y.; Langerak, A.W.; Muggen, A.F.; Claus, R.; et al. HLA class I-restricted MYD88 L265P-derived peptides as specific targets for lymphoma immunotherapy. OncoImmunology 2017, 6, e1219825. [Google Scholar] [CrossRef] [Green Version]
- Sotiropoulou, P.A.; Perez, S.A.; Voelter, V.; Echner, H.; Missitzis, I.; Tsavaris, N.B.; Papamichail, M.; Baxevanis, C.N. Natural CD8+ T-cell responses against MHC class I epitopes of the HER-2/neu oncoprotein in patients with epithelial tumors. Cancer Immunol. Immunother. 2003, 52, 771–779. [Google Scholar] [CrossRef]
- Miller, N.J.; Church, C.D.; Dong, L.; Crispin, D.; Fitzgibbon, M.P.; Lachance, K.; Jing, L.; Shinohara, M.; Gavvovidis, I.; Willimsky, G.; et al. Tumor-Infiltrating Merkel Cell Polyomavirus-Specific T Cells Are Diverse and Associated with Improved Patient Survival. Cancer Immunol. Res. 2017, 5, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A.; Byrd, K.; Vreeland, T.J.; Clifton, G.T.; Jackson, D.O.; Hale, D.F.; Herbert, G.S.; Myers, J.W.; Greene, J.M.; Berry, J.S.; et al. Final analysis of a phase I/IIa trial of the folate-binding protein-derived E39 peptide vaccine to prevent recurrence in ovarian and endometrial cancer patients. Cancer Med. 2019, 8, 4678–4687. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Yoshikawa, T.; Kojima, T.; Shoda, K.; Nosaka, K.; Mizuno, S.; Wada, S.; Fujimoto, Y.; Sasada, T.; Kohashi, K.; et al. Heat shock protein 105 peptide vaccine could induce antitumor immune reactions in a phase I clinical trial. Cancer Sci. 2019, 110, 3049–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.; Agnihotri, S.; Shoger, K.E.; Myers, M.I.; Smith, N.; Chaparala, S.; Villanueva, C.R.; Chattopadhyay, A.; Lee, A.V.; Butterfield, L.H.; et al. Peptide vaccine immunotherapy biomarkers and response patterns in pediatric gliomas. JCI Insight 2018, 3, e98791. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| HLA Type | Peptide Name | Peptide Sequence | HLA-Binding Affinity (%Rank) | Reactive Patients | Rate | |||
|---|---|---|---|---|---|---|---|---|
| NetMHCpan 4.0 | SYFPEITHI | IEDB | NetCTLpan 1.1 | |||||
| HLA-A*0201 | 169–177 | YTFGAALFV | 0.169 | 20 | 0.8 | 1 | 4 | 0.364 |
| 93–101 | GLLVSIFAL | 0.186 | 28 | 0.5 | 0.3 | 4 | 0.364 | |
| 118–126 | TLTSGIMFI | 0.127 | 23 | 1 | 0.8 | 7 | 0.636 | |
| 71–79 | GLPAMLQAV | 0.173 | 26 | 0.28 | 1 | 3 | 0.273 | |
| 118–127 | TLTSGIMFIV | 0.672 | 19 | 0.51 | 1 | 3 | 0.273 | |
| HLA-A*1101 | 228–236 | STGFGSNTK | 0.091 | 23 | 0.42 | 0.8 | 3 | 0.500 |
| 94–102 | LLVSIFALK | 0.446 | 20 | 0.86 | 0.3 | 1 | 0.167 | |
| 218–226 | VAYKPGGFK | 0.131 | 10 | 0.56 | 0.8 | 2 | 0.333 | |
| 217–226 | SVAYKPGGFK | 0.106 | 24 | 0.33 | 0.4 | 4 | 0.667 | |
| 42–51 | AVFNYQGLWR | 0.371 | 26 | 0.82 | 0.8 | 3 | 0.500 | |
| 212–221 | HASGHSVAYK | 0.483 | 12 | 0.73 | 1 | 2 | 0.333 | |
| 93–102 | GLLVSIFALK | 1.123 | 24 | 0.94 | 0.4 | 3 | 0.500 | |
| Factors | Total Number | Reactive | Not Reactive | p-Value | |
|---|---|---|---|---|---|
| Gender | Male | 23 | 11 | 12 | 0.525 |
| Female | 6 | 2 | 4 | ||
| Age | ≥60 | 17 | 11 | 6 | 0.01 |
| <60 | 12 | 2 | 10 | ||
| TNM stage | II | 2 | 1 | 1 | 0.824 |
| III | 13 | 5 | 8 | ||
| IV | 14 | 7 | 7 | ||
| Lauren classification | Intestinal type | 8 | 5 | 3 | 0.429 |
| Diffuse type | 5 | 2 | 3 | ||
| Missing | 16 | 8 | 8 | - | |
| Claudin18.2 expression | High | 9 | 8 | 1 | 0.002 |
| Low and negative | 11 | 2 | 9 | ||
| Missing | 9 | 3 | 6 | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, B.; Chen, F.; Zhang, X.; Wang, Z.; Che, K.; Wu, N.; Yu, L.; Fan, X.; Liu, B.; Wei, J. Antigen-Specific T Cell Immunotherapy Targeting Claudin18.2 in Gastric Cancer. Cancers 2022, 14, 2758. https://doi.org/10.3390/cancers14112758
Xu B, Chen F, Zhang X, Wang Z, Che K, Wu N, Yu L, Fan X, Liu B, Wei J. Antigen-Specific T Cell Immunotherapy Targeting Claudin18.2 in Gastric Cancer. Cancers. 2022; 14(11):2758. https://doi.org/10.3390/cancers14112758
Chicago/Turabian StyleXu, Bo, Fangjun Chen, Xin Zhang, Zhongda Wang, Keying Che, Nandie Wu, Lixia Yu, Xiangshan Fan, Baorui Liu, and Jia Wei. 2022. "Antigen-Specific T Cell Immunotherapy Targeting Claudin18.2 in Gastric Cancer" Cancers 14, no. 11: 2758. https://doi.org/10.3390/cancers14112758
APA StyleXu, B., Chen, F., Zhang, X., Wang, Z., Che, K., Wu, N., Yu, L., Fan, X., Liu, B., & Wei, J. (2022). Antigen-Specific T Cell Immunotherapy Targeting Claudin18.2 in Gastric Cancer. Cancers, 14(11), 2758. https://doi.org/10.3390/cancers14112758