
Targeted Therapy for Adrenocortical Carcinoma: A Genomic-Based Search for Available and Emerging Options

 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Linking FDA-Approved Targeted Therapy and Targeted Therapy in Clinical Trials to Their Genetic Targets
2.1.1. FDA-Approved Targeted Therapy
2.1.2. Targeted Therapy Drugs in Current Clinical Trials
2.2. Identification of Altered Druggable Genes in Adrenocortical Carcinoma (ACC)
2.2.1. Alterations in Druggable Genes of ACC Patients
2.2.2. Gain of Function in Druggable Oncogenes
2.2.3. CNV Amplification in Druggable Oncogenes
2.2.4. Mutation Hotspot Analysis
2.3. Drug Response Prediction
2.3.1. Gene Alterations Affecting Drug Response
2.3.2. Drug Response Prediction
- (a)
- Gain of function in an oncogene;
- (b)
- CNV amplification in an oncogene;
- (c)
- Specific genetic alterations in an oncogene;
- (d)
- Genetic alterations in tumor suppressor genes.
- (i)
- The gene underlying the drug target shows a copy number increase;
- (ii)
- The drug targets a gene whose product shows a gain of function;
- (iii)
- A specific alteration with known drug effectiveness is present.
2.4. Ethics
3. Results
3.1. Targeted Therapy and Linked Genetic Targets
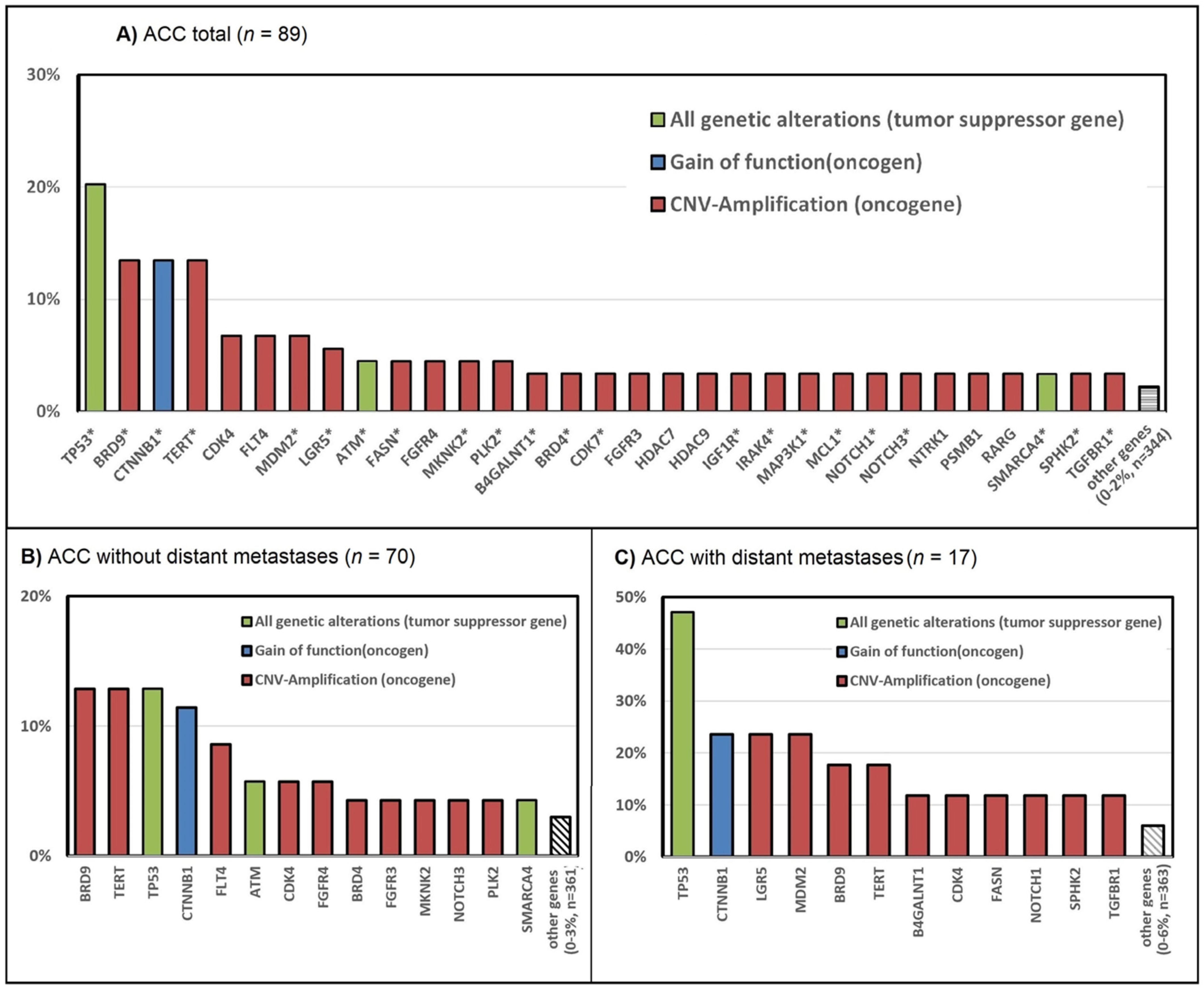
3.2. Genetic Alterations in ACC—The TCGA Dataset
3.3. Mutation Hotspots Based on the TCGA Data
3.4. CNV Co-Amplification Pairs Based on the TCGA Data
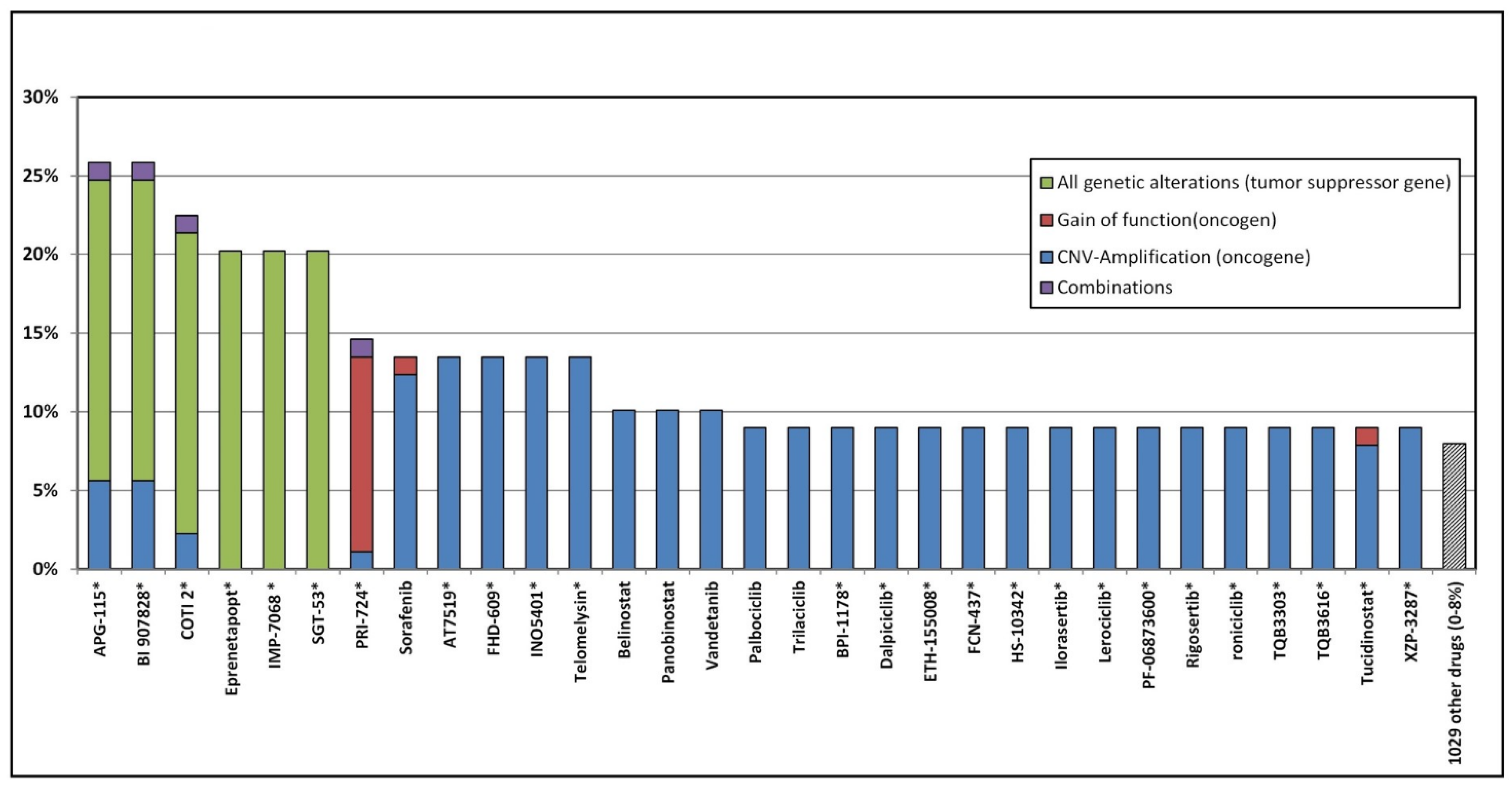
3.5. Potential Genomic Drug Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gross, D.J.; Munter, G.; Bitan, M.; Siegal, T.; Gabizon, A.; Weitzen, R.; Merimsky, O.; Ackerstein, A.; Salmon, A.; Sella, A.; et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr. Relat. Cancer 2006, 13, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Fay, A.P.; Elfiky, A.; Teló, G.H.; McKay, R.R.; Kaymakcalan, M.; Nguyen, P.L.; Vaidya, A.; Ruan, D.T.; Bellmunt, J.; Choueiri, T.K. Adrenocortical carcinoma: The management of metastatic disease. Crit. Rev. Oncol. Hematol. 2014, 92, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.; Habra, M.A. Update on adrenocortical carcinoma management and future directions. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Paragliola, R.M.; Torino, F.; Papi, G.; Locantore, P.; Pontecorvi, A.; Corsello, S.M. Role of Mitotane in Adrenocortical Carcinoma—Review and State of the art. Eur. Endocrinol. 2018, 14, 62–66. [Google Scholar] [CrossRef]
- Konda, B.; Kirschner, L.S. Novel targeted therapies in adrenocortical carcinoma. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 233–241. [Google Scholar] [CrossRef][Green Version]
- Alyateem, G.; Nilubol, N. Current Status and Future Targeted Therapy in Adrenocortical Cancer. Front. Endocrinol. 2021, 12, 613248. [Google Scholar] [CrossRef]
- Fassnacht, M.; Terzolo, M.; Allolio, B.; Baudin, E.; Haak, H.; Berruti, A.; Welin, S.; Schade-Brittinger, C.; Lacroix, A.; Jarzab, B.; et al. Combination chemotherapy in advanced adrenocortical carcinoma. N. Engl. J. Med. 2012, 366, 2189–2197. [Google Scholar] [CrossRef]
- Grisanti, S.; Cosentini, D.; Laganà, M.; Morandi, A.; Lazzari, B.; Ferrari, L.; Volta, A.D.; Ambrosini, R.; Ferrari, V.D.; Sigala, S.; et al. Clinical Prognostic Factors in Patients With Metastatic Adrenocortical Carcinoma Treated With Second Line Gemcitabine Plus Capecitabine Chemotherapy. Front. Endocrinol. 2021, 12, 624102. [Google Scholar] [CrossRef]
- Assié, G.; Letouzé, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; René-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Kamilaris, C.D.C.; Hannah-Shmouni, F.; Stratakis, C.A. Adrenocortical tumorigenesis: Lessons from genetics. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101428. [Google Scholar] [CrossRef] [PubMed]
- Gara, S.K.; Lack, J.; Zhang, L.; Harris, E.; Cam, M.; Kebebew, E. Metastatic adrenocortical carcinoma displays higher mutation rate and tumor heterogeneity than primary tumors. Nat. Commun. 2018, 9, 4172. [Google Scholar] [CrossRef] [PubMed]
- Pozdeyev, N.; Fishbein, L.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Ross, J.S.; Darabi, S.; Demeure, M.J.; Kar, A.; Foust, L.J.; et al. Targeted genomic analysis of 364 adrenocortical carcinomas. Endocr. Relat. Cancer 2021, 28, 671–681. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, C.; Edgerly, M.; Velarde, M.; Wilkerson, J.; Venkatesan, A.M.; Pittaluga, S.; Yang, S.X.; Nguyen, D.; Balasubramaniam, S.; Fojo, T. The VEGF inhibitor axitinib has limited effectiveness as a therapy for adrenocortical cancer. J. Clin. Endocrinol. Metab. 2014, 99, 1291–1297. [Google Scholar] [CrossRef]
- Samnotra, V.; Vassilopoulou-Sellin, R.; Fojo, A.T.; Oh, W.; LaRocca, R.; Ernstoff, M.; Memoli, V.; Cole, B.; Quinn, D.; Simmons, P.; et al. A phase II trial of gefitinib monotherapy in patients with unresectable adrenocortical carcinoma (ACC). J. Clin. Oncol. 2007, 25, 15527. [Google Scholar] [CrossRef]
- Quinkler, M.; Hahner, S.; Wortmann, S.; Johanssen, S.; Adam, P.; Ritter, C.; Strasburger, C.; Allolio, B.; Fassnacht, M. Treatment of advanced adrenocortical carcinoma with erlotinib plus gemcitabine. J. Clin. Endocrinol. Metab. 2008, 93, 2057–2062. [Google Scholar] [CrossRef]
- Fassnacht, M.; Berruti, A.; Baudin, E.; Demeure, M.J.; Gilbert, J.; Haak, H.; Kroiss, M.; Quinn, D.I.; Hesseltine, E.; Ronchi, C.L.; et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: A double-blind, randomised, phase 3 study. Lancet Oncol. 2015, 16, 426–435. [Google Scholar] [CrossRef]
- De Martino, M.C.; Feelders, R.A.; Pivonello, C.; Simeoli, C.; Papa, F.; Colao, A.; Pivonello, R.; Hofland, L.J. The role of mTOR pathway as target for treatment in adrenocortical cancer. Endocr. Connect. 2019, 8, R144–R156. [Google Scholar] [CrossRef]
- Fraenkel, M.; Gueorguiev, M.; Barak, D.; Salmon, A.; Grossman, A.B.; Gross, D.J. Everolimus therapy for progressive adrenocortical cancer. Endocrine 2013, 44, 187–192. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- My Cancer Genome. What is My Cancer Genome? Available online: https://www.mycancergenome.org/about/what-is-my-cancer-genome/ (accessed on 20 February 2022).
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef] [PubMed]
- The National Cancer Institute. NCI Drug Dictionary. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug (accessed on 2 March 2022).
- Hescheler, D.A.; Hartmann, M.J.M.; Riemann, B.; Michel, M.; Bruns, C.J.; Alakus, H.; Chiapponi, C. Anaplastic thyroid cancer: Genome-based search for new targeted therapy options. Endocr. Connect. 2022, 11, e210624. [Google Scholar] [CrossRef]
- The U.S. National Institute of Health. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 20 February 2022).
- Switzerland Springer Nature. AdisInsight International. Available online: https://adisinsight.springer.com/ (accessed on 1 March 2022).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Bhattarai, T.S.; Schram, A.M.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; Chakravarty, D.; Phillips, S.; Kandoth, C.; Penson, A.; et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov. 2018, 8, 174–183. [Google Scholar] [CrossRef]
- The Broad Institute. TARGET. Available online: https://software.broadinstitute.org/cancer/cga/target (accessed on 20 February 2022).
- Hescheler, D.A.; Plum, P.S.; Zander, T.; Quaas, A.; Korenkov, M.; Gassa, A.; Michel, M.; Bruns, C.J.; Alakus, H. Identification of targeted therapy options for gastric adenocarcinoma by comprehensive analysis of genomic data. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2020, 23, 627–638. [Google Scholar] [CrossRef]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical Carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef]
- Sharma, E.; Dahal, S.; Sharma, P.; Bhandari, A.; Gupta, V.; Amgai, B.; Dahal, S. The Characteristics and Trends in Adrenocortical Carcinoma: A United States Population Based Study. J. Clin. Med. Res. 2018, 10, 636–640. [Google Scholar] [CrossRef]
- Hescheler, D.; Riemann, B.; Hartmann, M.; Michel, M.; Faust, M.; Bruns, C.; Alakus, H.; Chiapponi, C. Targeted Therapy of Papillary Thyroid Cancer: A Comprehensive Genomic Analysis. Front. Endocrinol. 2021, 12, 748941. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.D.; Tang, Q.; Kong, Y.; Rong, T.; Wang, Q.; Li, N.; Fang, X.; Gu, J.; Xiong, D.; Yin, Y.; et al. MDM2 inhibitor APG-115 exerts potent antitumor activity and synergizes with standard-of-care agents in preclinical acute myeloid leukemia models. Cell Death Discov. 2021, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Cornillie, J.; Wozniak, A.; Li, H.; Gebreyohannes, Y.K.; Wellens, J.; Hompes, D.; Debiec-Rychter, M.; Sciot, R.; Schöffski, P. Anti-tumor activity of the MDM2-TP53 inhibitor BI-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring MDM2 amplification. Clin. Transl. Oncol. 2020, 22, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Chan, G.K.; Gamper, A.M. Targeting the DNA Damage Response for Cancer Therapy by Inhibiting the Kinase Wee1. Front. Oncol. 2022, 12, 828684. [Google Scholar] [CrossRef]
- Gabata, R.; Harada, K.; Mizutani, Y.; Ouchi, H.; Yoshimura, K.; Sato, Y.; Kitao, A.; Kimura, K.; Kouji, H.; Miyashita, T.; et al. Anti-tumor Activity of the Small Molecule Inhibitor PRI-724 Against β-Catenin-activated Hepatocellular Carcinoma. Anticancer Res. 2020, 40, 5211–5219. [Google Scholar] [CrossRef]
- Nasser, F.; Moussa, N.; Helmy, M.W.; Haroun, M. Dual targeting of Notch and Wnt/β-catenin pathways: Potential approach in triple-negative breast cancer treatment. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 481–490. [Google Scholar] [CrossRef]
- Bahrami, A.; Amerizadeh, F.; ShahidSales, S.; Khazaei, M.; Ghayour-Mobarhan, M.; Sadeghnia, H.R.; Maftouh, M.; Hassanian, S.M.; Avan, A. Therapeutic Potential of Targeting Wnt/β-Catenin Pathway in Treatment of Colorectal Cancer: Rational and Progress. J. Cell. Biochem. 2017, 118, 1979–1983. [Google Scholar] [CrossRef]
- Moroney, M.R.; Woodruff, E.; Qamar, L.; Bradford, A.P.; Wolsky, R.; Bitler, B.G.; Corr, B.R. Inhibiting Wnt/beta-catenin in CTNNB1-mutated endometrial cancer. Mol. Carcinog. 2021, 60, 511–523. [Google Scholar] [CrossRef]
- Martinez-Font, E.; Pérez-Capó, M.; Ramos, R.; Felipe, I.; Garcías, C.; Luna, P.; Terrasa, J.; Martín-Broto, J.; Vögler, O.; Alemany, R.; et al. Impact of Wnt/β-Catenin Inhibition on Cell Proliferation through CDC25A Downregulation in Soft Tissue Sarcomas. Cancers 2020, 12, 2556. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.F.; Spoettl, G.; Maurer, J.; Nölting, S.; Auernhammer, C.J. Inhibition of Wnt/β-Catenin Signaling in Neuroendocrine Tumors in vitro: Antitumoral Effects. Cancers 2020, 12, 345. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Vanderbilt, C.M.; Lin, Y.T.; Benhamida, J.K.; Jungbluth, A.A.; Rana, S.; Momeni-Boroujeni, A.; Chang, J.C.; McFarlane, T.; Salazar, P.; et al. A Pan-Cancer Study of Somatic TERT Promoter Mutations and Amplification in 30,773 Tumors Profiled by Clinical Genomic Sequencing. J. Mol. Diagn. 2021, 23, 253–263. [Google Scholar] [CrossRef]
- Liang, R.; Weigand, I.; Lippert, J.; Kircher, S.; Altieri, B.; Steinhauer, S.; Hantel, C.; Rost, S.; Rosenwald, A.; Kroiss, M.; et al. Targeted Gene Expression Profile Reveals CDK4 as Therapeutic Target for Selected Patients With Adrenocortical Carcinoma. Front. Endocrinol. 2020, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Fragni, M.; Tiberio, G.A.M.; Galli, D.; Roca, E.; Salvi, V.; Bosisio, D.; Missale, C.; Terzolo, M.; Memo, M.; et al. Palbociclib inhibits proliferation of human adrenocortical tumor cells. Endocrine 2018, 59, 213–217. [Google Scholar] [CrossRef]
- Wang, S.; Dong, D.; Zhang, W.; Hu, H.; Li, H.; Zhu, Y.; Zhou, J.; Shan, X.; Tian, J. Specific Borrmann classification in advanced gastric cancer by an ensemble multilayer perceptron network: A multicenter research. Med. Phys. 2021, 48, 5017–5028. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Berruti, A.; Sperone, P.; Ferrero, A.; Germano, A.; Ardito, A.; Priola, A.M.; De Francia, S.; Volante, M.; Daffara, F.; Generali, D.; et al. Phase II study of weekly paclitaxel and sorafenib as second/third-line therapy in patients with adrenocortical carcinoma. Eur. J. Endocrinol. 2012, 166, 451–458. [Google Scholar] [CrossRef]
- Van Erp, N.P.; Guchelaar, H.-J.; Ploeger, B.A.; Romijn, J.A.; Den Hartigh, J.; Gelderblom, H. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur. J. Endocrinol. 2011, 164, 621–626. [Google Scholar] [CrossRef]
- Cerquetti, L.; Bucci, B.; Raffa, S.; Amendola, D.; Maggio, R.; Lardo, P.; Petrangeli, E.; Torrisi, M.R.; Toscano, V.; Pugliese, G.; et al. Effects of Sorafenib, a Tyrosin Kinase Inhibitor, on Adrenocortical Cancer. Front. Endocrinol. 2021, 12, 667798. [Google Scholar] [CrossRef]
- Kroiss, M.; Quinkler, M.; Johanssen, S.; Van Erp, N.P.; Lankheet, N.; Pöllinger, A.; Laubner, K.; Strasburger, C.J.; Hahner, S.; Müller, H.H.; et al. Sunitinib in refractory adrenocortical carcinoma: A phase II, single-arm, open-label trial. J. Clin. Endocrinol. Metab. 2012, 97, 3495–3503. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Kim, E.S.; Nava-Parada, P.; Alam, S.; Johnson, F.M.; Stephens, A.W.; Simantov, R.; Poondru, S.; Gedrich, R.; Lippman, S.M.; et al. Phase I study of intermittent oral dosing of the insulin-like growth factor-1 and insulin receptors inhibitor OSI-906 in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; De Leo, S.; Di Stefano, M.; Vannucchi, G.; Persani, L.; Fugazzola, L. Primary Adrenal Insufficiency During Lenvatinib or Vandetanib and Improvement of Fatigue After Cortisone Acetate Therapy. J. Clin. Endocrinol. Metab. 2019, 104, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Poh, C.; Arora, M.; Ghuman, S.; Tuscano, J. Belinostat in Relapsed/Refractory T-Cell Large Granular Lymphocyte Leukemia. Acta Haematol. 2021, 144, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Eleutherakis-Papaiakovou, E.; Kanellias, N.; Kastritis, E.; Gavriatopoulou, M.; Terpos, E.; Dimopoulos, M.A. Efficacy of Panobinostat for the Treatment of Multiple Myeloma. J. Oncol. 2020, 2020, 7131802. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Spartalis, E.; Kotrotsios, K.; Chrysikos, D.; Spartalis, M.; Paschou, S.A.; Schizas, D.; Tsamakis, K.; Dimitroulis, D.; Troupis, T.; Nikiteas, N. Histone Deacetylase Inhibitors and Papillary Thyroid Cancer. Curr. Pharm. Des. 2021, 27, 2199–2208. [Google Scholar] [CrossRef]
- Cartron, A.M.; Nguyen, T.H.; Roh, Y.S.; Kwatra, M.M.; Kwatra, S.G. Janus kinase inhibitors for atopic dermatitis: A promising treatment modality. Clin. Exp. Dermatol. 2021, 46, 820–824. [Google Scholar] [CrossRef]
- Azaro, A.; Massard, C.; Tap, W.D.; Cassier, P.A.; Merchan, J.; Italiano, A.; Anderson, B.; Yuen, E.; Yu, D.; Oakley, G., 3rd; et al. A phase 1b study of the Notch inhibitor crenigacestat (LY3039478) in combination with other anticancer target agents (taladegib, LY3023414, or abemaciclib) in patients with advanced or metastatic solid tumors. Investig. New Drugs 2021, 39, 1089–1098. [Google Scholar] [CrossRef]
- Xue, D.; Li, D.; Dou, C.; Li, J. A Comprehensive Bioinformatic Analysis of NOTCH Pathway Involvement in Stomach Adenocarcinoma. Dis. Markers 2021, 2021, 4739868. [Google Scholar] [CrossRef]
- Zhdanovskaya, N.; Firrincieli, M.; Lazzari, S.; Pace, E.; Rossi, P.S.; Felli, M.P.; Talora, C.; Screpanti, I.; Palermo, R. Targeting Notch to Maximize Chemotherapeutic Benefits: Rationale, Advanced Strategies, and Future Perspectives. Cancers 2021, 13, 5106. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, C.L.; Sbiera, S.; Altieri, B.; Steinhauer, S.; Wild, V.; Bekteshi, M.; Kroiss, M.; Fassnacht, M.; Allolio, B. Notch1 pathway in adrenocortical carcinomas: Correlations with clinical outcome. Endocr. Relat. Cancer 2015, 22, 531–543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Le Tourneau, C.; Hoimes, C.; Zarwan, C.; Wong, D.J.; Bauer, S.; Claus, R.; Wermke, M.; Hariharan, S.; Von Heydebreck, A.; Kasturi, V.; et al. Avelumab in patients with previously treated metastatic adrenocortical carcinoma: Phase 1b results from the JAVELIN solid tumor trial. J. Immunother. Cancer 2018, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.; Zheng, Y.; Kelly, V.; Katz, S.S.; Chou, J.; Do, R.K.G.; Capanu, M.; Zamarin, D.; Saltz, L.B.; Ariyan, C.E.; et al. PD-1 Blockade in Advanced Adrenocortical Carcinoma. J. Clin. Oncol. 2020, 38, 71–80. [Google Scholar] [CrossRef]
- Muzzi, J.C.D.; Magno, J.M.; Cardoso, M.A.; De Moura, J.; Castro, M.A.A.; Figueiredo, B.C. Adrenocortical Carcinoma Steroid Profiles: In Silico Pan-Cancer Analysis of TCGA Data Uncovers Immunotherapy Targets for Potential Improved Outcomes. Front. Endocrinol. 2021, 12, 672319. [Google Scholar] [CrossRef]
- Fiorentini, C.; Grisanti, S.; Cosentini, D.; Abate, A.; Rossini, E.; Berruti, A.; Sigala, S. Molecular Drivers of Potential Immunotherapy Failure in Adrenocortical Carcinoma. J. Oncol. 2019, 2019, 6072863. [Google Scholar] [CrossRef]
- Kiesewetter, B.; Riss, P.; Scheuba, C.; Mazal, P.; Kretschmer-Chott, E.; Haug, A.; Raderer, M. Management of adrenocortical carcinoma: Are we making progress? Ther. Adv. Med. Oncol. 2021, 13, 17588359211038409. [Google Scholar] [CrossRef]
- Jänne, P.A.; Van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef]
- Imyanitov, E.N.; Levchenko, E.V.; Kuligina, E.S.; Orlov, S.V. Treating non-small cell lung cancer with selumetinib: An up-to-date drug evaluation. Expert Opin. Pharmacother. 2020, 21, 1943–1953. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef]
- Van Brummelen, E.M.J.; Huijberts, S.; Van Herpen, C.; Desar, I.; Opdam, F.; Van Geel, R.; Marchetti, S.; Steeghs, N.; Monkhorst, K.; Thijssen, B.; et al. Phase I Study of Afatinib and Selumetinib in Patients with KRAS-Mutated Colorectal, Non-Small Cell Lung, and Pancreatic Cancer. Oncologist 2021, 26, e290–e545. [Google Scholar] [CrossRef] [PubMed]
- Lippert, J.; Appenzeller, S.; Liang, R.; Sbiera, S.; Kircher, S.; Altieri, B.; Nanda, I.; Weigand, I.; Gehrig, A.; Steinhauer, S.; et al. Targeted Molecular Analysis in Adrenocortical Carcinomas: A Strategy Toward Improved Personalized Prognostication. J. Clin. Endocrinol. Metab. 2018, 103, 4511–4523. [Google Scholar] [CrossRef] [PubMed]
- Darabi, S.; Braxton, D.R.; Eisenberg, B.L.; Demeure, M.J. Molecular genomic profiling of adrenocortical cancers in clinical practice. Surgery 2021, 169, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Huff, L.; Litman, T.; Im, K.; Edgerly, M.; Del Rivero, J.; Pittaluga, S.; Merino, M.; Bates, S.E.; Dean, M. Metastatic and recurrent adrenocortical cancer is not defined by its genomic landscape. BMC Med. Genom. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug/ ClinicalTrials.gov ID/Reference | Mechanism of Action | Setting | Primary Outcome | Study Design | Status |
|---|---|---|---|---|---|
| Cabozantinib NCT03612232 | VEGFR 1/2/3, KIT, NTRK2, FLT-3, AXL, RET, MET, and TEK. | Relapsed/refractory advanced or metastatic ACC (mitotane discontinued, serum concentration < 2 mg/L) | PFS at 4 months | Single-arm Phase II | Recruiting |
| Cabozantinib NCT03370718 | Locally advanced or metastatic ACC (mitotane stopped for 1 month, serum concentration < 2 mg/L) | PFS at 4 months | Single-arm phase II | Active, not recruiting | |
| Camrelizumab/Apatinib NCT04318730 | Camrelizumab: PD-1 receptor Apatinib: VEGF2, KIT, SRC | Second-line treatment of recurrent or metastatic adrenocortical carcinoma | ORR | Single-arm phase II | not yet recruiting |
| IPI-549 (Eganelisib)/Nivolumab NCT02637531 | IPI-549: PIK3C Nivolumab: PD-1 receptor | ACC locally advanced or metastatic and other advanced and/or metastatic carcinoma or melanoma, excluding sarcoma | DLT, AE | Single Arm phase I/Ib | active, not recruiting |
| Relacorilant/pembrolizumab NCT04373265 | Relacorilant: SGRM Pembrolizumab: PD-1 receptor | Locally advanced or metastatic ACC with glucocorticoid excess (mitotane level ≤ 4 mg/L) | ORR, dose-limiting toxicities | Phase Ib | recruiting |
| Therapeutic vaccine (EO2401)/nivolumab NCT04187404 | Nivolumab: PD-1 receptor | ACC locally advanced or metastatic (also including pheochromocytoma or paraganglioma) | Safety | Phase I/II | recruiting |
| ONC201 NCT03034200 | MAPK1 | Unresectable, recurrent, locally advanced, refractory, or metastatic neuroendocrine tumors including cholangiocarcinoma and ACC (age 14 and older) | CR, PR | Single-arm phase II | active not recruiting |
| Nivolumab/ipilimumab NCT03333616 | Nivolumab: PD-1 receptor ipilimumab: CTLA-4 binding | Locally advanced or metastatic ACC (mitotane allowed for control or endocrine symptoms) and other rare genitourinary tumors | ORR | Single arm phase II | Recruiting |
| Nivolumab/ipilimumab NCT02834013 | Relapsed/refractory advanced or metastatic ACC or other rare tumors | ORR | Single-arm, phase II | Recruiting | |
| Pembrolizumab NCT02721732 | Pembrolizumab: PD-1 receptor | Relapsed/refractory advanced or metastatic ACC or other rare tumors | Non-progression at 27 weeks, adverse events | Single-arm phase II | active, not recruiting |
| Pembrolizumab/lenvatinib NCT05036434 | Pembrolizumab: PD-1 receptor Lenvatinib: VEGFR, PDGFR, EGFR, RET, KIT | Advanced ACC after failure of platinum- and mitotane-based chemotherapy | ORR | single-arm phase II | Not yet recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hescheler, D.A.; Hartmann, M.J.M.; Riemann, B.; Michel, M.; Bruns, C.J.; Alakus, H.; Chiapponi, C. Targeted Therapy for Adrenocortical Carcinoma: A Genomic-Based Search for Available and Emerging Options. Cancers 2022, 14, 2721. https://doi.org/10.3390/cancers14112721
Hescheler DA, Hartmann MJM, Riemann B, Michel M, Bruns CJ, Alakus H, Chiapponi C. Targeted Therapy for Adrenocortical Carcinoma: A Genomic-Based Search for Available and Emerging Options. Cancers. 2022; 14(11):2721. https://doi.org/10.3390/cancers14112721
Chicago/Turabian StyleHescheler, Daniel Alexander, Milan Janis Michael Hartmann, Burkhard Riemann, Maximilian Michel, Christiane Josephine Bruns, Hakan Alakus, and Costanza Chiapponi. 2022. "Targeted Therapy for Adrenocortical Carcinoma: A Genomic-Based Search for Available and Emerging Options" Cancers 14, no. 11: 2721. https://doi.org/10.3390/cancers14112721
APA StyleHescheler, D. A., Hartmann, M. J. M., Riemann, B., Michel, M., Bruns, C. J., Alakus, H., & Chiapponi, C. (2022). Targeted Therapy for Adrenocortical Carcinoma: A Genomic-Based Search for Available and Emerging Options. Cancers, 14(11), 2721. https://doi.org/10.3390/cancers14112721