
Dyslipidemia in Children Treated with a BRAF Inhibitor for Low-Grade Gliomas: A New Side Effect?

 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
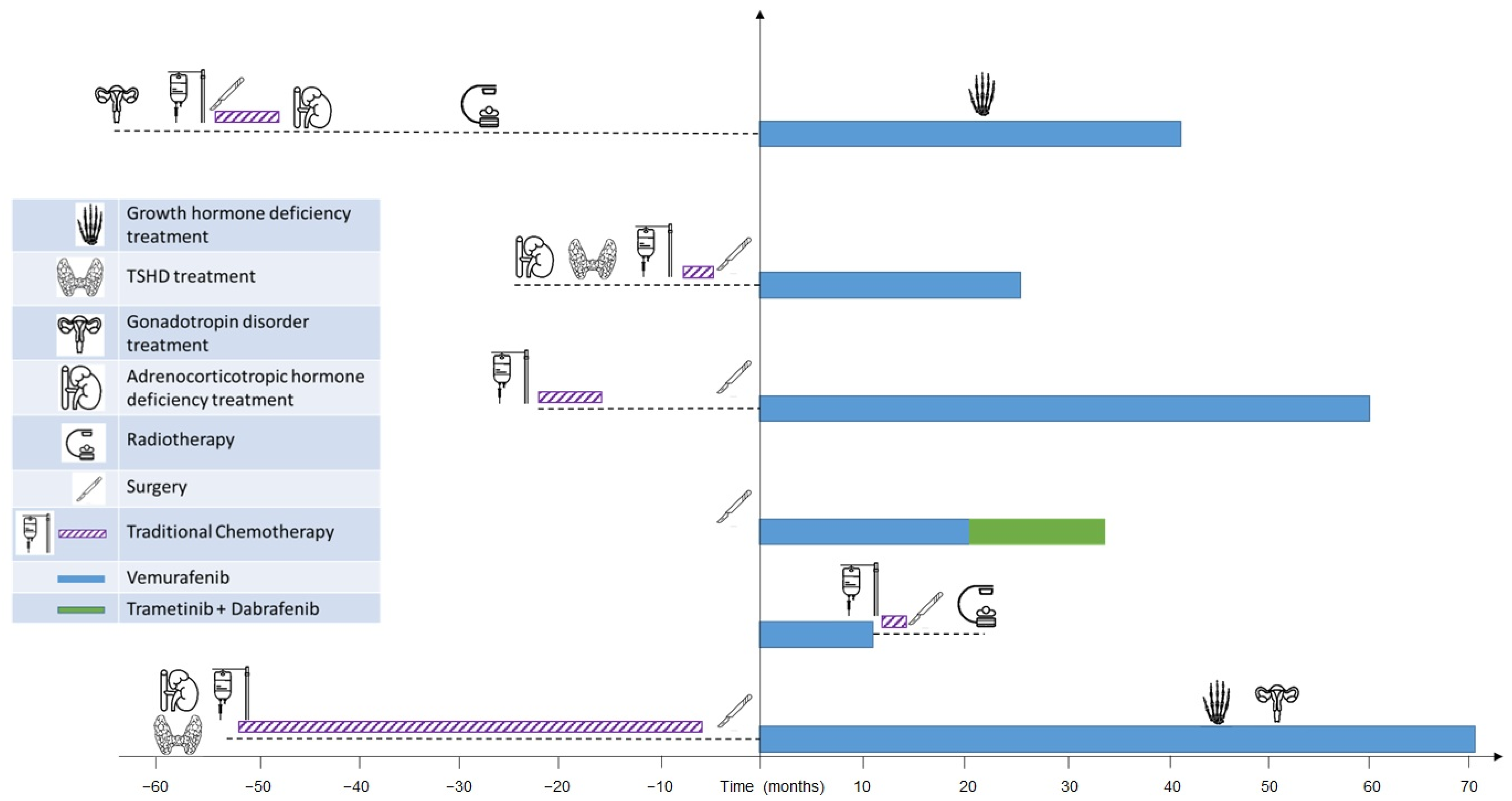
3.1. Study Population
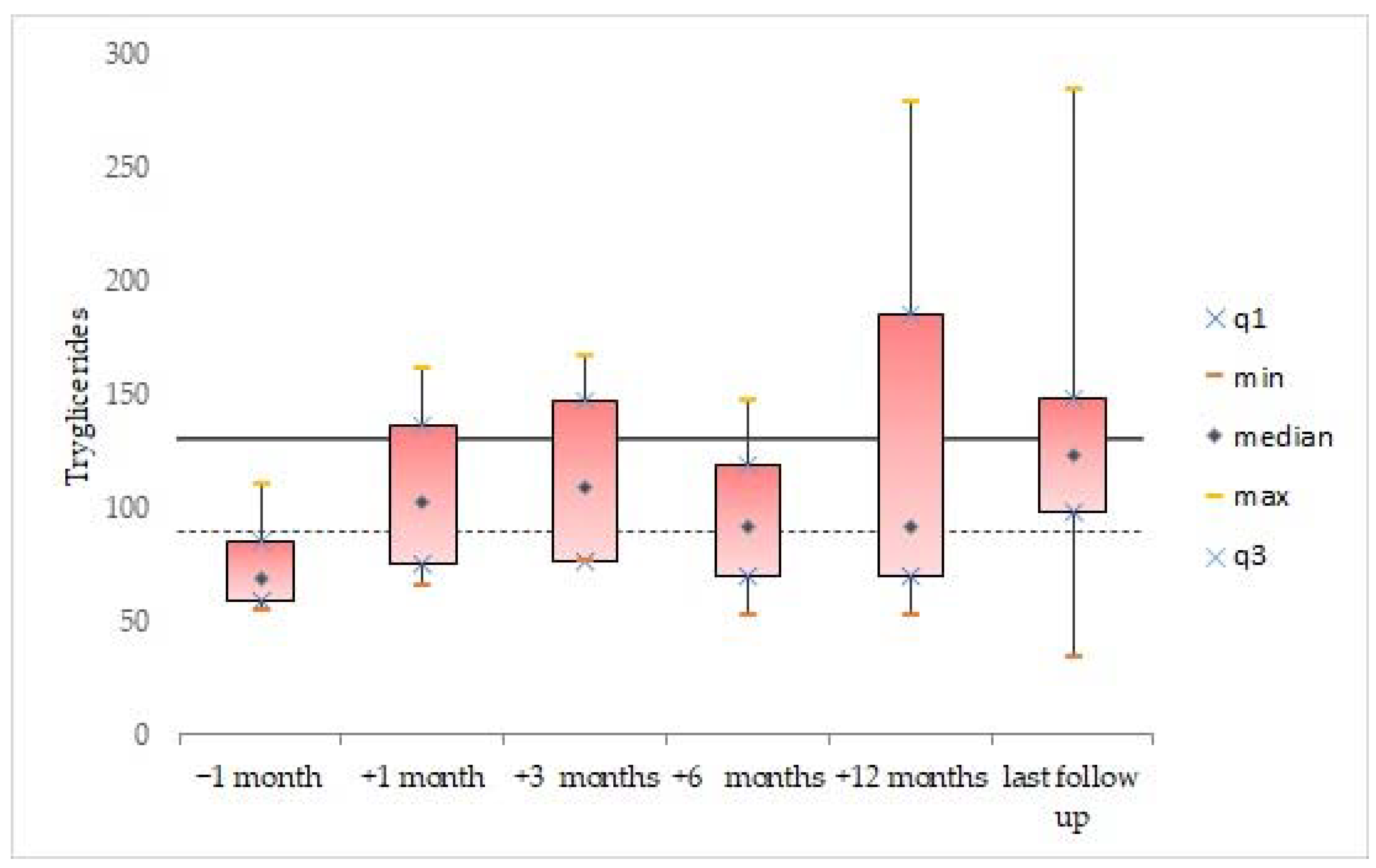
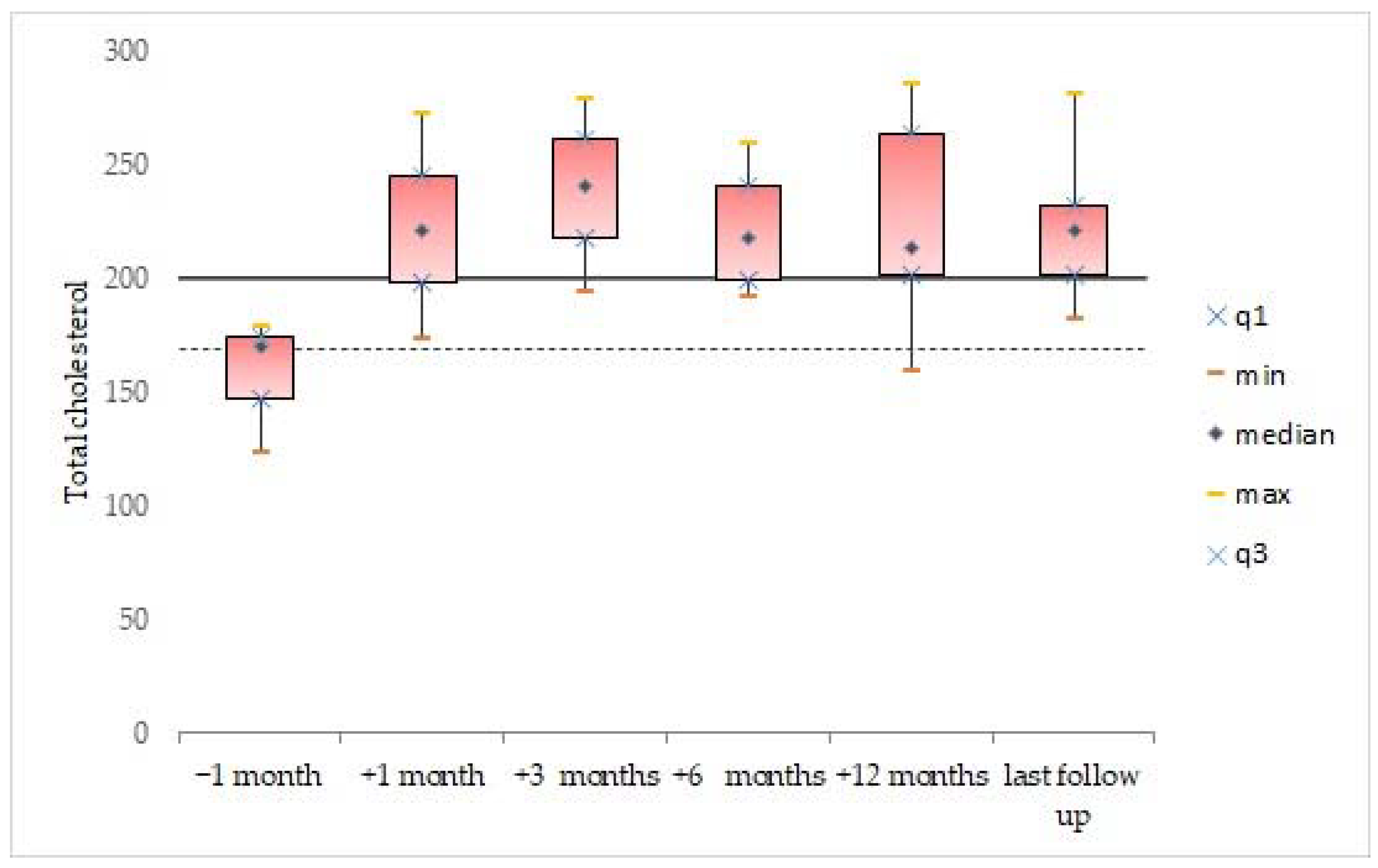
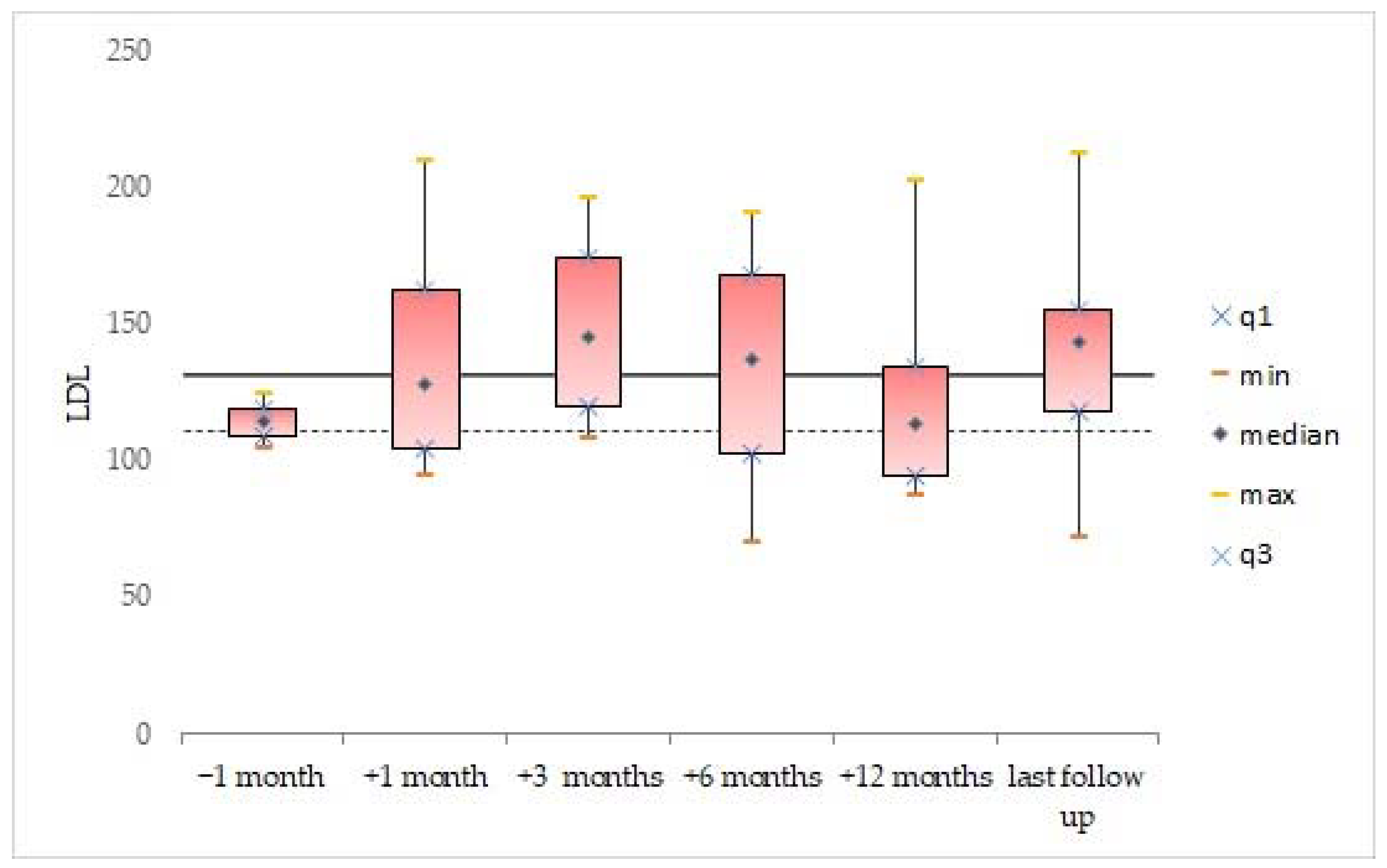
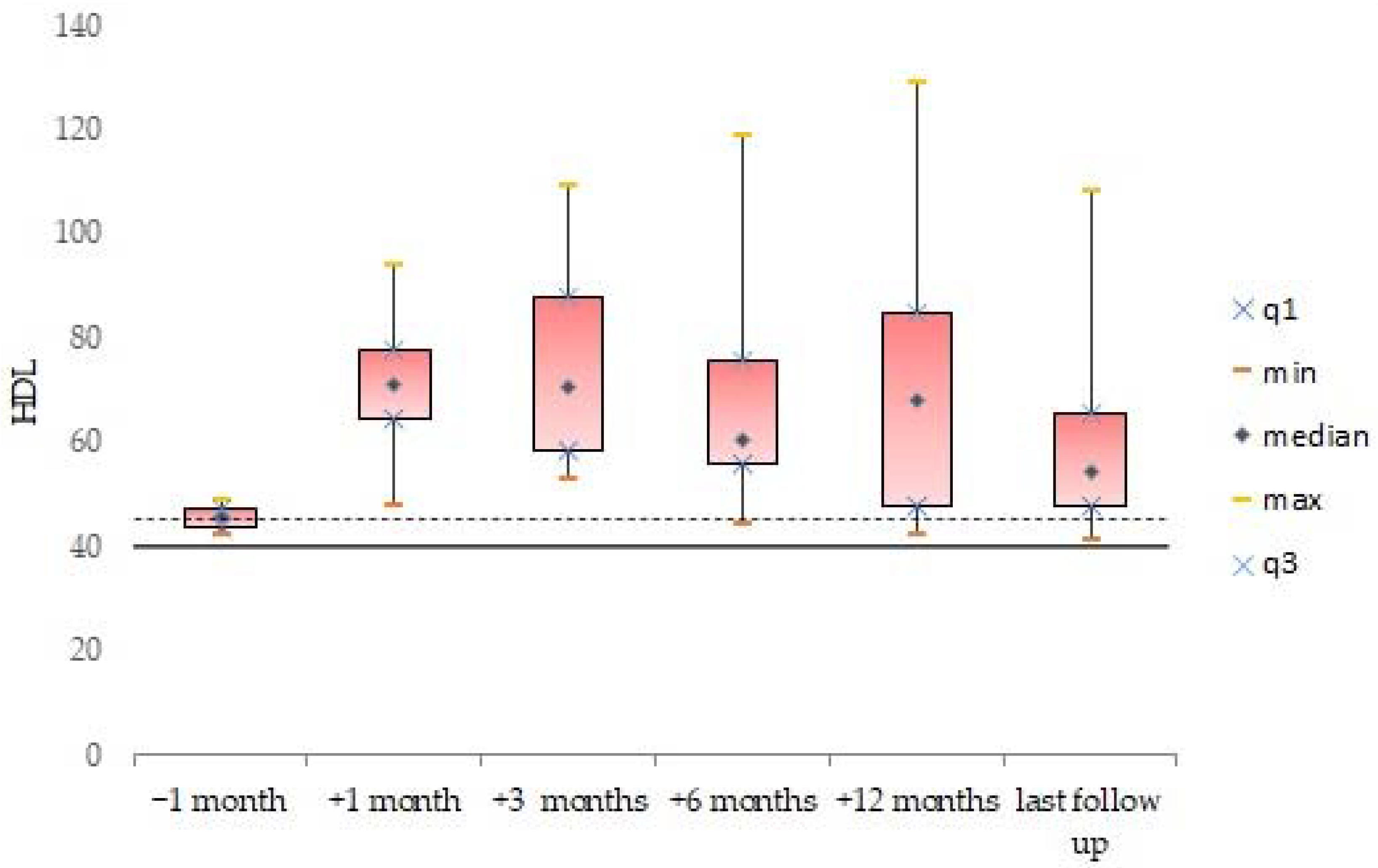
3.2. Blood Lipid Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, K.J.; Cullen, J.; Barnholtz-Sloan, J.S.; Ostrom, Q.T.; Langer, C.E.; Turner, M.C.; McKean-Cowdin, R.; Fisher, J.L.; Lupo, P.J.; Partap, S.; et al. Childhood brain tumor epidemiology: A brain tumor epidemiology consortium review. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2716–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Blank, P.; Bandopadhayay, P.; Haas-Kogan, D.; Fouladi, M.; Fangusaro, J. Management of pediatric low-grade glioma. Curr. Opin. Pediatr. 2019, 31, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Merchant, T.E.; Conklin, H.M.; Wu, S.; Lustig, R.H.; Xiong, X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: Prospective evaluation of cognitive, endocrine, and hearing deficits. J. Clin. Oncol. 2009, 27, 3691–3697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, G.T.; Liu, Q.; Yasui, Y.; Huang, S.; Ness, K.K.; Leisenring, W.; Hudson, M.M.; Donaldson, S.S.; King, A.A.; Stovall, M.; et al. Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2009, 101, 946–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloi, D.; Belgioia, L.; Barra, S.; Giannelli, F.; Cavagnetto, F.; Gallo, F.; Milanaccio, C.; Garrè, M.; Di Profio, S.; Di Iorgi, N.; et al. Neuroendocrine late effects after tailored photon radiotherapy for children with low grade gliomas: Long term correlation with tumour and treatment parameters. Radiother. Oncol. 2017, 125, 241–247. [Google Scholar] [CrossRef]
- Lassaletta, A.; Zapotocky, M.; Mistry, M.; Ramaswamy, V.; Honnorat, M.; Krishnatry, R.; Stucklin, A.G.; Zhukova, N.; Arnoldo, A.; Ryall, S.; et al. Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J. Clin. Oncol. 2017, 35, 2934–2941. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef]
- Behling, F.; Barrantes-Freer, A.; Skardelly, M.; Nieser, M.; Christians, A.; Stockhammer, F.; Rohde, V.; Tatagiba, M.; Hartmann, C.; Stadelmann, C.; et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn. Pathol. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Bar, E.E.; Lin, A.; Tihan, T.; Burger, P.C.; Eberhart, C.G. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J. Neuropathol. Exp. Neurol. 2008, 67, 878–887. [Google Scholar] [CrossRef] [Green Version]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar] [CrossRef]
- Ho, C.Y.; Mobley, B.C.; Gordish-Dressman, H.; Vandenbussche, C.J.; Mason, G.E.; Bornhorst, M.; Esbenshade, A.J.; Tehrani, M.; Orr, B.A.; LaFrance, D.R.; et al. A clinicopathologic study of diencephalic pediatric low-grade gliomas with BRAF V600 mutation. Acta Neuropathol. 2015, 130, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Mistry, M.; Zhukova, N.; Merico, D.; Rakopoulos, P.; Krishnatry, R.; Shago, M.; Stavropoulos, J.; Alon, N.; Pole, J.D.; Ray, P.N.; et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup ofchildhood secondary high-grade glioma. J. Clin. Oncol. 2015, 33, 1015–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selt, F.; van Tilburg, C.M.; Bison, B.; Sievers, P.; Harting, I.; Ecker, J.; Pajtler, K.W.; Sahm, F.; Bahr, A.; Simon, M.; et al. Response to trametinib treatment in progressive pediatric low-grade glioma patients. J. Neurooncol. 2020, 149, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Nicolaides, T.; Nazemi, K.J.; Crawford, J.; Kilburn, L.; Minturn, J.; Gajjar, A.; Gauvain, K.; Leary, S.; Dhall, G.; Aboian, M.; et al. Phase I study of vemurafenib in children with recurrent or progressive BRAFV600E mutant brain tumors: Pacific Pediatric Neuro-Oncology Consortium study (PNOC-002). Oncotarget 2020, 11, 1942–1952. [Google Scholar] [CrossRef]
- Hargrave, D.R.; Bouffet, E.; Tabori, U.; Broniscer, A.; Cohen, K.J.; Hansford, J.R.; Geoerger, B.; Hingorani, P.; Dunkel, I.J.; Russo, M.W.; et al. Efficacy and Safety of Dabrafenib in Pediatric Patients with BRAF V600 Mutation-Positive Relapsed or Refractory Low-Grade Glioma: Results from a Phase I/IIa Study. Clin. Cancer Res. 2019, 25, 7303–7311. [Google Scholar] [CrossRef]
- Marks, A.M.; Bindra, R.S.; DiLuna, M.L.; Huttner, A.; Jairam, V.; Kahle, K.T.; Kieran, M.W. Response to the BRAF/MEK inhibitors dabrafenib/trametinib in an adolescent with a BRAF V600E mutated anaplastic ganglioglioma intolerant to vemurafenib. Pediatr. Blood Cancer. 2018, 65, 26969. [Google Scholar] [CrossRef]
- Touat, M.; Gratieux, J.; Condette Auliac, S.; Sejean, K.; Aldea, S.; Savatovsky, J.; Perkins, G.; Blons, H.; Ligon, K.L.; Idbaih, A.; et al. Vemurafenib and cobimetinib overcome resistance to vemurafenib in BRAF-mutant ganglioglioma. Neurology 2018, 91, 523–525. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef] [Green Version]
- Garnier, L.; Ducray, F.; Verlut, C.; Mihai, M.I.; Cattin, F.; Petit, A.; Curtit, E. Prolonged Response Induced by Single Agent Vemurafenib in a BRAF V600E Spinal Ganglioglioma: A Case Report and Review of the Literature. Front. Oncol. 2019, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, F.; Ceglie, G.; Cacchione, A.; Alessi, I.; Colafati, G.S.; Carai, A.; Diomedi-Camassei, F.; De Billy, E.; Agolini, E.; Mastronuzzi, A.; et al. BRAF V600E Inhibitor (Vemurafenib) for BRAF V600E Mutated Low Grade Gliomas. Front. Oncol. 2018, 8, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobre, L.; Zapotocky, M.; Ramaswamy, V.; Ryall, S.; Bennett, J.; Alderete, D.; Guill, J.B.; Baroni, L.; Bartels, U.; Bavle, A.; et al. Outcomes of BRAF V600E Pediatric Gliomas Treated with Targeted BRAF Inhibition. JCO Precis. Oncol. 2020, 4, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, D.; Ruggiero, A.; Amato, M.; Maurizi, P.; Riccardi, R. BRAF and MEK inhibitors in pediatric glioma: New therapeutic strategies, new toxicities. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Safety of BRAF+MEK Inhibitor Combinations: Severe Adverse Event Evaluation. Cancers 2020, 12, 1650. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- WHO Multicentre Growth Reference Study Group. WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr. Suppl. 2006, 450, 76–85. [Google Scholar] [CrossRef]
- Willett, W.C.; Sacks, F.; Trichopoulou, A.; Drescher, G.; Ferro-Luzzi, A.; Helsing, E.; Trichopoulos, D. Mediterranean diet pyramid: A cultural model for healthy eating. Am. J. Clin. Nutr. 1995, 61, 1402–1406. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: Summary report. Pediatrics 2011, 128, 213–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCI. National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v.5.0. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_5x7.pdf (accessed on 1 April 2022).
- Cohen-Aubart, F.; Guerin, M.; Poupel, L.; Cluzel, P.; Saint-Charles, F.; Charlotte, F.; Arsafi, Y.; Emile, J.F.; Frisdal, E.; Le Goff, C.; et al. Hypoalphalipoproteinemia and BRAFV600E Mutation Are Major Predictors of Aortic Infiltration in the Erdheim-Chester Disease. Arter. Thromb. Vasc. Biol. 2018, 38, 1913–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergès, B.; Walter, T.; Cariou, B. Endocrine side effects of anti-cancer drugs: Effects of anti-cancer targeted therapies on lipid and glucose metabolism. Eur. J. Endocrinol. 2014, 170, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bai, L.; Li, W. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 2020, 8, 605154. [Google Scholar] [CrossRef]
- Stamatakos, S.; Beretta, G.L.; Vergani, E.; Dugo, M.; Corno, C.; Corna, E.; Tinelli, S.; Frigerio, S.; Ciusani, E.; Rodolfo, M.; et al. Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations. Cancers 2021, 13, 2284. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology 2020, 9, 474. [Google Scholar] [CrossRef]
- Hong, X.; Roh, W.; Sullivan, R.J.; Wong, K.H.K.; Wittner, B.S.; Guo, H.; Dubash, T.D.; Sade-Feldman, M.; Wesley, B.; Horwitz, E.; et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discov. 2021, 11, 678–695. [Google Scholar] [CrossRef]
- Valvo, V.; Iesato, A.; Kavanagh, T.R.; Priolo, C.; Zsengeller, Z.; Pontecorvi, A.; Stillman, I.E.; Burke, S.D.; Liu, X.; Nucera, C. Fine-Tuning Lipid Metabolism by Targeting Mitochondria-Associated Acetyl-CoA-Carboxylase 2 in BRAFV600E Papillary Thyroid Carcinoma. Thyroid 2021, 31, 1335–1358. [Google Scholar] [CrossRef]
- Kang, H.B.; Fan, J.; Lin, R.; Elf, S.; Ji, Q.; Zhao, L.; Jin, L.; Seo, J.H.; Shan, C.; Arbiser, J.L.; et al. Metabolic Rewiring by Oncogenic BRAF V600E Links Ketogenesis Pathway to BRAF-MEK1 Signaling. Mol. Cell 2015, 6, 345–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Lin, R.; Jin, L.; Zhao, L.; Kang, H.B.; Pan, Y.; Liu, S.; Qian, G.; Qian, Z.; Konstantakou, E.; et al. Prevention of Dietary-Fat-Fueled Ketogenesis Attenuates BRAF V600E Tumor Growth. Cell Metab. 2017, 25, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Mishra, R.; Patel, H.; Alanazi, S.; Wei, X.; Ma, Z.; Garrett, J.T. BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels. Cancers 2020, 12, 1661. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive Oxygen and Nitrogen Species-Induced Protein Modifications: Implication in Carcinogenesis and Anticancer Therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef] [Green Version]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Innocenzi, D.; Alò, P.L.; Balzani, A.; Sebastiani, V.; Silipo, V.; La Torre, G.; Ricciardi, G.; Bosman, C.; Calvieri, S. Fatty acid synthase expression in melanoma. J. Cutan. Pathol. 2003, 30, 23–28. [Google Scholar] [CrossRef]
- Kapur, P.; Rakheja, D.; Roy, L.C.; Hoang, M.P. Fatty acid synthase expression in cutaneous melanocytic neoplasms. Mod. Pathol. 2005, 18, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 2020, 159, 245–293. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, B.; Lewis, C.A.; Bensaad, K.; Ros, S.; Zhang, Q.; Ferber, E.C.; Konisti, S.; Peck, B.; Miess, H.; East, P. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van den Eynde, K.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 9, 2500. [Google Scholar] [CrossRef]
- Garandeau, D.; Noujarède, J.; Leclerc, J.; Imbert, C.; Garcia, V.; Bats, M.L.; Rambow, F.; Gilhodes, J.; Filleron, T.; Meyer, N.; et al. Targeting the Sphingosine 1-Phosphate Axis Exerts Potent Antitumor Activity in BRAFi-Resistant Melanomas. Mol. Cancer Ther. 2019, 18, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Kotzka, J.; Müller-Wieland, D.; Koponen, A.; Njamen, D.; Kremer, L.; Roth, G.; Munck, M.; Knebel, B.; Krone, W. ADD1/SREBP-1c mediates insulin-induced gene expression linked to the MAP kinase pathway. Biochem. Biophys. Res. Commun. 1998, 249, 375–379. [Google Scholar] [CrossRef]
- Fleischmann, M.; Iynedjian, P.B. Regulation of sterol regulatory-element binding protein 1 gene expression in liver: Role of insulin and protein kinase B/cAkt. Biochem. J. 2000, 349, 13–17. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Vemurafenib Group (n = 6) |
|---|---|
| Sex number: | |
| male/female | 3/3 |
| Age at diagnosis: | |
| mean years ± SD | 5.5 ± 5.6 |
| median | 3.4 (0.3; 13.8) |
| Age at the start of vemurafenib: | |
| mean years ± SD | 8.4 ± 6.1 |
| median | 7.1 (2.8; 18.8) |
| Tumor site: | |
| hypothalamic/chiasmatic | 4 |
| basal ganglia | 1 |
| spinal cord | 1 |
| Tumor histology: | |
| ganglioglioma | 4 |
| pilocytic astrocytoma | 2 |
| Previous treatment: | |
| surgery or biopsy only | 1 |
| one line of chemotherapy | 2 |
| two lines of chemotherapy | 1 |
| ≥three lines of chemotherapy | 1 |
| ≥three lines of chemotherapy + RT | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crocco, M.; Verrico, A.; Milanaccio, C.; Piccolo, G.; De Marco, P.; Gaggero, G.; Iurilli, V.; Di Profio, S.; Malerba, F.; Panciroli, M.; et al. Dyslipidemia in Children Treated with a BRAF Inhibitor for Low-Grade Gliomas: A New Side Effect? Cancers 2022, 14, 2693. https://doi.org/10.3390/cancers14112693
Crocco M, Verrico A, Milanaccio C, Piccolo G, De Marco P, Gaggero G, Iurilli V, Di Profio S, Malerba F, Panciroli M, et al. Dyslipidemia in Children Treated with a BRAF Inhibitor for Low-Grade Gliomas: A New Side Effect? Cancers. 2022; 14(11):2693. https://doi.org/10.3390/cancers14112693
Chicago/Turabian StyleCrocco, Marco, Antonio Verrico, Claudia Milanaccio, Gianluca Piccolo, Patrizia De Marco, Gabriele Gaggero, Valentina Iurilli, Sonia Di Profio, Federica Malerba, Marta Panciroli, and et al. 2022. "Dyslipidemia in Children Treated with a BRAF Inhibitor for Low-Grade Gliomas: A New Side Effect?" Cancers 14, no. 11: 2693. https://doi.org/10.3390/cancers14112693
APA StyleCrocco, M., Verrico, A., Milanaccio, C., Piccolo, G., De Marco, P., Gaggero, G., Iurilli, V., Di Profio, S., Malerba, F., Panciroli, M., Giordano, P., Calevo, M. G., Casalini, E., Di Iorgi, N., & Garrè, M. L. (2022). Dyslipidemia in Children Treated with a BRAF Inhibitor for Low-Grade Gliomas: A New Side Effect? Cancers, 14(11), 2693. https://doi.org/10.3390/cancers14112693