1. Introduction
The voltage-gated potassium channel K
V1.3 is a transmembrane protein belonging to the K
V1 (Shaker) subfamily, which is the largest subfamily of voltage-gated potassium channels with eight members (K
V1.1–K
V1.8). K
V1.3 is formed by four monomers, each containing six transmembrane segments (S1–S6) [
1]. The S5–S6 domain of each monomer assembles to form the K
+ conducting pore that is surrounded by four voltage sensor domains (VSD), each consisting of segments S1 to S4. The VSDs detect depolarization of the membrane and initiate conformational changes that lead to the opening of the pore. The selectivity filter in the K
+ conducting pore consists of oxygen cages that mimic the hydration shell of potassium in solution and allow the rapid and selective passage of potassium ions [
2,
3,
4].
The K
V1.3 channel has only recently emerged as a molecular target in cancer therapy. Cancer cells display mutations enabling extensive proliferation and resistance to apoptosis. K
V1.3 channel activity has been found to be involved in cell proliferation, migration, and invasion, which are among the most important processes in cancer progression. Moreover, overexpression of K
V1.3 enhances tumorigenic processes and promotes tumor progression [
5,
6,
7,
8]. The use of small-molecule K
V1.3 inhibitors could selectively suppress the proliferation of cancer cells, thus providing a new potential therapeutic approach [
9]. Although K
V1.3 has been recognized as a tumor marker in cancer tissues with predominantly higher K
V1.3 expression, a clear pattern of altered K
V1.3 expression in cancer cells compared with healthy cells has not yet been found. The type and stage of disease also influence K
V1.3 expression. Up-regulated K
V1.3 expression was detected in breast, colon, and prostate tumors, in smooth muscle and skeletal muscle cancers, and in mature neoplastic B cells in chronic lymphocytic leukemia [
10].
To date, several small-molecule K
V1.3 inhibitors have been discovered (
Figure 1) [
11,
12,
13]. Originally, K
V1.3 inhibitors were extensively studied to selectively target the proliferation of effector memory T (T
EM) cells in the immune system and to develop a new therapy for T-cell—mediated autoimmune and chronic inflammatory diseases [
14]. However, none of these compounds have been optimised to have suitable properties for clinical trials. Since K
V1 family channels have high subtype homology, it is very difficult to find a potent isoform-selective K
V1.3 inhibitor [
15]. For most of the described compounds, only low or moderate selectivity towards other Kv1.x channels was achieved, preventing subsequent optimization. Cryogenic electron microscopy (cryoEM) was recently used to determine the structures of human K
V1.3 alone and bound to dalazatide, which may aid the structure-based design of new inhibitors in the future. To date, the exact binding site for most known K
V1.3 inhibitors has not been experimentally demonstrated, and these new cryoEM structures may help to solve this problem [
16,
17].
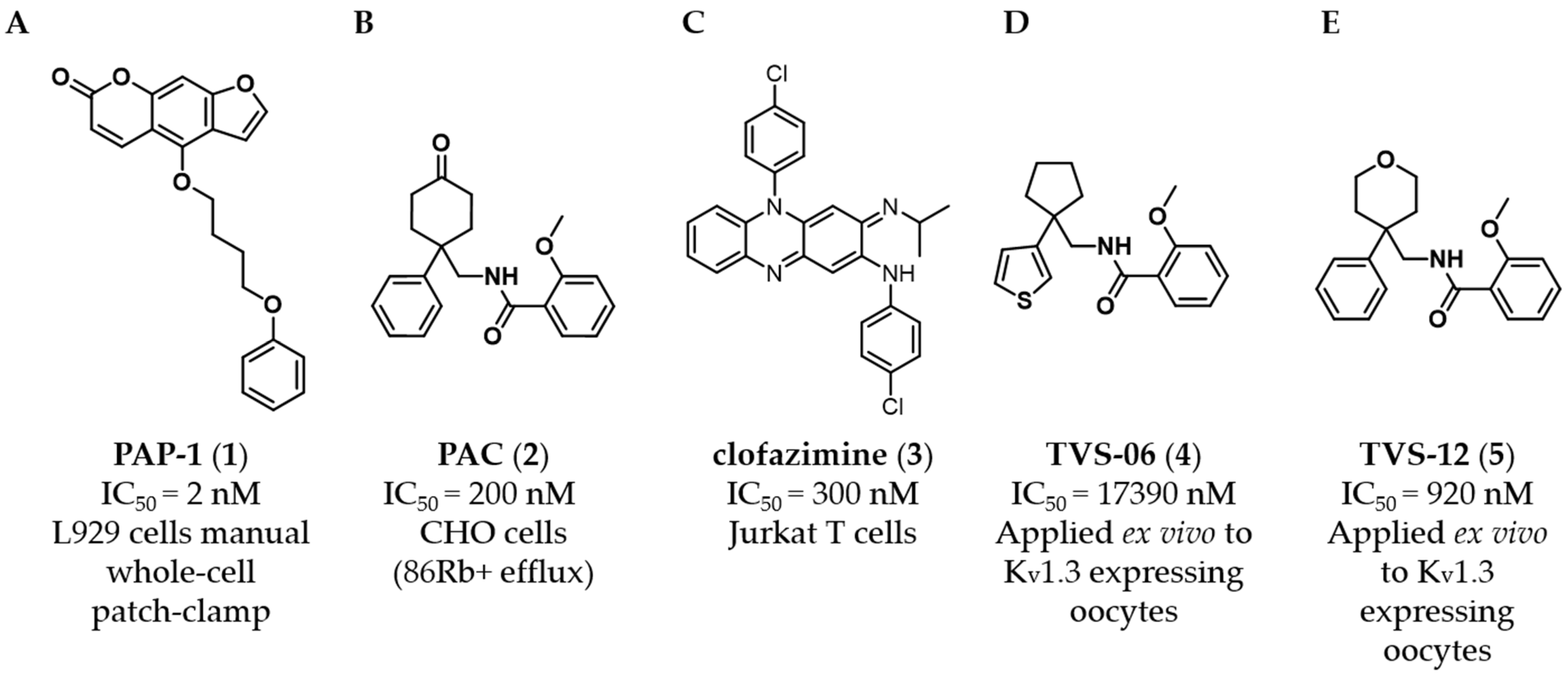
The psoralen derivative PAP-1 (
1,
Figure 1A) is the most potent (IC
50 = 2 nM, L929 cells, manual whole-cell patch clamp) and selective (i.e., 23-fold over K
V1.5) small-molecule K
V1.3 inhibitor known [
11]. Benzamide PAC (
2,
Figure 1B), which inhibits K
V1.3 with an IC
50 of 200 nM (CHO cells,
86Rb
+ efflux), is not selective toward K
V1.x family channels [
12]. The antimycobacterial drug clofazimine (
3,
Figure 1C,) is also a K
V1.3 inhibitor (IC
50 = 300 nM, Jurkat T cells), inhibiting K
V1.3 with tenfold higher potency than K
V1.1, K
V1.2, K
V1.5 and K
V3.1 [
13]. PAP-1 (
1) at a concentration of 20 µM, and clofazimine (
3) at a concentration of 1 or 10 µM in combination with the multi-drug resistance (MDR) pump inhibitors (MDRi) cyclosporine H (4 µM) and probenecid (100 µM), which induced mild apoptosis in cancer cells (human SAOS2, mouse B16F10 melanoma, and human B-CLL) by specifically inhibiting K
V1.3. Similarly, clofazimine (
3) induced significant apoptosis in several pancreatic ductal adenocarcinoma (PDAC) cell lines with EC
50 values in the micromolar range and significantly reduced primary tumor weight in an orthotopic PDAC xenotransplantation model in SCID beige mice [
18,
19,
20].
In previous work, we identified two new K
V1.3 inhibitors by virtual screening based on a 3D similarity search using the K
V1.3 inhibitor PAC (
2,
Figure 1B) as a query [
21]. Hit compound
4 (
Figure 1D), with an IC
50 of 17.4 µM, was a much weaker K
V1.3 inhibitor than hit compound
5 (
Figure 1E), with an IC
50 of 920 nM (applied ex vivo in
Xenopus oocytes expressing K
V1.3). However, Kv1.3 inhibitor
4 exhibited a good selectivity profile and did not affect other K
V1.x family channels or more distantly related channels. In this paper, we describe the optimization of new thiophene-based Kv1.3 inhibitors, their selectivity profile against K
V1.x and some other selected ion channels, and the potential of this new class of Kv1.3 inhibitors for antiproliferative activity in 2D and 3D cell models.
2. Materials and Methods
2.1. Oocyte Collection, Oocyte Injection, Drug Solutions, Electrophysiological Recordings, and Statistical Analysis in Xenopus Laevis Oocytes
Stage V–VI oocytes [
21] were isolated by partial ovariectomy from
Xenopus laevis frogs (African clawed frogs). Mature female frogs were purchased from CRB Xénopes (Rennes, France) and were housed in the Aquatic Facility (KU Leuven) in compliance with the regulations of the European Union (EU) concerning the welfare of laboratory animals as declared in Directive 2010/63/EU. The use of
Xenopus laevis was approved by the Animal Ethics Committee of the KU Leuven (Project nr. P186/2019). After anaesthetizing the frogs by a 15-min submersion in 0.1% tricaine methanesulfonate (amino benzoic acid ethyl ester; Merck, Kenilworth, NJ, USA), pH 7.0, the oocytes were collected. The isolated oocytes were then washed with a 1.5 mg/mL collagenase solution for 2 h to remove the follicle layer.
Ion channels (Kv1.x, Kv2.1, Kv4.2, Kv10.1) were expressed in Xenopus laevis oocytes by linearization of the plasmids and subsequent in vitro transcription using a commercial T7 or SP6 mMESSAGE mMACHINE transcription kit (Ambion, Carlsbad, California, USA). Defolliculated Xenopus oocytes were then injected with 20–50 nL of the cRNA at a concentration of 1 ng/nL using a micro-injector (Drummond Scientific1, Broomall, PA, USA). The oocytes were incubated in a solution containing (in mM): NaCl, 96; KCl, 2; CaCl2, 1.8; MgCl2, 2 and HEPES, 5 (pH 7.5), supplemented with 50 mg/L gentamycin sulfate and 90 mg/L theophylline. After ex vivo translation, the ion channels were correctly inserted in the cell membrane of the oocytes.
Two-electrode voltage-clamp recordings were performed at room temperature (18–22 °C) using a Geneclamp 500 amplifier (Molecular Devices, San Jose, CA, USA) and pClamp data acquisition (Axon Instruments, Union City, CA, USA) and using an integrated digital TEVC amplifier controlled by HiClamp, an automated Voltage-Clamp Screening System (Multi Channel Systems MCS GmbH, Reutlingen, Germany). Whole-cell currents from oocytes were recorded 1–4 days after injection. The bath solution composition was ND96 (in mM): NaCl, 96; KCl, 2; CaCl
2, 1.8; MgCl
2, 2 and HEPES, 5 (pH 7.5). Voltage and current electrodes were filled with a 3 M solution of KCl in H
2O. Resistances of both electrodes were kept between 0.5 and 1.5 MΩ. The elicited Kv1.x, Kv2.1, Kv4.2 and Kv10.1 currents were filtered at 0.5 kHz and sampled at 2 kHz using a four-pole low-pass Bessel filter. Leak subtraction was performed using a P/4 protocol [
21].
For the electrophysiological analysis of the compounds, a number of protocols were applied from a holding potential of −90 mV. Currents for Kv1.x, Kv2.1, Kv4.2 and Kv10.1 were evoked by 1 s depolarizing pulses either to 0 mV or to a range of voltage steps between −80 mV and +40 mV. For the analysis of the data, only the inhibition at the 0 mV step was used. The concentration dependency of all compounds was assessed by measuring the current inhibition in the presence of increasing compound concentrations. To this end, a stock solution of the compounds was prepared in 100% DMSO for the sake of solubility. From this stock solution, adequate dilutions with a maximum of 0.5% DMSO were made for testing. The data of the concentration-response curves were fitted with the Hill equation: y = 100/{1 + (IC50/[compound])h}, where y is the amplitude of the compound-induced effect, IC50 is the compound concentration at half-maximal efficacy, [compound] is the compound concentration, and h is the Hill coefficient.
All electrophysiological data (
Figure S1) are presented as means ± S.E.M of n ≥ three independent experiments unless otherwise indicated. All data were analyzed using pClamp Clampfit 10.4 (Molecular Devices, Downingtown, PA, USA), OriginPro 9 (Originlab, Northampton, MA, USA), GraphPad Prism 8 software (GraphPad Software, Inc., San Diego, CA, USA) and DataMining (Multi Channel Systems MCS GmbH, Reutlingen, Germany). The Dunnett test and one-way ANOVA were performed to calculate the significance of the induced inhibition compared to the control.
2.2. Cell Culture, Transfection, Drug Solutions, Electrophysiological Recordings, and Statistical Analysis in Ltk− Cells
Ltk− cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% horse serum and 1% penicillin/streptomycin (Invitrogen, Waltham, CA, USA). Human hKV1.3 channels were transiently expressed in these Ltk− cells by transfecting subconfluent 60 × 15 mm cell culture dishes (Corning, NY, USA) with 1-1.5 µg plasmid DNA containing the hKv1.3 sequence (KCNA3) using lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. The coding sequence of hKv1.3 was cloned in a pEGFP plasmid without removing the stop codon (i.e., eGFP was not transcribed). Therefore, during transfection 0.5 µg of pEGFP plasmid (expressing eGFP) was added as transfection marker Cells were collected 24h post-transfection using a 0.05% trypsin-EDTA solution (Thermo Fisher Scientific, Waltham, MA, USA) and transferred to the recording chamber mounted on the stage of an inverted Nikon Eclipse TE2000 fluorescence microscope (Nikon, Minato, Japan). The compounds were dissolved in 100% DMSO and stock solutions were stored at -20°C. Before use, the stock concentrations were diluted with extracellular recording solution to appropriate concentrations, making sure that the final DMSO concentration never exceeded 0.1%. A 0.1% DMSO solution was prepared as a vehicle control.
All recordings were conducted at room temperature (20–23 °C) in the whole-cell configuration using an axopatch 200b amplifier (Molecular Devices, San Jose, CA, USA). Applied voltage pulse protocols and current recordings were controlled with pClamp 10 software and digitized using an axon digidata 1440 (Molecular Devices, San Jose, CA, USA). The cells in the recording chamber were continuously superfused with an extracellular solution (containing in mM): NaCl 145, KCl 4, MgCl
2 1, CaCl
2 1.8, HEPES 10, and glucose 10 (adjusted to pH 7.35 with NaOH). Patch pipettes were pulled from borosilicate glass (World Precision Instruments, Sarasota, FL, USA), using a P-2000 puller (Sutter Instruments, Novato, CA, USA), with resistances ranging from 1.5–2 MΩ. These pipettes were backfilled with an intracellular solution (containing in mM): KCl 110, K
2ATP 5, MgCl
2 2, HEPES 10, and K
4BAPTA 5 (adjusted to pH 7.2 with KOH). Junction potentials were zeroed with the filled pipette in the extracellular solution of the recording chamber. A series resistance compensation of 80% was employed. Recordings were passed through a 5 kHz low-pass filter while being sampled at 10 kHz. A single step from holding potential (−80 mV) to +40 mV was utilized to monitor K
V1.3 current inhibition. The pulse duration was 200 ms with an interpulse interval of 15 s. As PAP-1 blocks K
V1.3 in a use-dependent manner, by preferentially binding to the channels C-type inactivated state, the protocol was slightly modified: the pulse duration and interval between pulses were increased to 2 s and 30 s, respectively [
11]. A longer pulse duration maximizes the number of channels in their inactivated state. Different concentrations of the compounds were independently added to the recording chamber in the vicinity of the cell investigated using a pressurized fast-perfusion system (custom built with electro-fluidic valves from the Lee Company, Westbrook, CT, USA).
The data was analyzed with pClamp 10 software (Molecular Devices, San Jose, CA, USA), and the dose-response curves shown were made using Sigmaplot 11.0 (Systat software, Palo Alto, CA USA). Data was excluded when the estimated voltage error exceeded 5mV after series resistance compensation. Dose-response curves were obtained by plotting y, the normalized current, as a function of compound concentration. Results are expressed as mean ± SEM, with n being the number of cells analyzed. EC50 values were determined by fitting the Hill equation to the dose-response curve.
2.3. Cell Culture for Apoptosis and Proliferation Assays
Cell lines Panc-1 (ACC 783) and Colo-357 (CVCL_0221) were obtained through the validated collection of the pHioniC consortium (MSC-ITN 813834). Both cell lines were grown in RPMI1640, supplemented with 1 mM sodium pyruvate and 1% Glutamax (all Gibco Thermo-Fisher, Waltham, MA USA) 10%FBS (BioChrom, Berlin, Germany), at 5% CO2 and 37 °C in a humidified atmosphere.
2.4. Apoptosis and Proliferation Assays
For cell proliferation assays, cells were seeded at a density of 10,000 cells/well in 96-well flat bottom culture plates (Corning, Kaiseslautern, Germany) and proliferation was measured through culture confluency using an IncuCyte device (Sartorius, Göttingen, Germany). Every hour, two phase contrast images per well were acquired, and a confluency mask was generated by training the analysis algorithm using representative images. Every image was then analyzed using the obtained parameters to determine culture confluency. Treatments were then added when cells reached a confluency of ~30% (Panc-1) or ~55% (Colo-357), and proliferation was determined for the following 60 h. Proliferation is reported as confluency increase with respect to the start of the treatment.
Spheroids were cultured in round bottom ultra-low attachment 96-well plates (Corning). The optimal seeding densities were empirically determined (8000 cells/well for Colo-357 or 10,000 cells/well for Panc-1 spheroids. The cells were suspended in 2% Matrigel (Corning), centrifuged at 1000× g for 10 min and the spheroids were allowed to form in the incubator. Once the spheroids were formed, the treatments indicated together with 8.9 µM cycloheximide (as apoptosis sensitizer) were added and apoptosis was determined by live imaging in the Incucyte system in real time for approximately 60 h using Caspase-3/7 green reagent (Sartorius) as a reporter of apoptosis, which crosses the cell membrane and is cleaved by the activated Caspase-3/7, resulting in the fluorescent staining of nuclear DNA. Apoptosis on Panc-1 tumor spheroids is presented as integrated green fluorescence in the whole spheroid.
2.5. Ligand-Based Pharmacophore Modeling
Nine K
V1.3 inhibitors with inhibition greater than 75% at 10 µM were used as a training set for the generation of ten pharmacophore models in LigandScout 4.4 Expert (Inte:Ligand GmbH, Vienna, Austria) [
22]. For each inhibitor, a maximum of 200 conformations were computed using iCon algorithm in LigandScout [
23] with default “BEST” settings. Ten ligand-based pharmacophore models were created using the default settings. For each of the models, the creation of exclusion volumes coat around the alignment of the ligands was enabled. For the interaction analyses and image preparation, the highest ranked ligand-based pharmacophore model was used.
2.6. Turbidimetric Solubility Assay
For the phosphate-buffered saline, we dissolved 2.38 g of disodium hydrogen phosphate dodecahydrate, 0.19 g of potassium dihydrogen phosphate, and 8.0 g of sodium chloride in water and diluted it to 1000 mL with the same solvent. Then we adjusted the pH to 7.4. The stock DMSO solutions of the compounds were prepared at concentrations of 50 mM, 20 mM, 10 mM, and 1 mM. The final solutions (500 µM, 200 µM, 100 µM, and 10 µM) were prepared by adding 2 µL of the stock DMSO solution to 198 µL of phosphate-buffered saline. The absorbance (area scan) was measured at 620 nm for sample volume of 200 µL and the appearance of the solutions was checked by visual inspection.
4. Discussion
With the structure-activity relationship studies of novel structural classes K
V1.3 inhibitors, we identified structural parts that are important for K
V1.3 inhibition. With the structural optimization of hit compounds
4 and
5 we developed new K
V1.3 inhibitors (e.g.,
14,
43, and
44) with improved potency compared to hit compounds. To explain the structural requirements for K
V1.3 inhibition, we created a pharmacophore model of the new K
V1.3 inhibitors in Ligandscout 4.4 Expert (Inte:Ligand GmbH) using inhibitors that achieved K
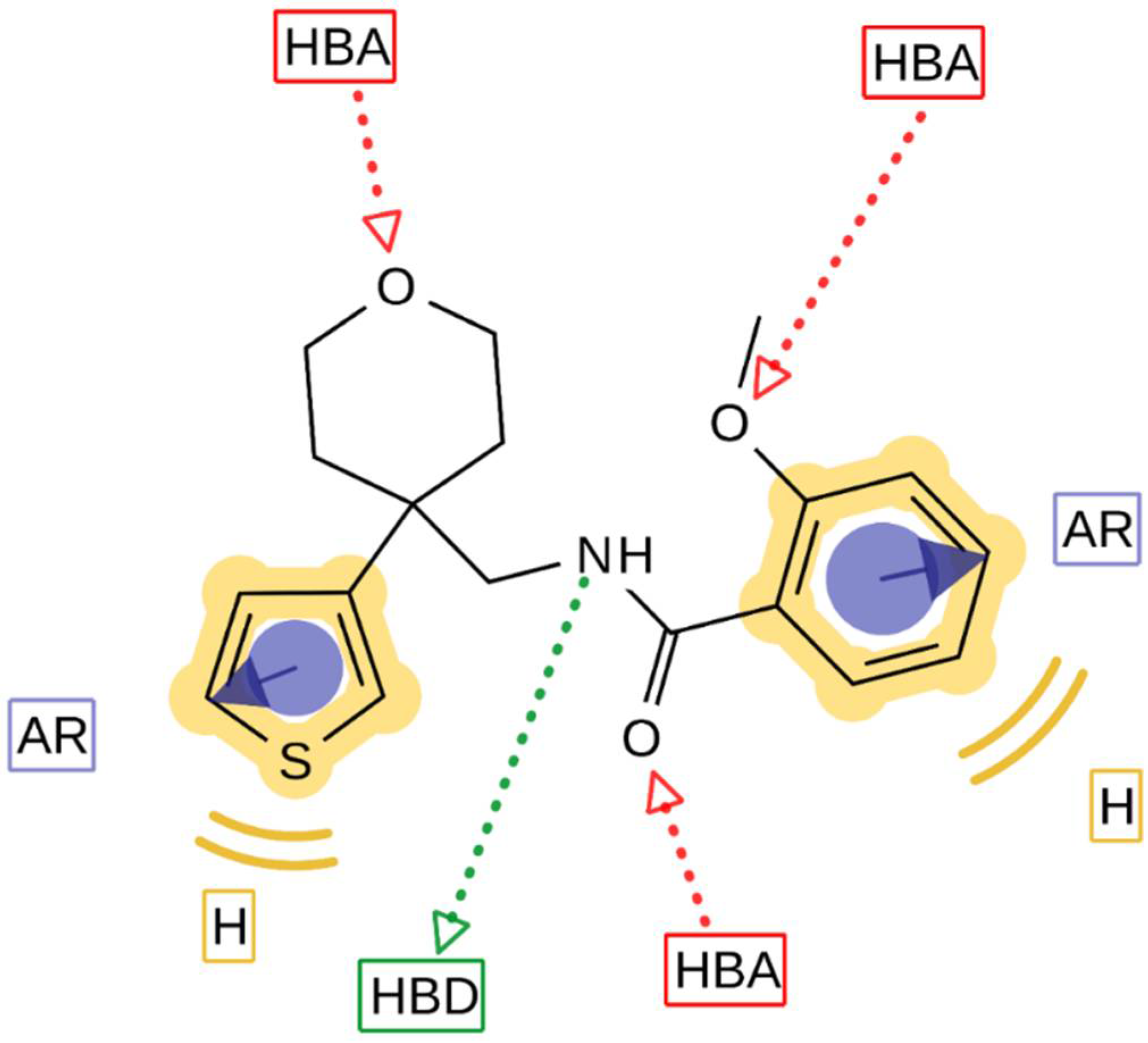
V1.3 inhibition greater than 75% at a concentration of 10 µM. The ligand-based pharmacophore model (
Figure 7) is presented with the structure of the most potent K
V1.3 inhibitor
44. Structural elements in the most potent analogs that were required for high-affinity interaction with K
V1.3 were the amide bond (hydrogen bond acceptor, HBA, hydrogen bond donor, HBD), two aromatic features, oxygen in the 2-methoxy group as HBA, and oxygen in the rings of tetrahydrofuran (Type I) or tetrahydropyran (Type III-VI) rings as HBA. 2-Methoxybenzamide was present in the first aromatic feature (Types I, III and FThe second aromatic feature of most potent inhibitors included 3-substituted (Type III,
28 and
29) or unsubstituted benzene (Type IV,
36,
37), unsubstituted 2-thiophene (Type I,
14 and Type V,
43) or unsubstituted 3-thiophene (Type V,
44 and Type VI,
51).
Several known small molecule K
V1.3 inhibitors lack selectivity for K
V1.3 over the closely related K
V1.x family channels, which have high subtype homology. The lack of selectivity for K
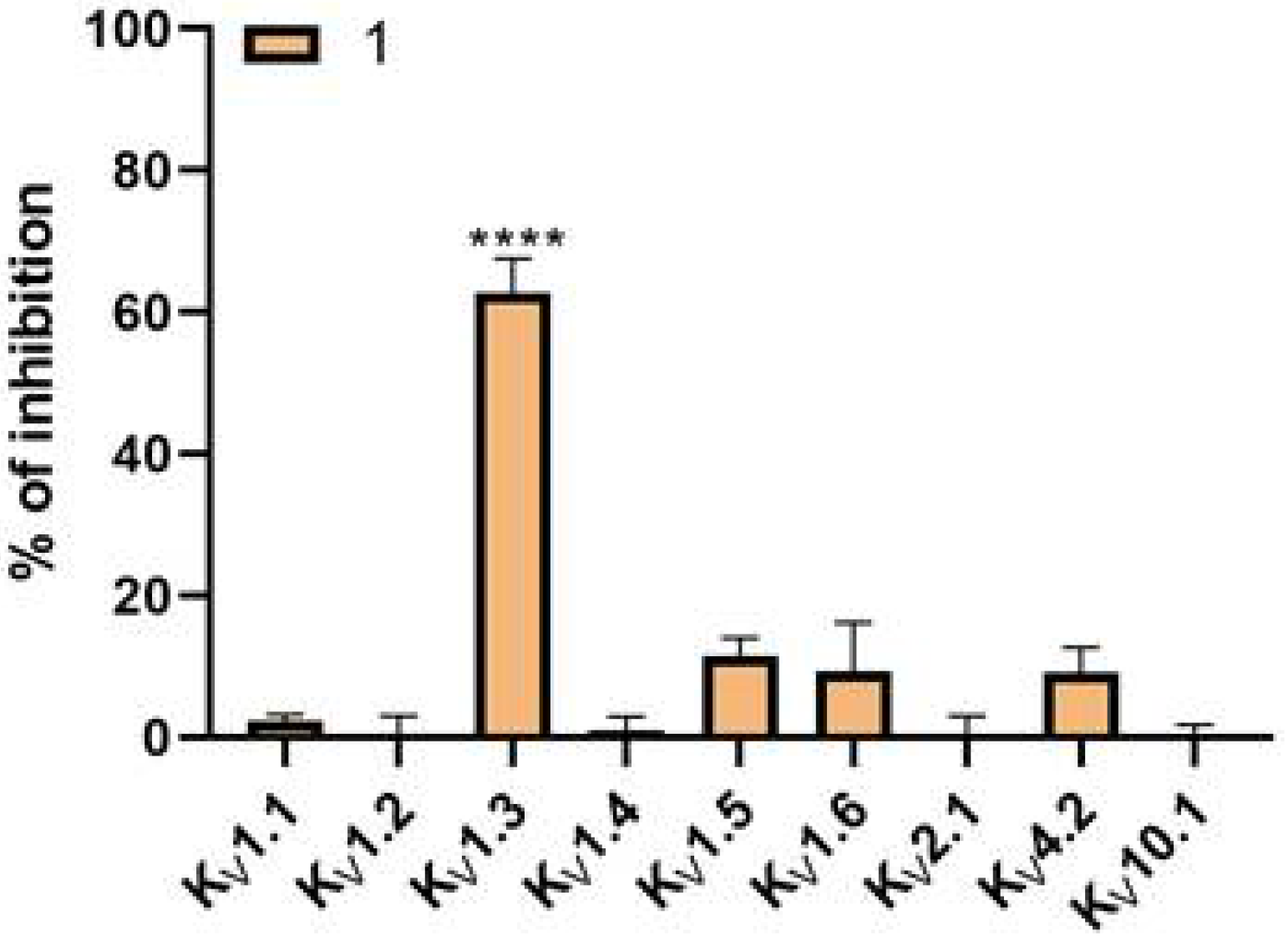
V1.5 raises many concerns regarding potential acute cardiac toxicity. Obtaining a selective small molecule inhibitor remains a major challenge, and often the lack of selectivity prevents subsequent optimization. Based on literature data, PAP-1 (
Figure 1,
1) is selective toward K
V1 channels, whereas it has the lowest selectivity toward the K
V1.5 channel (i.e., 23-fold over K
V1.5). We also included into our testing the reference compound PAP-1 to have a direct comparison of potency and selectivity with newly designed compounds. We determined IC
50 values for PAP-1 using three independent methods: manual voltage clamp on oocytes (IC
50 = 0.78 ± 0.01 µM), HiClamp system on oocytes (IC
50 = 2.67 ± 0.30 µM), and manual patch clamp on Ltk
− cells (IC
50 = 0.0004 ± 0.00002 µM). Surprisingly, the IC
50 values determined for PAP-1 on oocytes were approximately 390- to 1335-fold higher than the literature IC
50 value of 2 nM (L929 cells, manual whole-cell patch-clamp) [
24]. Regarding selectivity, the PAP-1 tested at a concentration of 10 µM showed no significant effects on channels K
V1.1, K
V1.2, K
V1.4, K
V1.5, K
V1.6, K
V2.1, K
V4.2, and K
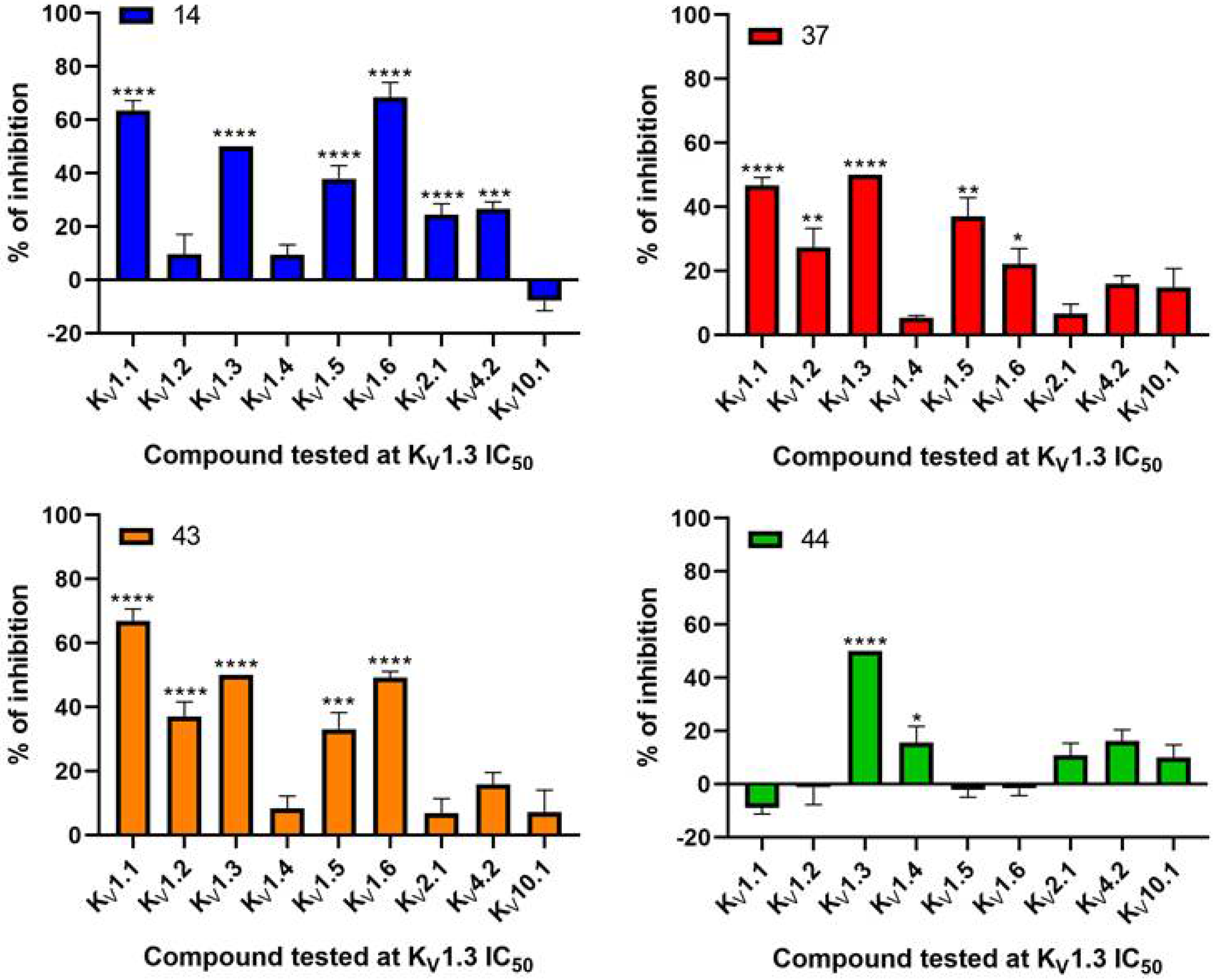
V10.1 using the HiClamp system on oocytes.
Comparing the IC
50 values in
Table 3, we can see some differences between the different test systems can be seen. The IC
50 values determined based on oocytes using the manual and HiClamp methods are in the same order of magnitude (middle-nanomolar to low-micromolar range), but the IC
50 values determined based on mammalian cells are in the low-nanomolar range. These differences can be attributed to several factors:
First, both mammalian and amphibian cells were used, which differ in size, composition, ion channel expression, and permeability to compounds [
25].
Second, when comparing the two different methods used on oocytes (manual voltage clamp and HiClamp), it seems that IC50 values are generally slightly higher for the HiClamp method than for the two manual techniques. This may be due to differences in perfusion between the three systems. In the manual setup, oocytes are manually impaled in the recording chamber with two electrodes, and the test solution is either externally applied to the bath filled with ND96 (in the case of the manual voltage-clamp experiments) or applied by continuous extracellular perfusion using a pressurized fast-perfusion system (in the case of the manual patch-clamp experiments), while KV1.3 currents are measured. The HiClamp, on the other hand, is a semi-automatic system, in which the oocyte is picked up from a 96-well plate, deposited in a basket and automatically impaled by two electrodes in the washing chamber. Next, the basket will submerge the oocyte in the test solution in another 96-well plate while KV1.3 currents are measured. In this plate, magnets are continuously stirring the test solutions to assure homogenous perfusion. The higher IC50s observed at the HiClamp could be due to adhesion of the compound to the walls of the 96-well plate or because of the continuous stirring by the magnet, which is not present in the two other experimental setups.
Third, the voltage protocols used are slightly different. In the voltage-clamp experiments, depolarizing pulses of 1 s duration were used at intervals of 1 s, whereas in the patch-clamp experiments, depolarizing pulses of 200 ms duration were applied at intervals of 15 s. The voltage protocol used in the patch-clamp experiments is slightly different. The voltage protocol can affect the IC
50 value depending on the state dependence and kinetics of the compound [
26,
27]. If the compound prefers to bind to a certain state of the channel and the voltage-protocol favors this state, then the observed IC
50 will be lower compared to when a voltage-protocol that does not favor this state is used. Similarly, lower affinities will be observed if compounds with slow binding kinetics are measured in a voltage-protocol with a short occupancy period of the open or inactivated state, and vice-versa [
28].
Because the solubility of the new drug candidates in aqueous media is relatively low in many cases, we performed a turbidimetric solubility test (
Table S1) in phosphate-buffered saline (with a final 1% DMSO concentration) for compounds
37,
43, and
44. Compounds
43 and
44 were soluble at a concentration of 500 µM and compound
37 was soluble at a concentration of 200 µM. For the determination of IC
50, the concentrations indicated on the x-axis in logarithmic scale were 0.01 µM, 0.1 µM, 1 µM, 10 µM, and 100 µM. At these concentrations, compounds
37,
43 and
44 were soluble, therefore the IC
50 values determined are not affected by the solubility of these compounds.
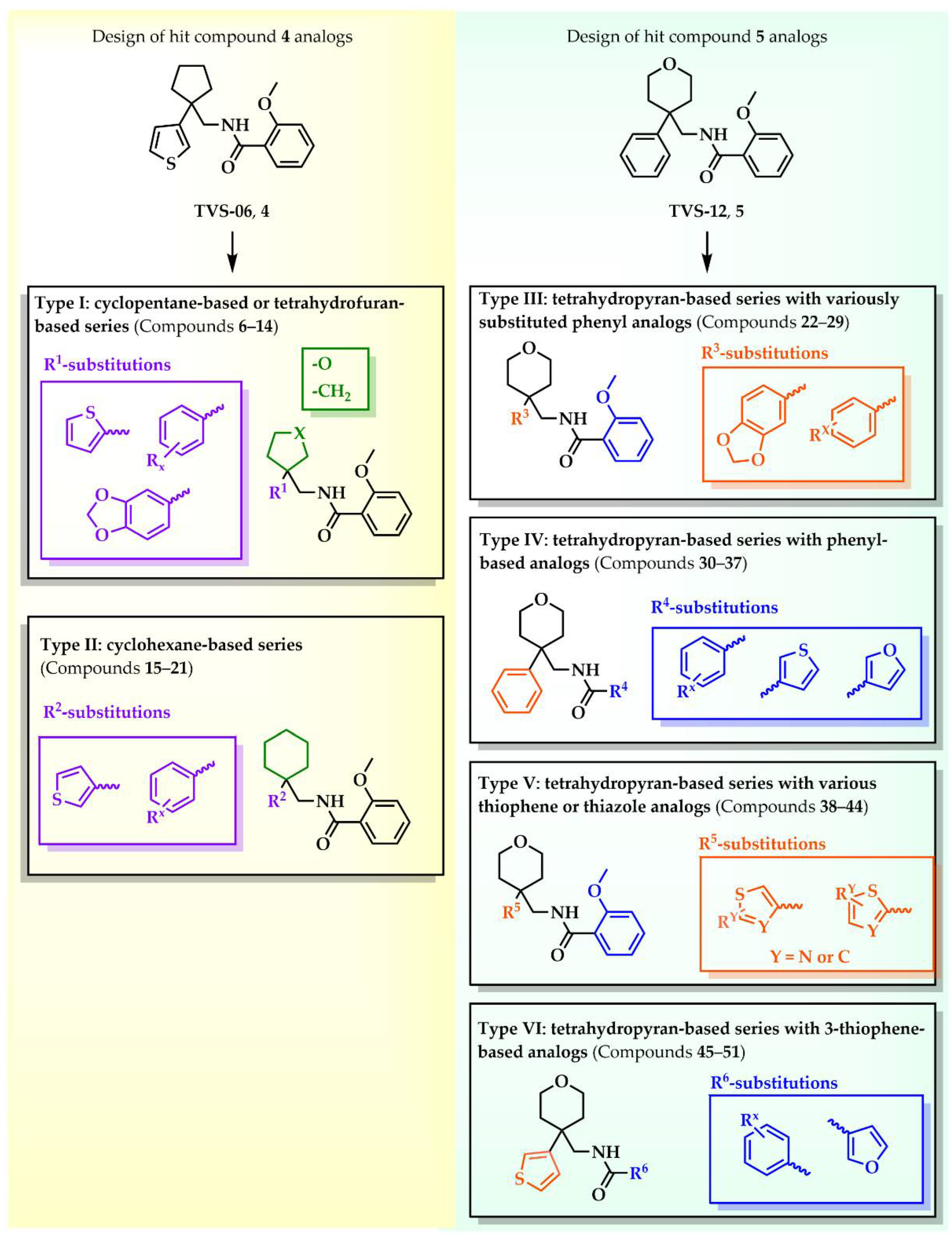
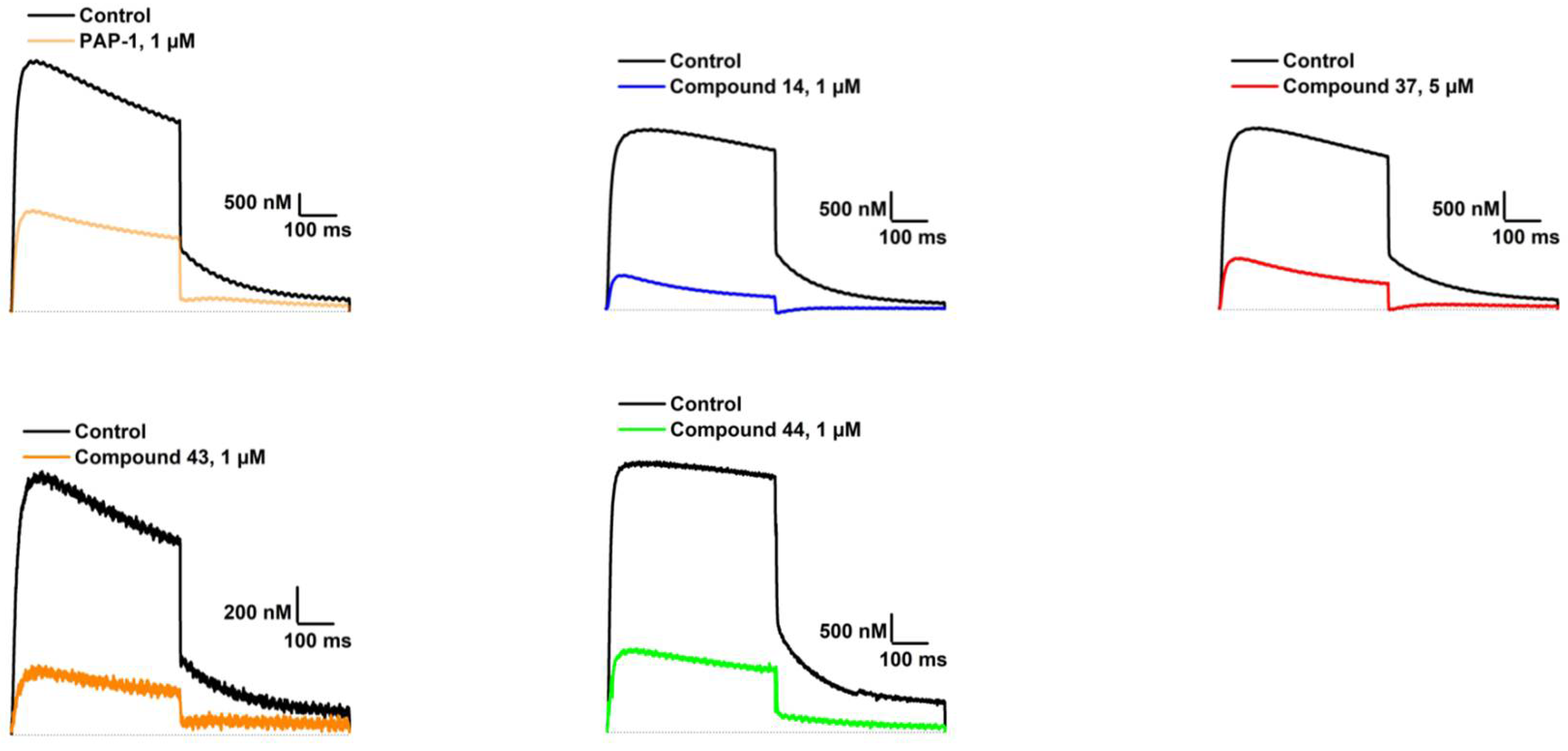
Our aim was to design novel potent and selective KV1.3 inhibitors based on previously determined hit compounds 4 and 5. Based on selectivity data for hit compounds 4 and 5, we hypothesized that the 3-thiophene scaffold and/or the 2-methoxybenzamide moiety together might be responsible for the high selectivity of hit compound 4. To test this hypothesis, we designed six types of novel analogs (Types I-VI) that incorporated 3-thiophene (Types V and VI), 2-thiophene (Types I and V), or benzene (Types III or IV) as scaffolds in the amine portion of the molecules. Compounds 43 (Type I) and 44 (Type V) had very similar affinity for KV1.3 (IC50 = 0.59 ± 0.15 µM and 0.47 ± 0.02 µM, respectively), but interestingly, the 2- or 3-thiophene position leads to a completely different selectivity profile. Among the new KV1.3 inhibitors, 44 (Type V) with 3-thiophene scaffold and 2-methoxybenzamide moiety showed the best selectivity profile showing no significant inhibition of KV1.x channels, except it inhibited KV1.4 with IC50 value of 8.48 ± 2.21 µM. In contrast, inhibitors 14, and 43 with 2-thiophene scaffold and 2-methoxy benzamide moiety lacked selectivity toward KV1.1, KV1.2, KV1.5, and KV1.6. It appears that both the 3-thiophene scaffold and the 2-methoxybenzamide moiety are responsible for the high selectivity of hit compound 4 and the novel benzamide analog 44, but the new structural elements of tetrahydropyran (37, 43 and 44) and tetrahydrofuran (14) allow higher potency for KV1.3.
K
V1.3 activity is required for cell proliferation, migration, and invasion, which are very important events in cancer progression [
9]. To date, there are several lines of evidence that cell-permeable inhibitors of K
V1.3, PAP-1 (
1) and clofazimine (
3), specifically target K
V1.3 to mediate apoptosis. However, PAP-1-based mitochondria-targeted derivatives (PAPTP and PCARBTP) bound to the lipophilic triphenylphosphonium cation (TPP) were found to be very effective in several cancer models, including pancreatic ductal adenocarcinoma, melanoma, and glioblastoma [
30,
31]. PAPTP and PCARBTP were able to decrease the cell survival with EC
50 of approximately 3 and 6 µM in Panc-1 cell line. The decrease in MTS levels when 10 µM PAPTP or PCARBTP was used was due to the apoptosis of Panc-1, while clofazimine only induced cell death of less than 30% even at a concentration of 20 µM. On the other hand, cell death of Colo-357 was achieved only after treatment with high concentrations of PAPTP, PCARBTP, and clofazimine (EC
50s of 3.7, 2, and 1.5 µM, respectively) [
31].
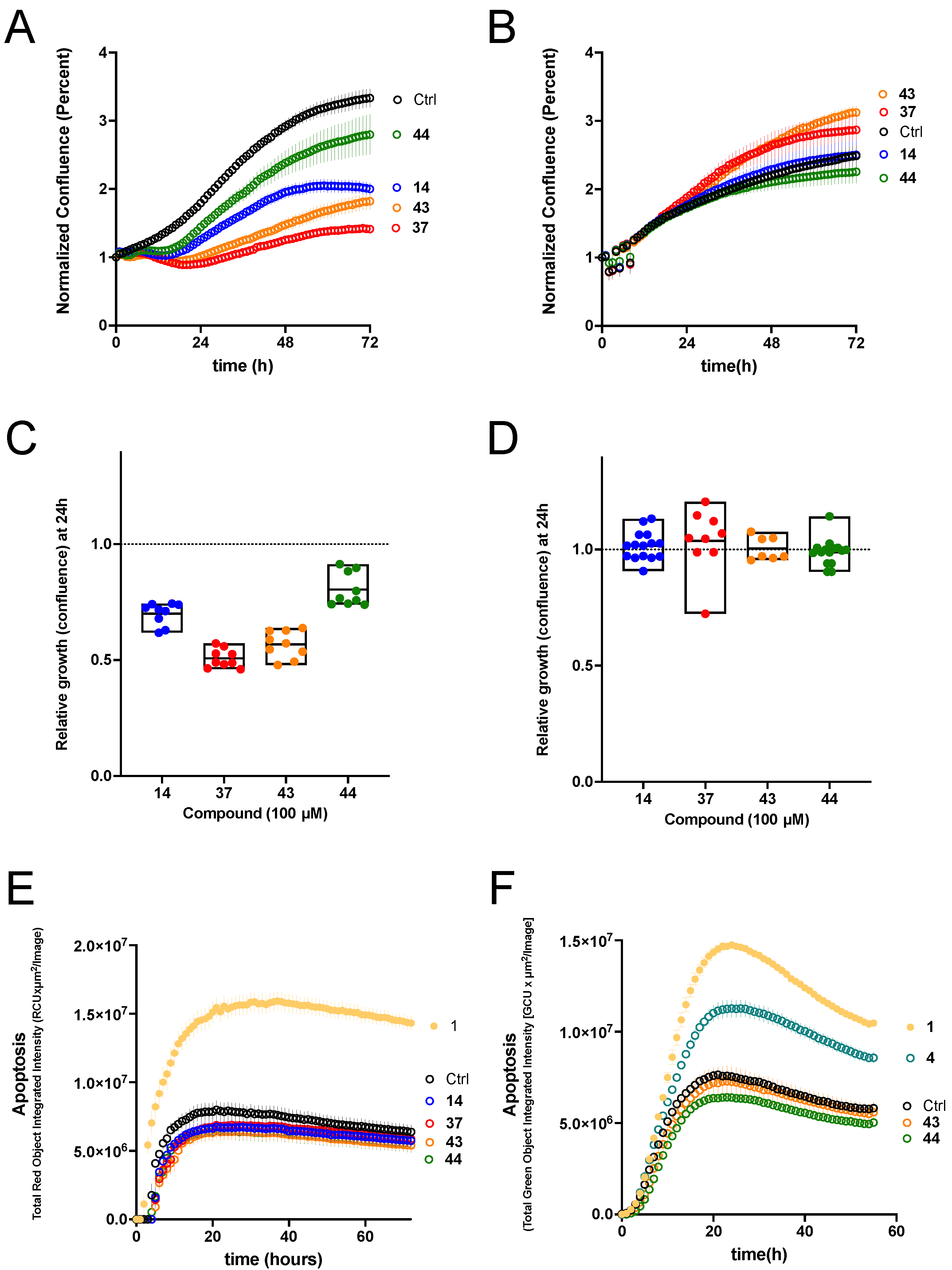
We tested the ability of our new potent K
V1.3 inhibitors to inhibit the proliferation of two K
V1.3-expressing PDAC cell lines, Colo-357 (metastatic) and Panc-1 (from the pancreatic duct). Growth of Panc-1 cells was inhibited by K
V1.3 inhibitors
14,
37,
43, and
44, with
37 in particular causing strong inhibition. Colo-357 growth was unaffected in the presence of these compounds. The fact that Colo-357 cells were unaffected, together with the modest effects observed in Panc-1, indicate that plasma membrane K
V1.3 is dispensable for the proliferation of these cell types. The discrepancy between cell lines can be attributed to the selectivity profiles of the compounds, since K
V1.1 and K
V1.5 have also been implicated in cell proliferation (see for example REF) [
32] and are targets for compounds
14,
37 and
43, while the most selective of the new compounds (
44) had the smallest effect. In Colo-357 tumor spheroids, starting hit compound
4 induced a significant amount of apoptosis, and the degree of apoptosis achieved by 100 µM of
4 was similar to that achieved by 50 µM PAP-1 (
1). All compounds were ineffective as apoptosis inducers in Panc-1 tumor spheroids, whereas PAP-1 was able to induce significant apoptosis. Based on our efficacy data in PDAC cell lines and Colo-357 tumor spheroids, we hypothesize that inhibition of mitochondrial K
V1.3 ion channels is required for significant anticancer activity, as previously demonstrated with PAP-1-based mitochondrial K
V1.3 inhibitors (PAPTP and PCARBTP). Whether the ability of compound
4, which is specific against K
V1.3 [
21], is due to its ability to reach mitochondrial K
V1.3 channels remains to be investigated. Our results, however, highlight the potential to develop new mitochondrial Kv1.3 inhibitors by adding a TPP moiety to our potent and selective thiophene-based K
V1.3 inhibitors.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















































