
Searching for SARS-CoV-2 in Cancer Tissues: Results of an Extensive Methodologic Approach based on ACE2 and Furin Expression

 , , , , , , , , and
, , , , , , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Cell Line Infection
2.3. Electron Microscopy
2.4. Immunohistochemistry
2.5. RNA in Situ Hybridization
2.6. RNA Extraction and RT-qPCR
2.7. Statistical Analysis
3. Results
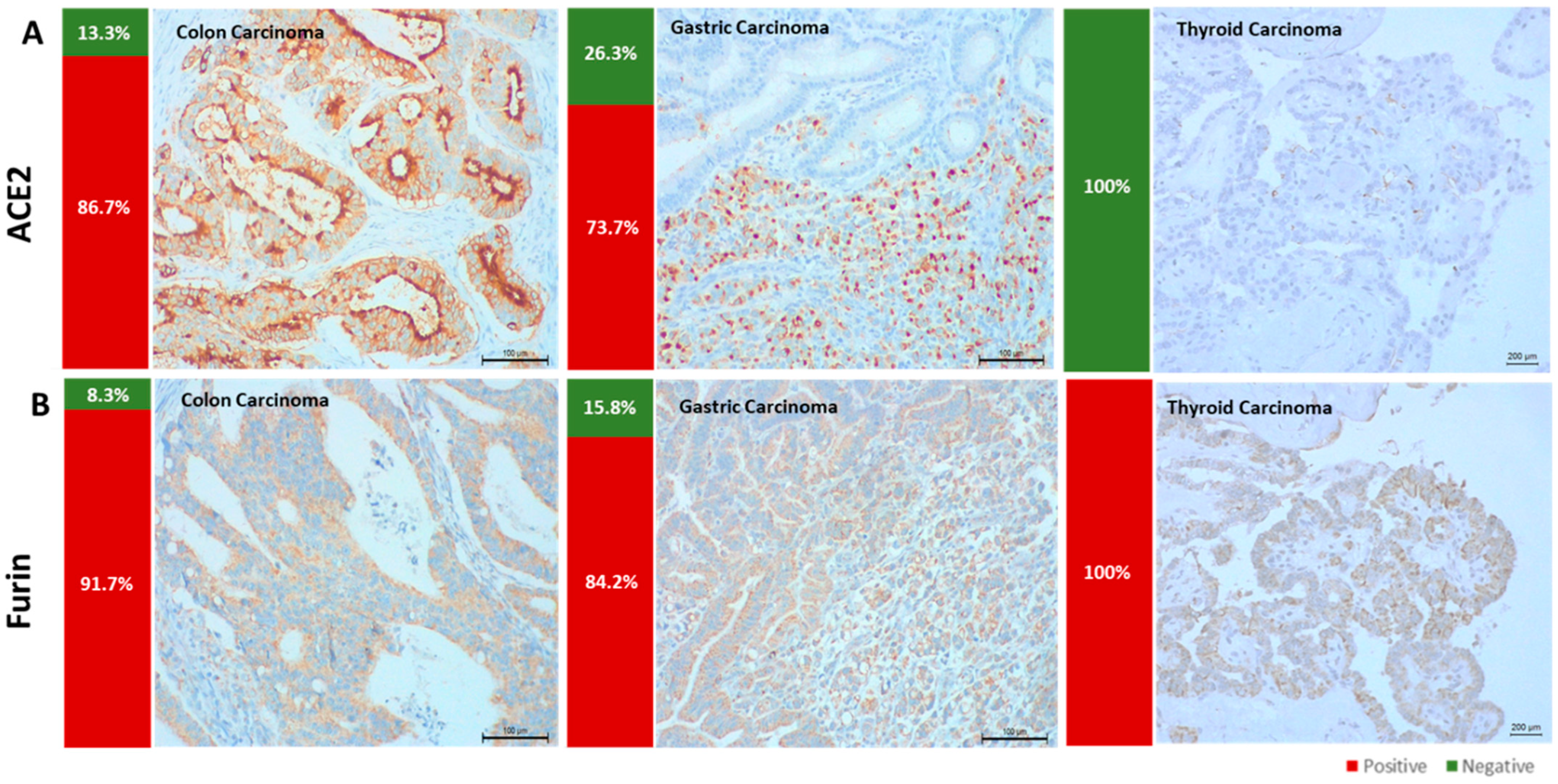
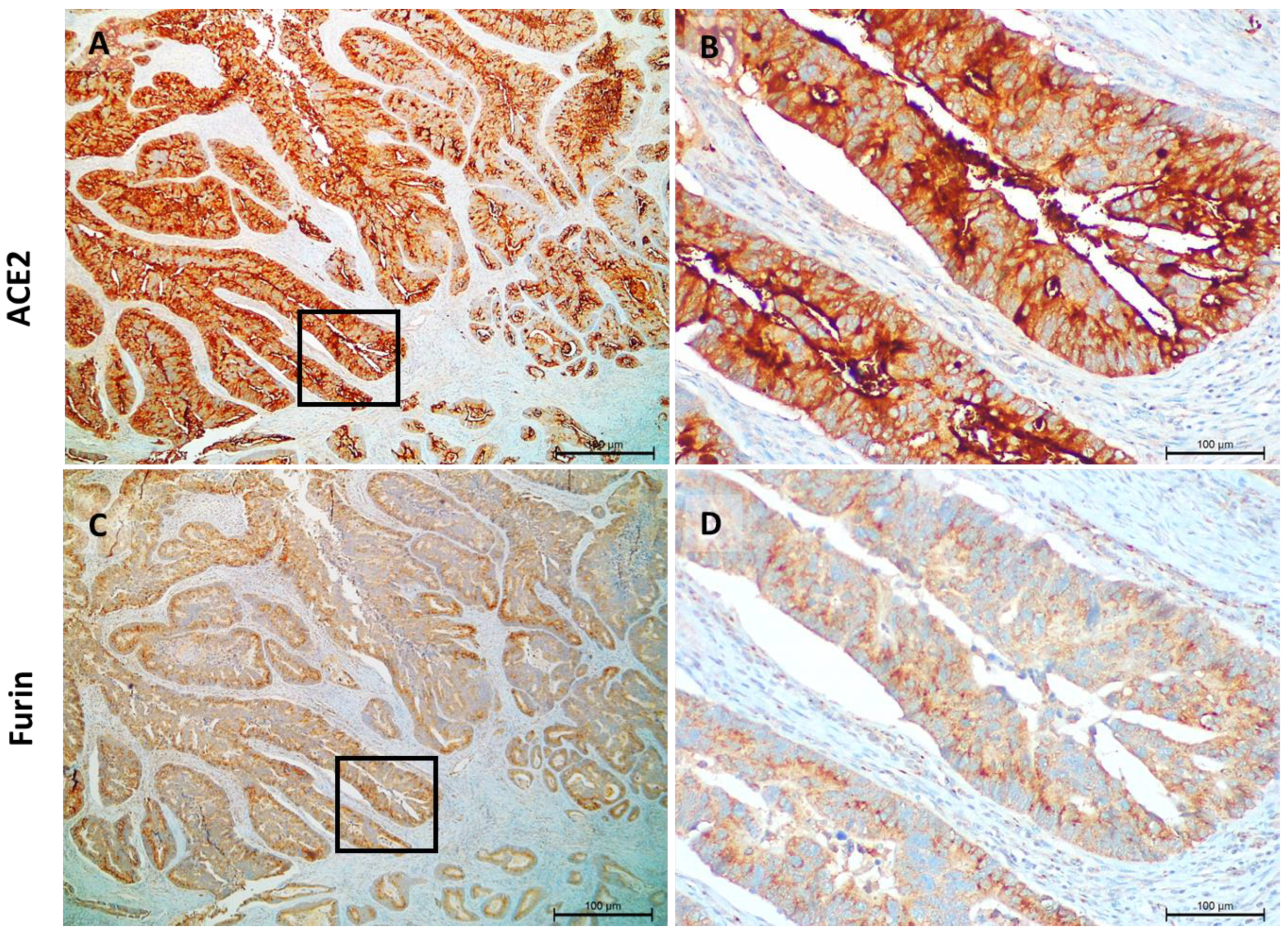
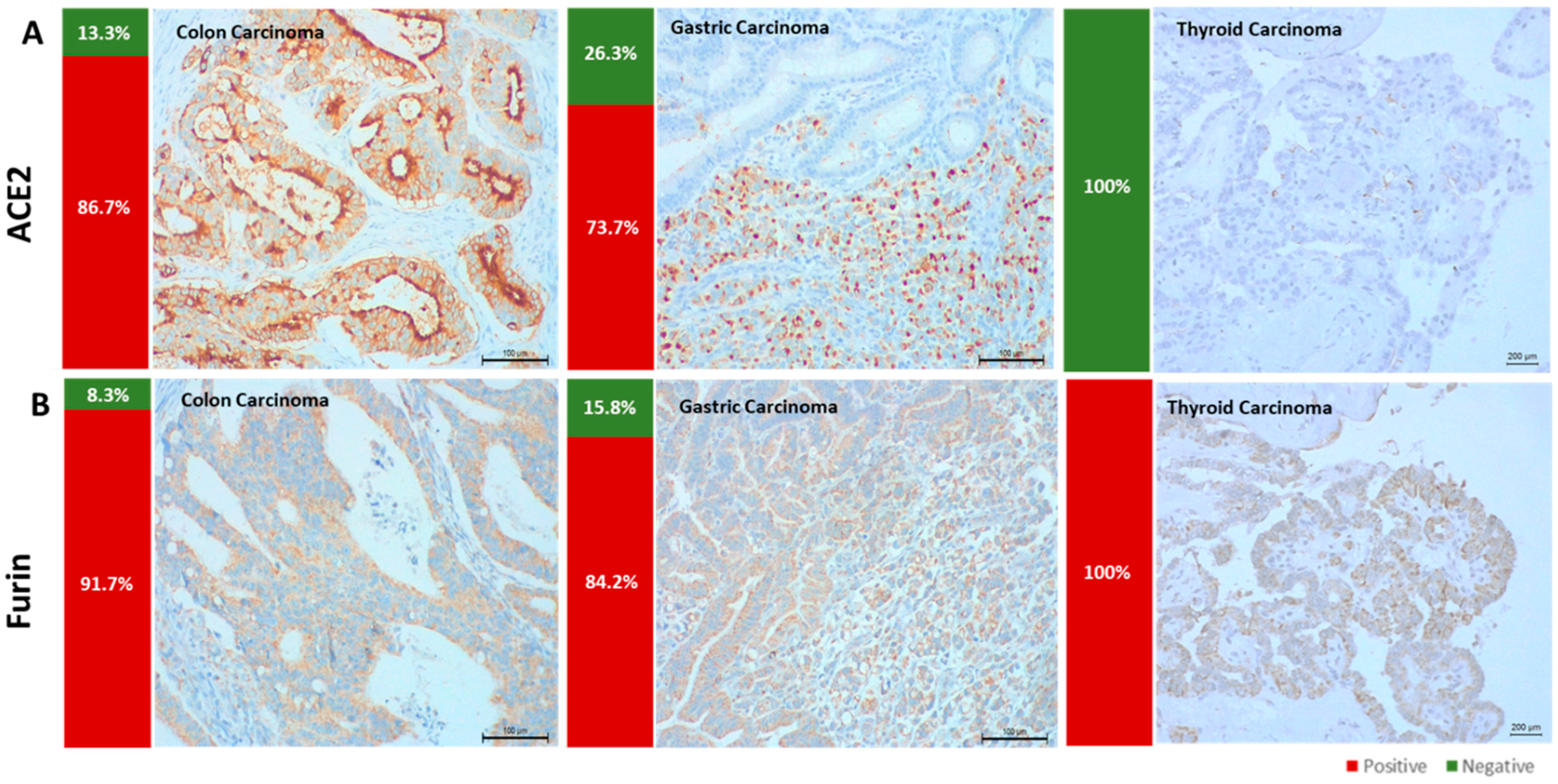
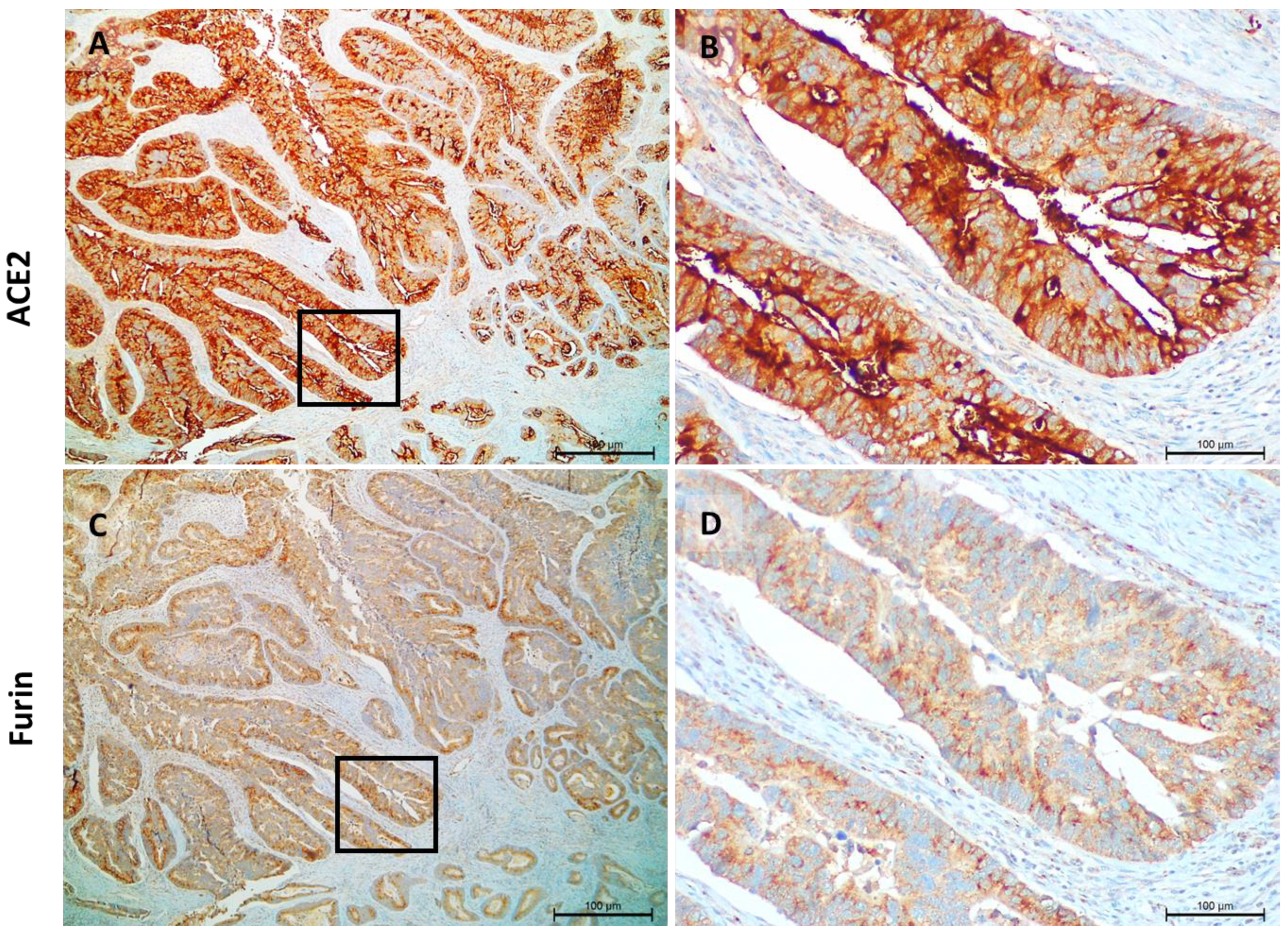
3.1. ACE2 and Furin Are Highly Expressed in Colon and Gastric Carcinomas
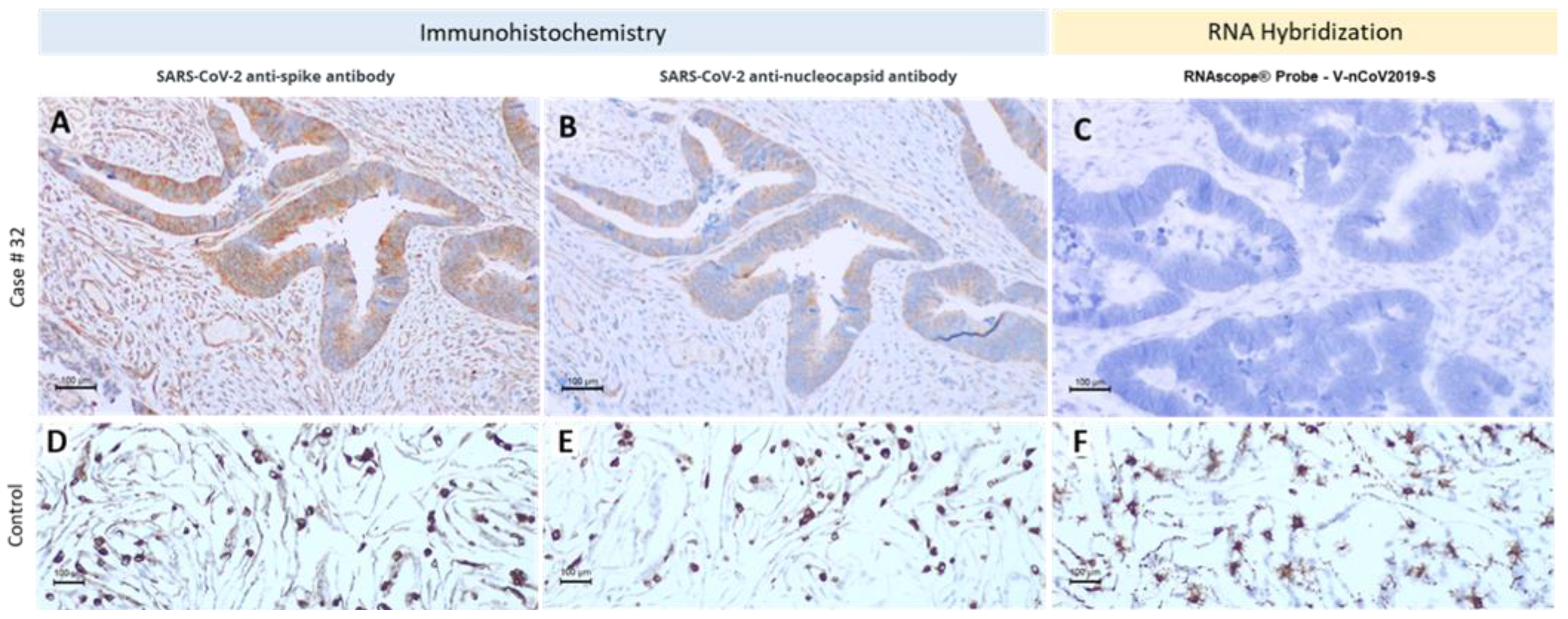
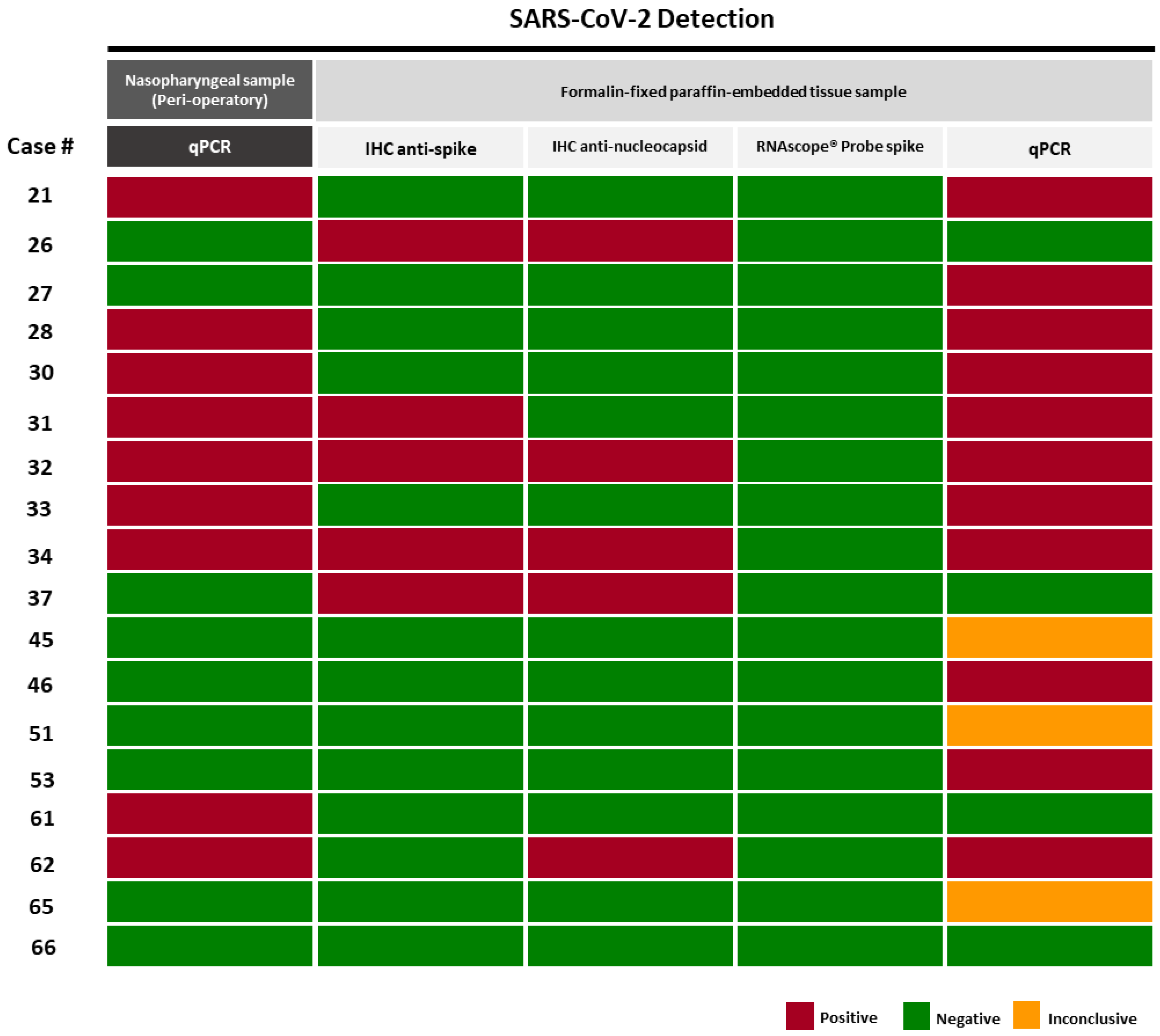
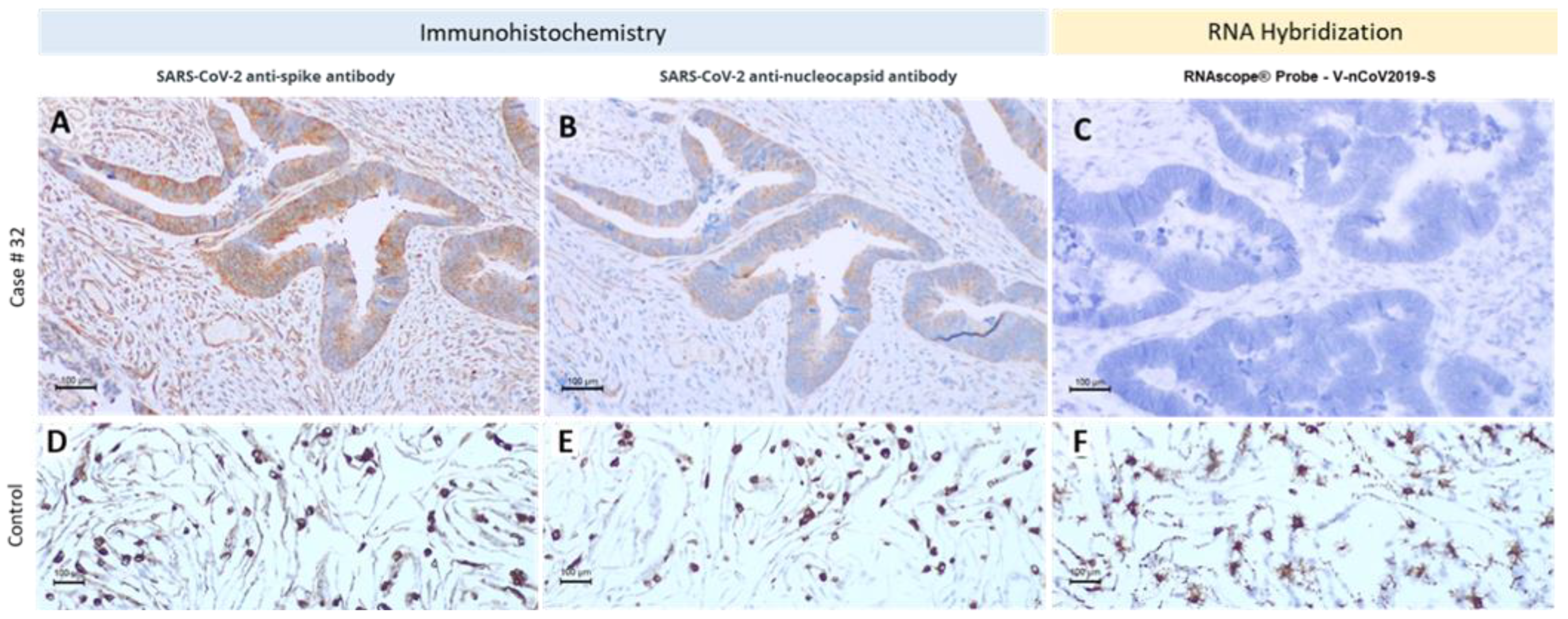
3.2. Different Methodologic Approaches to Detect SARS-CoV-2 in Tumor Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Escobedo, R.A.; Kaushal, D.; Singh, D.K. Insights Into the Changing Landscape of Coronavirus Disease 2019. Front. Cell. Infect. Microbiol. 2021, 11, 761521. [Google Scholar] [CrossRef] [PubMed]
- Hannah, H.E.M.; Rodés-Guirao, L.; Appel, C.; Giattino, C.; Ortiz-Ospina, E.; Hasell, J.; Macdonald, B.; Beltekian, D.; Roser, M. Coronavirus Pandemic (COVID-19). Available online: https://ourworldindata.org/coronavirus (accessed on 1 April 2022).
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Palit, P.; Chattopadhyay, D.; Thomas, S.; Kundu, A.; Kim, H.S.; Rezaei, N. Phytopharmaceuticals mediated Furin and TMPRSS2 receptor blocking: Can it be a potential therapeutic option for Covid-19? Phytomedicine 2021, 85, 153396. [Google Scholar] [CrossRef]
- Palit, P.; Chattopadhyay, D.; Thomas, S.; Kundu, A.; Kim, H.S.; Rezaei, N. Molecular detection of SARS-CoV-2 in formalin-fixed, paraffin-embedded specimens. JCI Insight 2020, 5, 153396. [Google Scholar]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Chen, R.; Ren, L.; Min, J.; Deng, F.; et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. 2021, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, A.C.; Ricardo, S.; Moreira, A.C.; Nunes, M.; Tavares, M.; Pinto, R.J.; Gomes, M.S.; Pereira, L. InfectionCMA: A Cell MicroArray Approach for Efficient Biomarker Screening in In Vitro Infection Assays. Pathogens 2022, 11, 313. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, Z.; Liao, H.; Marley, G.; Wu, D.; Tang, W. Epidemiologic, clinical, and laboratory findings of the COVID-19 in the current pandemic: Systematic review and meta-analysis. BMC Infect. Dis. 2020, 20, 640. [Google Scholar] [CrossRef]
- Zhong, P.; Xu, J.; Yang, D.; Shen, Y.; Wang, L.; Feng, Y.; Du, C.; Song, Y.; Wu, C.; Hu, X.; et al. COVID-19-associated gastrointestinal and liver injury: Clinical features and potential mechanisms. Signal Transduct. Target. Ther. 2020, 5, 256. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Zhang, M.; Feng, C.; Zhang, X.; Hu, S.; Zhang, Y.; Min, M.; Liu, B.; Ying, X.; Liu, Y. Susceptibility Factors of Stomach for SARS-CoV-2 and Treatment Implication of Mucosal Protective Agent in COVID-19. Front. Med. 2020, 7, 597967. [Google Scholar] [CrossRef]
- Baldelli, R.; Nicastri, E.; Petrosillo, N.; Marchioni, L.; Gubbiotti, A.; Sperduti, I.; Di Giacinto, P.; Rizza, L.; Rota, F.; Franco, M.; et al. Thyroid dysfunction in COVID-19 patients. J. Endocrinol. Investig. 2021, 44, 2735–2739. [Google Scholar] [CrossRef]
- Murugan, A.K.; Alzahrani, A.S. SARS-CoV-2 plays a pivotal role in inducing hyperthyroidism of Graves’ disease. Endocrine 2021, 73, 243–254. [Google Scholar] [CrossRef]
- Rotondi, M.; Coperchini, F.; Ricci, G.; Denegri, M.; Croce, L.; Ngnitejeu, S.T.; Villani, L.; Magri, F.; Latrofa, F.; Chiovato, L. Detection of SARS-COV-2 receptor ACE-2 mRNA in thyroid cells: A clue for COVID-19-related subacute thyroiditis. J. Endocrinol. Investig. 2020, 44, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, R.M.; Campennì, A.; Deandreis, D.; Siracusa, M.; Tozzoli, R.; Ovčariček, P.P.; Giovanella, L. SARS-COV-2-related immune-inflammatory thyroid disorders: Facts and perspectives. Expert Rev. Clin. Immunol. 2021, 17, 737–759. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.S.; Lorenz, K.; Ukkat, J.; Hoang-Vu, C.; Trojanowicz, B. Angiotensin converting enzymes ACE and ACE2 in thyroid cancer progression. Neoplasma 2020, 67, 402–409. [Google Scholar] [CrossRef]
- Liu, C.; Wang, K.; Zhang, M.; Hu, X.; Hu, T.; Liu, Y.; Hu, Q.; Wu, S.; Yue, J. High expression of ACE2 and TMPRSS2 and clinical characteristics of COVID-19 in colorectal cancer patients. NPJ Precis. Oncol. 2021, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Chai, P.; Yu, J.; Ge, S.; Jia, R.; Fan, X. Genetic alteration, RNA expression, and DNA methylation profiling of coronavirus disease 2019 (COVID-19) receptor ACE2 in malignancies: A pan-cancer analysis. J. Hematol. Oncol. 2020, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Bagalkot, T.; Fitzgerald, W.; Sadovsky, E.; Chu, T.; Martínez-Marchal, A.; Brieño-Enríquez, M.; Su, E.J.; Margolis, L.; Sorkin, A.; et al. Term Human Placental Trophoblasts Express SARS-CoV-2 Entry Factors ACE2, TMPRSS2, and Furin. mSphere 2021, 6, e00250-21. [Google Scholar] [CrossRef]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological Features of Covid-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef]
- Chong, P.Y.; Iqbal, J.; Yeong, J.; Aw, T.C.; Chan, K.S.; Chui, P. Immune Response in Myocardial Injury: In Situ Hybridization and Immunohistochemistry Techniques for SARS-CoV-2 Detection in COVID-19 Autopsies. Front. Mol. Biosci. 2021, 8, 658932. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACE2 Expression | |||||
|---|---|---|---|---|---|
| Positive (n) | Negative (n) | p Value | |||
| Furin expression | Colon carcinoma | Positive | 49 | 6 | 0.128 |
| Negative | 3 | 2 | |||
| Gastric carcinoma | Positive | 13 | 14 | 0.552 | |
| Negative | 1 | 2 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricardo, S.; Canão, P.; Martins, D.; Magalhães, A.C.; Pereira, M.; Ribeiro-Junior, U.; de Mello, E.S.; Alves, V.A.; Pinto, R.; Leitão, D.; et al. Searching for SARS-CoV-2 in Cancer Tissues: Results of an Extensive Methodologic Approach based on ACE2 and Furin Expression. Cancers 2022, 14, 2582. https://doi.org/10.3390/cancers14112582
Ricardo S, Canão P, Martins D, Magalhães AC, Pereira M, Ribeiro-Junior U, de Mello ES, Alves VA, Pinto R, Leitão D, et al. Searching for SARS-CoV-2 in Cancer Tissues: Results of an Extensive Methodologic Approach based on ACE2 and Furin Expression. Cancers. 2022; 14(11):2582. https://doi.org/10.3390/cancers14112582
Chicago/Turabian StyleRicardo, Sara, Pedro Canão, Diana Martins, Ana C. Magalhães, Marina Pereira, Ulysses Ribeiro-Junior, Evandro Sobroza de Mello, Venâncio A. Alves, Regina Pinto, Dina Leitão, and et al. 2022. "Searching for SARS-CoV-2 in Cancer Tissues: Results of an Extensive Methodologic Approach based on ACE2 and Furin Expression" Cancers 14, no. 11: 2582. https://doi.org/10.3390/cancers14112582
APA StyleRicardo, S., Canão, P., Martins, D., Magalhães, A. C., Pereira, M., Ribeiro-Junior, U., de Mello, E. S., Alves, V. A., Pinto, R., Leitão, D., Alves, G., Oliveira, R., Wilton, J., Costelha, S., Meireles, D., Cabanes, D., David, L., & Schmitt, F. (2022). Searching for SARS-CoV-2 in Cancer Tissues: Results of an Extensive Methodologic Approach based on ACE2 and Furin Expression. Cancers, 14(11), 2582. https://doi.org/10.3390/cancers14112582