
Chromosomal Instability in Chronic Myeloid Leukemia: Mechanistic Insights and Effects


{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
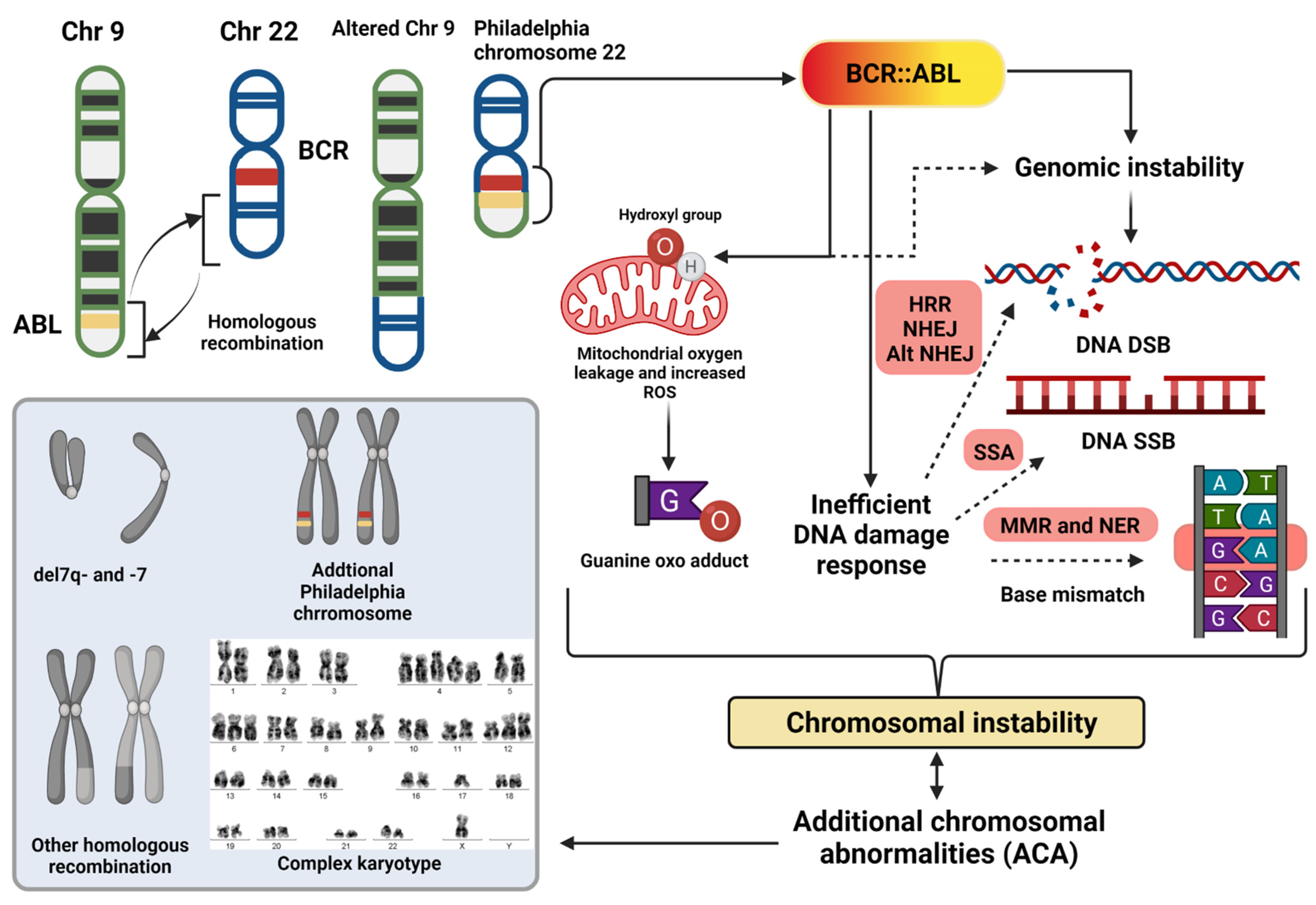
2. Chromosomal Instability in CML
2.1. Beyond the Philadelphia Chromosome: Additional Chromosomal Abnormalities
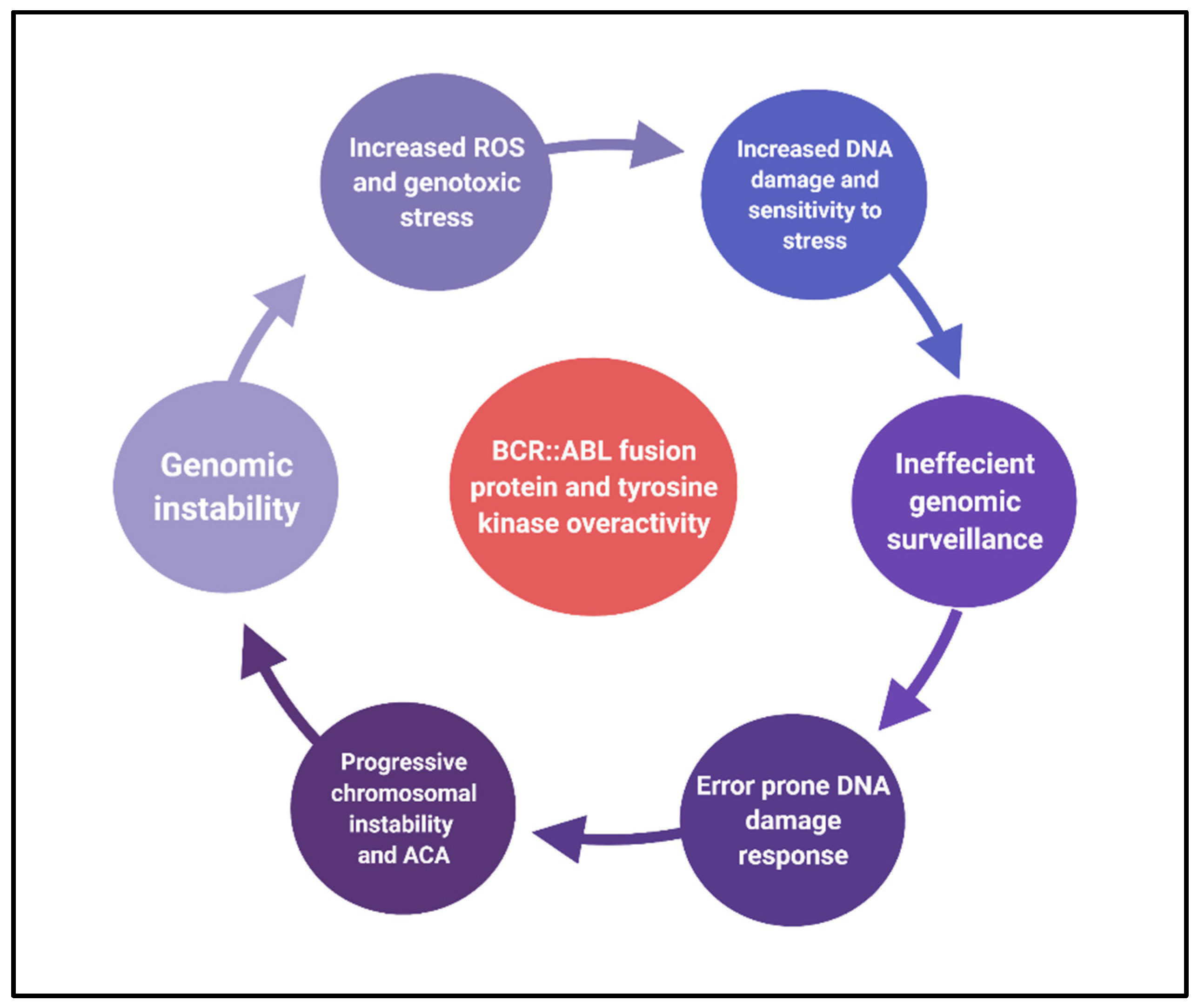
2.2. Causes of Chromosomal Instability
2.2.1. Reactive Oxygen Species
2.2.2. Inefficient Recognition of Genotoxic Stress
2.2.3. Error-Prone DNA Repair System
2.2.4. Centrosomal Aberration
2.3. Effects of Chromosomal Instability
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, C.A.; Rubin, C.M.; Carrino, J.J.; Le Beau, M.M.; Bernards, A.; Rowley, J.D. Long-Range Mapping of the Philadelphia Chromosome by Pulsed-Field Gel Electrophoresis. Blood 1988, 71, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The Biology of Chronic Myeloid Leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Lebecque, B.; Bourgne, C.; Vidal, V.; Berger, M.G. DNA Methylation and Intra-Clonal Heterogeneity: The Chronic Myeloid Leukemia Model. Cancers 2021, 13, 3587. [Google Scholar] [CrossRef] [PubMed]
- Ochi, Y.; Yoshida, K.; Huang, Y.-J.; Kuo, M.-C.; Nannya, Y.; Sasaki, K.; Mitani, K.; Hosoya, N.; Hiramoto, N.; Ishikawa, T.; et al. Clonal evolution and clinical implications of genetic abnormalities in blastic transformation of chronic myeloid leukaemia. Nat. Commun. 2021, 12, 2833. [Google Scholar] [CrossRef]
- Tobin, L.A.; Robert, C.; Rapoport, A.P.; Gojo, I.; Baer, M.R.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene 2013, 32, 1784–1793. [Google Scholar] [CrossRef]
- Pawlowska, E.; Blasiak, J. DNA Repair—A Double-Edged Sword in the Genomic Stability of Cancer Cells—The Case of Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2015, 16, 27535–27549. [Google Scholar] [CrossRef]
- Heller, G.; Topakian, T.; Altenberger, C.; Cerny-Reiterer, S.; Herndlhofer, S.; Ziegler, B.; Datlinger, P.; Byrgazov, K.; Bock, C.; Mannhalter, C.; et al. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia 2016, 30, 1861–1868. [Google Scholar] [CrossRef]
- Maupetit-Mehouas, S.; Court, F.; Bourgne, C.; Guerci-Bresler, A.; Cony-Makhoul, P.; Johnson, H.; Etienne, G.; Rousselot, P.; Guyotat, D.; Janel, A.; et al. DNA methylation profiling reveals a pathological signature that contributes to transcriptional defects of CD34(+) CD15(−) cells in early chronic-phase chronic myeloid leukemia. Mol. Oncol. 2018, 12, 814–829. [Google Scholar] [CrossRef]
- Koschmieder, S.; Vetrie, D. Epigenetic dysregulation in chronic myeloid leukaemia: A myriad of mechanisms and therapeutic options. Semin. Cancer Biol. 2018, 51, 180–197. [Google Scholar] [CrossRef]
- Saxena, K.; Jabbour, E.; Issa, G.; Sasaki, K.; Ravandi, F.; Maiti, A.; Daver, N.; Kadia, T.; DiNardo, C.D.; Konopleva, M.; et al. Impact of frontline treatment approach on outcomes of myeloid blast phase CML. J. Hematol. Oncol. 2021, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Mueller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kantarjian, H.M.; Yang, Y.; Sasaki, K.; Jain, P.; DellaSala, S.; Ravandi, F.; Kadia, T.; Pemmaraju, N.; Daver, N.; et al. A propensity score matching analysis of dasatinib and nilotinib as a frontline therapy for patients with chronic myeloid leukemia in chronic phase. Cancer 2016, 122, 3336–3343. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Kantarjian, H.; Alattar, M.L.; Jabbour, E.; Sasaki, K.; Nogueras Gonzalez, G.; Dellasala, S.; Pierce, S.; Verstovsek, S.; Wierda, W.; et al. Long-term molecular and cytogenetic response and survival outcomes with imatinib 400 mg, imatinib 800 mg, dasatinib, and nilotinib in patients with chronic-phase chronic myeloid leukaemia: Retrospective analysis of patient data from five clinical trials. Lancet Haematol. 2015, 2, e118–e128. [Google Scholar] [CrossRef]
- Réa, D.; Mauro, M.J.; Boquimpani, C.; Minami, Y.; Lomaia, E.; Voloshin, S.; Turkina, A.G.; Kim, D.-W.; Apperley, J.F.; Abdo, A.; et al. A phase 3, open-label, randomized study of asciminib, a STAMP inhibitor, vs bosutinib in CML after 2 or more prior TKIs. Blood 2021, 138, 2031–2041. [Google Scholar] [CrossRef]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Hughes, T.P.; Larson, R.A.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Boquimpani, C.; Pasquini, R.; Clark, R.E.; et al. Long-term outcomes with frontline nilotinib versus imatinib in newly diagnosed chronic myeloid leukemia in chronic phase: ENESTnd 10-year analysis. Leukemia 2021, 35, 440–453. [Google Scholar] [CrossRef]
- Cortes, J.E.; Saglio, G.; Kantarjian, H.M.; Baccarani, M.; Mayer, J.; Boqué, C.; Shah, N.P.; Chuah, C.; Casanova, L.; Bradley-Garelik, B.; et al. Final 5-Year Study Results of DASISION: The Dasatinib Versus Imatinib Study in Treatment-Naïve Chronic Myeloid Leukemia Patients Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2333–2340. [Google Scholar] [CrossRef]
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.-W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; Le Coutre, P.; et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 231–237. [Google Scholar] [CrossRef]
- Sasaki, K.; Strom, S.S.; O’Brien, S.; Jabbour, E.; Ravandi, F.; Konopleva, M.; Borthakur, G.; Pemmaraju, N.; Daver, N.; Jain, P.; et al. Relative survival in patients with chronic-phase chronic myeloid leukaemia in the tyrosine-kinase inhibitor era: Analysis of patient data from six prospective clinical trials. Lancet Haematol. 2015, 2, e186–e193. [Google Scholar] [CrossRef]
- Nowell, P.C.; Hungerford, D.A. Chromosome Studies on Normal and Leukemic Human Leukocytes. JNCI J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Alhuraiji, A.; Kantarjian, H.; Boddu, P.; Ravandi, F.; Borthakur, G.; DiNardo, C.; Daver, N.; Kadia, T.; Pemmaraju, N.; Pierce, S.; et al. Prognostic significance of additional chromosomal abnormalities at the time of diagnosis in patients with chronic myeloid leukemia treated with frontline tyrosine kinase inhibitors. Am. J. Hematol. 2018, 93, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.R.; Geetha, N.; Sakthivel, K.M.; Kumar, S.R.; Krishna, J.K.M.N.; Sreedharan, H. Impact of Additional Chromosomal Aberrations on the Disease Progression of Chronic Myelogenous Leukemia. Front. Oncol. 2019, 9, 88. [Google Scholar] [CrossRef]
- Wang, W.; Cortes, J.E.; Tang, G.; Khoury, J.D.; Wang, S.; Bueso-Ramos, C.E.; DiGiuseppe, J.A.; Chen, Z.; Kantarjian, H.M.; Medeiros, L.J.; et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood 2016, 127, 2742–2750. [Google Scholar] [CrossRef]
- Clark, R.E.; Apperley, J.F.; Copland, M.; Cicconi, S. Additional chromosomal abnormalities at chronic myeloid leukemia diagnosis predict an increased risk of progression. Blood Adv. 2021, 5, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Sasaki, K. Current status and novel strategy of CML. Int. J. Hematol. 2021, 113, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Jabbour, E.; Ravandi, F.; Borthakur, G.; Khoury, J.D.; Hu, S.; Garcia-Manero, G.; Wierda, W.; Issa, G.; Daver, N.; et al. Clinical Outcomes of Patients With Chronic Myeloid Leukemia With Concurrent Core Binding Factor Rearrangement and Philadelphia Chromosome. Clin. Lymphoma Myeloma Leuk. 2021, 21, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Kantarjian, H.M.; Gonzalez, G.N.; Borthakur, G.; Tang, G.; Wierda, W.; Sasaki, K.; Short, N.; Ravandi, F.; Kadia, T.; et al. Clonal chromosomal abnormalities appearing in Philadelphia chromosome-negative metaphases during CML treatment. Blood 2017, 130, 2084–2091. [Google Scholar] [CrossRef]
- Morita, K.; Kantarjian, H.M.; Sasaki, K.; Issa, G.C.; Jain, N.; Konopleva, M.; Short, N.J.; Takahashi, K.; DiNardo, C.D.; Kadia, T.M.; et al. Outcome of patients with chronic myeloid leukemia in lymphoid blastic phase and Philadelphia chromosome-positive acute lymphoblastic leukemia treated with hyper-CVAD and dasatinib. Cancer 2021, 127, 2641–2647. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.M.; Ghorab, A.; Sasaki, K.; Jabbour, E.J.; Nogueras Gonzalez, G.; Kanagal-Shamanna, R.; Issa, G.C.; Garcia-Manero, G.; Kc, D.; et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: Cohort study of 477 patients. Cancer 2017, 123, 4391–4402. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.; Sasaki, K.; Jabbour, E.; Dasarathula, J.; Nogueras Gonzalez, G.; Verstovsek, S.; Borthakur, G.; Wierda, W.; Kadia, T.; et al. Analysis of 2013 European LeukaemiaNet (ELN) responses in chronic phase CML across four frontline TKI modalities and impact on clinical outcomes. Br. J. Haematol. 2016, 173, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Kantarjian, H.; Patel, K.P.; Gonzalez, G.N.; Luthra, R.; Kanagal Shamanna, R.; Sasaki, K.; Jabbour, E.; Romo, C.G.; Kadia, T.M.; et al. Impact of BCR-ABL transcript type on outcome in patients with chronic-phase CML treated with tyrosine kinase inhibitors. Blood 2016, 127, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Dierov, J.; Sanchez, P.V.; Burke, B.A.; Padilla-Nash, H.; Putt, M.E.; Ried, T.; Carroll, M. BCR/ABL induces chromosomal instability after genotoxic stress and alters the cell death threshold. Leukemia 2009, 23, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Kopinski, P.K.; Ray, R.; Hoser, G.; Ngaba, D.; Flis, S.; Cramer, K.; Reddy, M.M.; Koptyra, M.; Penserga, T.; et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012, 119, 4253–4263. [Google Scholar] [CrossRef]
- Popp, H.D.; Kohl, V.; Naumann, N.; Flach, J.; Brendel, S.; Kleiner, H.; Weiss, C.; Seifarth, W.; Saussele, S.; Hofmann, W.-K.; et al. DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 1177. [Google Scholar] [CrossRef]
- Bolton-Gillespie, E.; Schemionek, M.; Klein, H.-U.; Flis, S.; Hoser, G.; Lange, T.; Nieborowska-Skorska, M.; Maier, J.; Kerstiens, L.; Koptyra, M.; et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood 2013, 121, 4175–4183. [Google Scholar] [CrossRef]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J. Clin. Investig. 2010, 120, 2254–2264. [Google Scholar] [CrossRef]
- Verma, D.; Kantarjian, H.; Shan, J.; O’Brien, S.; Estrov, Z.; Garcia-Manero, G.; Koller, C.; Borthakur, G.; Cortes, J. Survival outcomes for clonal evolution in chronic myeloid leukemia patients on second generation tyrosine kinase inhibitor therapy. Cancer 2010, 116, 2673–2681. [Google Scholar] [CrossRef]
- Schoch, C.; Haferlach, T.; Kern, W.; Schnittger, S.; Berger, U.; Hehlmann, R.; Hiddemann, W.; Hochhaus, A. Occurrence of additional chromosome aberrations in chronic myeloid leukemia patients treated with imatinib mesylate. Leukemia 2003, 17, 461–463. [Google Scholar] [CrossRef][Green Version]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; De Santis, S.; Monaldi, C.; Bruno, S.; Mancini, M. Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort? Int. J. Mol. Sci. 2021, 22, 7093. [Google Scholar] [CrossRef] [PubMed]
- Fabarius, A.; Leitner, A.; Hochhaus, A.; Müller, M.C.; Hanfstein, B.; Haferlach, C.; Göhring, G.; Schlegelberger, B.; Jotterand, M.; Reiter, A.; et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: Long-term observation of 1151 patients from the randomized CML Study IV. Blood 2011, 118, 6760–6768. [Google Scholar] [CrossRef]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Oehler, V.G. First-generation vs second-generation tyrosine kinase inhibitors: Which is best at diagnosis of chronic phase chronic myeloid leukemia? Hematol. Am Soc Hematol Educ Program 2020, 2020, 228–236. [Google Scholar] [CrossRef]
- Kim, T.D.; Türkmen, S.; Schwarz, M.; Koca, G.; Nogai, H.; Bommer, C.; Dörken, B.; Daniel, P.; le Coutre, P. Impact of additional chromosomal aberrations and BCR-ABL kinase domain mutations on the response to nilotinib in Philadelphia chromosome-positive chronic myeloid leukemia. Haematologica 2010, 95, 582–588. [Google Scholar] [CrossRef]
- Gong, Z.; Medeiros, L.J.; Cortes, J.E.; Chen, Z.; Zheng, L.; Li, Y.; Bai, S.; Lin, P.; Miranda, R.N.; Jorgensen, J.L.; et al. Cytogenetics-based risk prediction of blastic transformation of chronic myeloid leukemia in the era of TKI therapy. Blood Adv. 2017, 1, 2541–2552. [Google Scholar] [CrossRef]
- Amare, P.S.K.; Jain, H.; Kabre, S.; Walke, D.; Menon, H.; Sengar, M.; Khatri, N.; Bagal, B.; Dangi, U.; Subramanian, P.G.; et al. Characterization of Genomic Events Other than Ph and Evaluation of Prognostic Influence on Imatinib in Chronic Myeloid Leukemia (CML): A Study on 1449 Patients from India. J. Cancer Ther. 2016, 7, 285–296. [Google Scholar] [CrossRef]
- Chen, Z.; Cortes, J.E.; Jorgensen, J.L.; Wang, W.; Yin, C.C.; You, M.J.; Jabbour, E.; Kantarjian, H.M.; Medeiros, L.J.; Hu, S. Differential impact of additional chromosomal abnormalities in myeloid vs lymphoid blast phase of chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Leukemia 2016, 30, 1606–1609. [Google Scholar] [CrossRef][Green Version]
- Flis, K.; Irvine, D.; Copland, M.; Bhatia, R.; Skorski, T. Chronic myeloid leukemia stem cells display alterations in expression of genes involved in oxidative phosphorylation. Leuk. Lymphoma 2012, 53, 2474–2478. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nieborowska-Skorska, M.; Flis, S.; Skorski, T. AKT-induced reactive oxygen species generate imatinib-resistant clones emerging from chronic myeloid leukemia progenitor cells. Leukemia 2014, 28, 2416–2418. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Chu, S.C.; Gramlich, J.L.; Pride, Y.B.; Babendreier, E.; Chauhan, D.; Salgia, R.; Podar, K.; Griffin, J.D.; Sattler, M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 2005, 105, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef]
- Trombetti, S.; Cesaro, E.; Catapano, R.; Sessa, R.; Lo Bianco, A.; Izzo, P.; Grosso, M. Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 2470. [Google Scholar] [CrossRef]
- Koptyra, M.; Falinski, R.; Nowicki, M.O.; Stoklosa, T.; Majsterek, I.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006, 108, 319–327. [Google Scholar] [CrossRef]
- Dierov, J.; Dierova, R.; Carroll, M. BCR/ABL translocates to the nucleus and disrupts an ATR-dependent intra-S phase checkpoint. Cancer Cell 2004, 5, 275–285. [Google Scholar] [CrossRef]
- Shafman, T.; Khanna, K.K.; Kedar, P.; Spring, K.; Kozlov, S.; Yen, T.; Hobson, K.; Gatei, M.; Zhang, N.; Watters, D.; et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature 1997, 387, 520–523. [Google Scholar] [CrossRef]
- Yoshida, K.; Komatsu, K.; Wang, H.-G.; Kufe, D. c-Abl tyrosine kinase regulates the human Rad9 checkpoint protein in response to DNA damage. Mol. Cell. Biol. 2002, 22, 3292–3300. [Google Scholar] [CrossRef][Green Version]
- Wolanin, K.; Magalska, A.; Kusio-Kobialka, M.; Podszywalow-Bartnicka, P.; Vejda, S.; McKenna, S.L.; Mosieniak, G.; Sikora, E.; Piwocka, K. Expression of oncogenic kinase Bcr-Abl impairs mitotic checkpoint and promotes aberrant divisions and resistance to microtubule-targeting agents. Mol. Cancer Ther. 2010, 9, 1328–1338. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stetka, J.; Gursky, J.; Liñan Velasquez, J.; Mojzikova, R.; Vyhlidalova, P.; Vrablova, L.; Bartek, J.; Divoky, V. Role of DNA Damage Response in Suppressing Malignant Progression of Chronic Myeloid Leukemia and Polycythemia Vera: Impact of Different Oncogenes. Cancers 2020, 12, 903. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Follows, G.A.; Beer, P.A.; Scott, L.M.; Huntly, B.J.P.; Green, A.R.; Alexander, D.R. Inhibition of the Bcl-xL Deamidation Pathway in Myeloproliferative Disorders. N. Engl. J. Med. 2008, 359, 2778–2789. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.T.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008, 68, 6884–6888. [Google Scholar] [CrossRef]
- Waters, C.A.; Strande, N.T.; Wyatt, D.W.; Pryor, J.M.; Ramsden, D.A. Nonhomologous end joining: A good solution for bad ends. DNA Repair. 2014, 17, 39–51. [Google Scholar] [CrossRef]
- Sallmyr, A.; Rassool, F.V. Up-Regulated WRN and DNA Ligase IIIα Are Involved in Alternative NHEJ Repair Pathway of DNA Double Strand Breaks (DSB) in Chronic Myeloid Leukemia (CML). Blood 2007, 110, 1016. [Google Scholar] [CrossRef]
- Newman, E.A.; Lu, F.; Bashllari, D.; Wang, L.; Opipari, A.W.; Castle, V.P. Alternative NHEJ Pathway Components Are Therapeutic Targets in High-Risk Neuroblastoma. Cell Growth Differ. 2015, 13, 470–482. [Google Scholar] [CrossRef]
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers 2021, 13, 1392. [Google Scholar] [CrossRef]
- Salles, D.; Mencalha, A.L.; Ireno, I.C.; Wiesmüller, L.; Abdelhay, E. BCR-ABL stimulates mutagenic homologous DNA double-strand break repair via the DNA-end-processing factor CtIP. Carcinogenesis 2011, 32, 27–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Muvarak, N.; Kelley, S.; Robert, C.; Baer, M.R.; Perrotti, D.; Gambacorti-Passerini, C.; Civin, C.; Scheibner, K.; Rassool, F.V. c-MYC Generates Repair Errors via Increased Transcription of Alternative-NHEJ Factors, LIG3 and PARP1, in Tyrosine Kinase–Activated Leukemias. Cell Growth Differ. 2015, 13, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.A.; Chukkapalli, S.; Bashllari, D.; Thomas, T.T.; Van Noord, R.A.; Lawlor, E.R.; Hoenerhoff, M.J.; Opipari, A.W.; Opipari, V.P. Alternative NHEJ pathway proteins as components of MYCN oncogenic activity in human neural crest stem cell differentiation: Implications for neuroblastoma initiation. Cell Death Dis. 2017, 8, 3208. [Google Scholar] [CrossRef]
- Chakraborty, S.; Stark, J.M.; Sun, C.-L.; Modi, H.; Chen, W.; O’Connor, T.R.; Forman, S.J.; Bhatia, S.; Bhatia, R. Chronic myelogenous leukemia stem and progenitor cells demonstrate chromosomal instability related to repeated breakage-fusion-bridge cycles mediated by increased nonhomologous end joining. Blood 2012, 119, 6187–6197. [Google Scholar] [CrossRef]
- Zakov, S.; Kinsella, M.; Bafna, V. An algorithmic approach for breakage-fusion-bridge detection in tumor genomes. Proc. Natl. Acad. Sci. USA 2013, 110, 5546–5551. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Jonson, T.; Petersén, Å.; Strömbeck, B.; Cin, P.D.; Höglund, M.; Mitelman, F.; Mertens, F.; Mandahl, N. Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 12683–12688. [Google Scholar] [CrossRef]
- Valikhani, M.; Rahimian, E.; Ahmadi, S.E.; Chegeni, R.; Safa, M. Involvement of classic and alternative non-homologous end joining pathways in hematologic malignancies: Targeting strategies for treatment. Exp. Hematol. Oncol. 2021, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Liu, L.; Sun, S.; Sun, S. Centrosome dysfunction: A link between senescence and tumor immunity. Signal Transduct. Target. Ther. 2020, 5, 107. [Google Scholar] [CrossRef]
- Vitre, B.; Holland, A.J.; Kulukian, A.; Shoshani, O.; Hirai, M.; Wang, Y.; Maldonado, M.; Cho, T.; Boubaker, J.; Swing, D.A.; et al. Chronic centrosome amplification without tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E6321–E6330. [Google Scholar] [CrossRef]
- Giehl, M.; Fabarius, A.; Frank, O.; Hochhaus, A.; Hafner, M.; Hehlmann, R.; Seifarth, W. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia 2005, 19, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Giehl, M.; Leitner, A.; Haferlach, C.; Duesberg, P.; Hofmann, W.K.; Hofheinz, R.; Seifarth, W.; Hochhaus, A.; Fabarius, A. Detection of centrosome aberrations in disease-unrelated cells from patients with tumor treated with tyrosine kinase inhibitors. Eur. J. Haematol. 2010, 85, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Fabarius, A.; Giehl, M.; Rebacz, B.; Krämer, A.; Frank, O.; Haferlach, C.; Duesberg, P.; Hehlmann, R.; Seifarth, W.; Hochhaus, A. Centrosome aberrations and G1 phase arrest after in vitro and in vivo treatment with the SRC/ABL inhibitor dasatinib. Haematologica 2008, 93, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Haaß, W.; Kleiner, H.; Weiß, C.; Haferlach, C.; Schlegelberger, B.; Müller, M.C.; Hehlmann, R.; Hofmann, W.-K.; Fabarius, A.; Seifarth, W.; et al. Clonal Evolution and Blast Crisis Correlate with Enhanced Proteolytic Activity of Separase in BCR-ABL b3a2 Fusion Type CML under Imatinib Therapy. PLoS ONE. 2015, 10, e0129648. [Google Scholar] [CrossRef] [PubMed]
- Spiess, B.; Kleiner, H.; Flach, J.; Fabarius, A.; Saussele, S.; Hofmann, W.-K.; Seifarth, W. Separase activity distribution can be a marker of major molecular response and proliferation of CD34(+) cells in TKI-treated chronic myeloid leukemia patients. Ann. Hematol. 2020, 99, 991–1006. [Google Scholar] [CrossRef]
- Jabbour, E.J.; Hughes, T.P.; Cortés, J.E.; Kantarjian, H.M.; Hochhaus, A. Potential mechanisms of disease progression and management of advanced-phase chronic myeloid leukemia. Leuk. Lymphoma 2014, 55, 1451–1462. [Google Scholar] [CrossRef]
- Alves, R.; Gonçalves, A.C.; Rutella, S.; Almeida, A.M.; De Las Rivas, J.; Trougakos, I.P.; Ribeiro, A.B.S. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia-From Molecular Mechanisms to Clinical Relevance. Cancers 2021, 13, 4820. [Google Scholar] [CrossRef] [PubMed]
- Bavaro, L.; Martelli, M.; Cavo, M.; Soverini, S. Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update. Int. J. Mol. Sci. 2019, 20, 6141. [Google Scholar] [CrossRef]
- Aitken, M.J.L.; Benton, C.B.; Issa, G.C.; Sasaki, K.; Yilmaz, M.; Short, N.J. Two Cases of Possible Familial Chronic Myeloid Leukemia in a Family with Extensive History of Cancer. Acta Haematol. 2021, 144, 585–590. [Google Scholar] [CrossRef]
- Warsch, W.; Walz, C.; Sexl, V. JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia. Blood 2013, 122, 2167–2175. [Google Scholar] [CrossRef]
- Kurosu, T.; Nagao, T.; Wu, N.; Oshikawa, G.; Miura, O. Inhibition of the PI3K/Akt/GSK3 Pathway Downstream of BCR/ABL, Jak2-V617F, or FLT3-ITD Downregulates DNA Damage-Induced Chk1 Activation as Well as G2/M Arrest and Prominently Enhances Induction of Apoptosis. PLoS ONE 2013, 8, e79478. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Rana, A.; Datoguia, T.S.; Hamerschlak, N.; Brumatti, G. BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia. Pharmaceutics 2022, 14, 215. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, X.; Kim, Y.; Zhang, R.; Li, S.; Lee, J.Y. Acquired genomic copy number changes in CML patients with the Philadelphia chromosome (Ph+). Cancer Genet. 2012, 205, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, M.; Wang, X.; Ren, Y.; Kim, Y.M.; Wang, X.; Lu, X.; Pang, H.; Liu, G.; Gu, Y.; et al. Genomic Copy Number Variants in CML Patients with the Philadelphia Chromosome (Ph+): An Update. Front. Genet. 2021, 12, 1471. [Google Scholar] [CrossRef] [PubMed]
- Gribble, S.M.; Sinclair, P.B.; Grace, C.; Green, A.R.; Nacheva, E.P. Comparative Analysis of G-Banding, Chromosome Painting, Locus-Specific Fluorescence In Situ Hybridization, and Comparative Genomic Hybridization in Chronic Myeloid Leukemia Blast Crisis. Cancer Genet. Cytogenet. 1999, 111, 7–17. [Google Scholar] [CrossRef]
- Greulich-Bode, K.M.; Heinze, B. On the Power of Additional and Complex Chromosomal Aberrations in CML. Curr Genom. 2012, 13, 471–476. [Google Scholar] [CrossRef]
- Maiti, A.; Franquiz, M.J.; Ravandi, F.; Cortes, J.E.; Jabbour, E.J.; Sasaki, K.; Marx, K.; Daver, N.G.; Kadia, T.M.; Konopleva, M.Y.; et al. Venetoclax and BCR-ABL Tyrosine Kinase Inhibitor Combinations: Outcome in Patients with Philadelphia Chromosome-Positive Advanced Myeloid Leukemias. Acta Haematol. 2020, 143, 567–573. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.; Boddu, P.C.; Nogueras-González, G.M.; Verstovsek, S.; Garcia-Manero, G.; Borthakur, G.; Sasaki, K.; Kadia, T.M.; Sam, P.; et al. Analysis of cardiovascular and arteriothrombotic adverse events in chronic-phase CML patients after frontline TKIs. Blood Adv. 2019, 3, 851–861. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; O’Brien, S.; Ravandi, F.; Konopleva, M.; Borthakur, G.; Garcia-Manero, G.; Wierda, W.G.; Daver, N.; Ferrajoli, A.; et al. Incidence of second malignancies in patients with chronic myeloid leukemia in the era of tyrosine kinase inhibitors. Int. J. Hematol. 2019, 109, 545–552. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; Jain, P.; Jabbour, E.J.; Ravandi, F.; Konopleva, M.; Borthakur, G.; Takahashi, K.; Pemmaraju, N.; Daver, N.; et al. Conditional survival in patients with chronic myeloid leukemia in chronic phase in the era of tyrosine kinase inhibitors. Cancer 2016, 122, 238–248. [Google Scholar] [CrossRef]
- Sasaki, K.; Lahoti, A.; Jabbour, E.; Jain, P.; Pierce, S.; Borthakur, G.; Daver, N.; Kadia, T.; Pemmaraju, N.; Ferrajoli, A.; et al. Clinical Safety and Efficacy of Nilotinib or Dasatinib in Patients with Newly Diagnosed Chronic-Phase Chronic Myelogenous Leukemia and Pre-Existing Liver and/or Renal Dysfunction. Clin. Lymphoma Myeloma Leuk. 2016, 16, 152–162. [Google Scholar] [CrossRef]
- Sasaki, K.; Jabbour, E.J.; Ravandi, F.; Konopleva, M.; Borthakur, G.; Wierda, W.G.; Daver, N.; Takahashi, K.; Naqvi, K.; DiNardo, C.; et al. The LEukemia Artificial Intelligence Program (LEAP) in chronic myeloid leukemia in chronic phase: A model to improve patient outcomes. Am. J. Hematol. 2021, 96, 241–250. [Google Scholar] [CrossRef]
- Shoukier, M.; Borthakur, G.; Jabbour, E.; Ravandi, F.; Garcia-Manero, G.; Kadia, T.; Matthews, J.; Masarova, L.; Naqvi, K.; Sasaki, K.; et al. The effect of eltrombopag in managing thrombocytopenia associated with tyrosine kinase therapy in patients with chronic myeloid leukemia and myelofibrosis. Haematologica 2021, 106, 2853–2858. [Google Scholar]
- Haddad, F.; Kantarjian, H.; Jabbour, E.J.; Issa, G.C.; Garcia-Manero, G.; Ravandi, F.; Konopleva, M.; Ferrajoli, A.; Kadia, T.M.; Pemmaraju, N.; et al. Treatment-free remission in patients with chronic myeloid leukemia following the discontinuation of tyrosine kinase inhibitors. Blood 2021, 138, 1480. [Google Scholar] [CrossRef]
- Alfayez, M.; Richard-Carpentier, G.; Jabbour, E.; Vishnu, P.; Naqvi, K.; Sasaki, K.; Cortes, J.; Pemmaraju, N. Sudden blastic transformation in treatment-free remission chronic myeloid leukaemia. Br. J. Haematol. 2019, 187, 543–545. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K. Chronic myeloid leukemia: Update on treatment and survival prediction. Jpn. J. Clin. Hematol. 2020, 61, 1179–1186. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senapati, J.; Sasaki, K. Chromosomal Instability in Chronic Myeloid Leukemia: Mechanistic Insights and Effects. Cancers 2022, 14, 2533. https://doi.org/10.3390/cancers14102533
Senapati J, Sasaki K. Chromosomal Instability in Chronic Myeloid Leukemia: Mechanistic Insights and Effects. Cancers. 2022; 14(10):2533. https://doi.org/10.3390/cancers14102533
Chicago/Turabian StyleSenapati, Jayastu, and Koji Sasaki. 2022. "Chromosomal Instability in Chronic Myeloid Leukemia: Mechanistic Insights and Effects" Cancers 14, no. 10: 2533. https://doi.org/10.3390/cancers14102533
APA StyleSenapati, J., & Sasaki, K. (2022). Chromosomal Instability in Chronic Myeloid Leukemia: Mechanistic Insights and Effects. Cancers, 14(10), 2533. https://doi.org/10.3390/cancers14102533