
Pediatric Sarcomas: The Next Generation of Molecular Studies


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Clinical Challenges of Pediatric Sarcomas
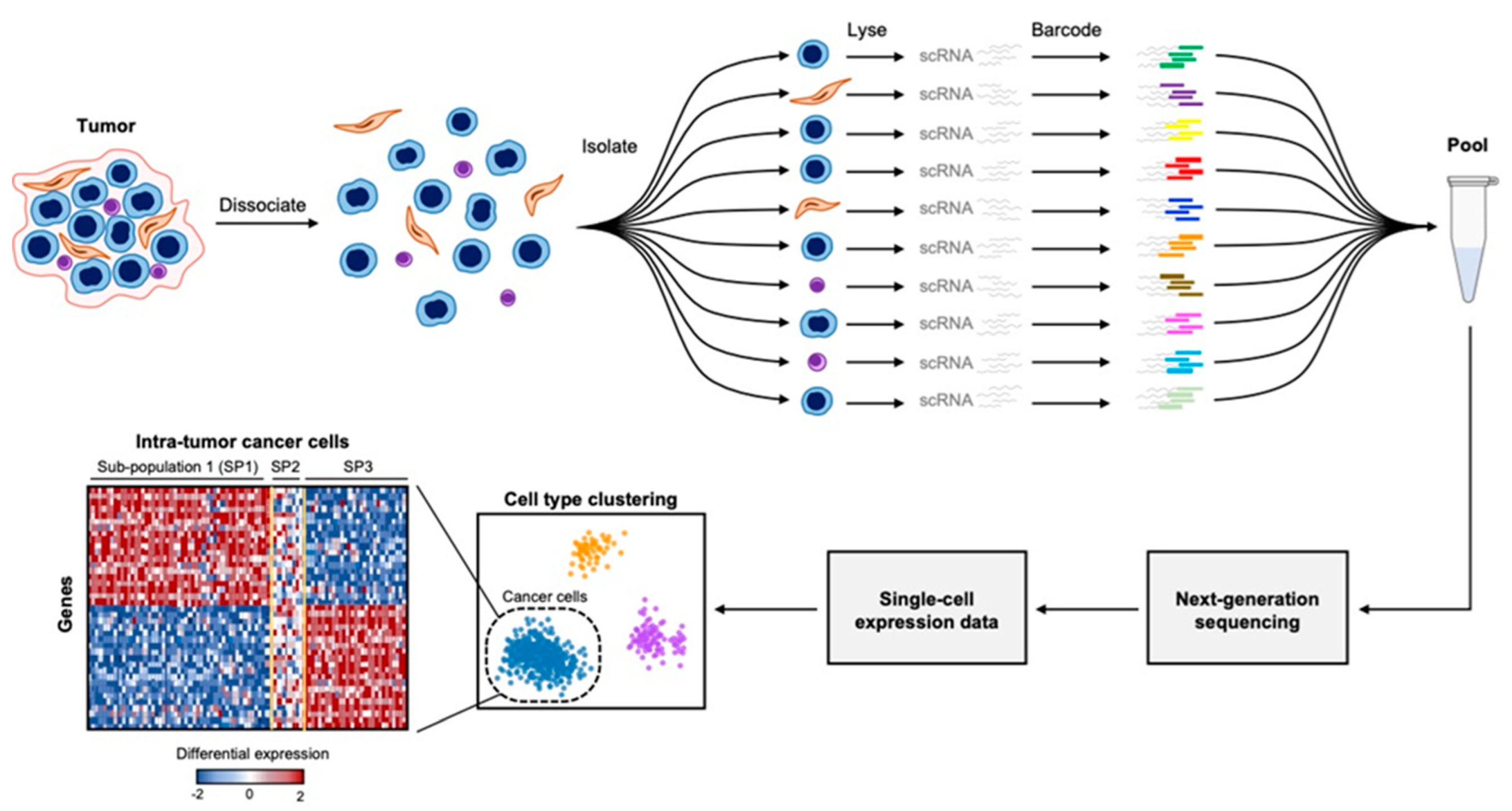
3. Comprehensive Molecular Profiling at Single-Cell Resolution
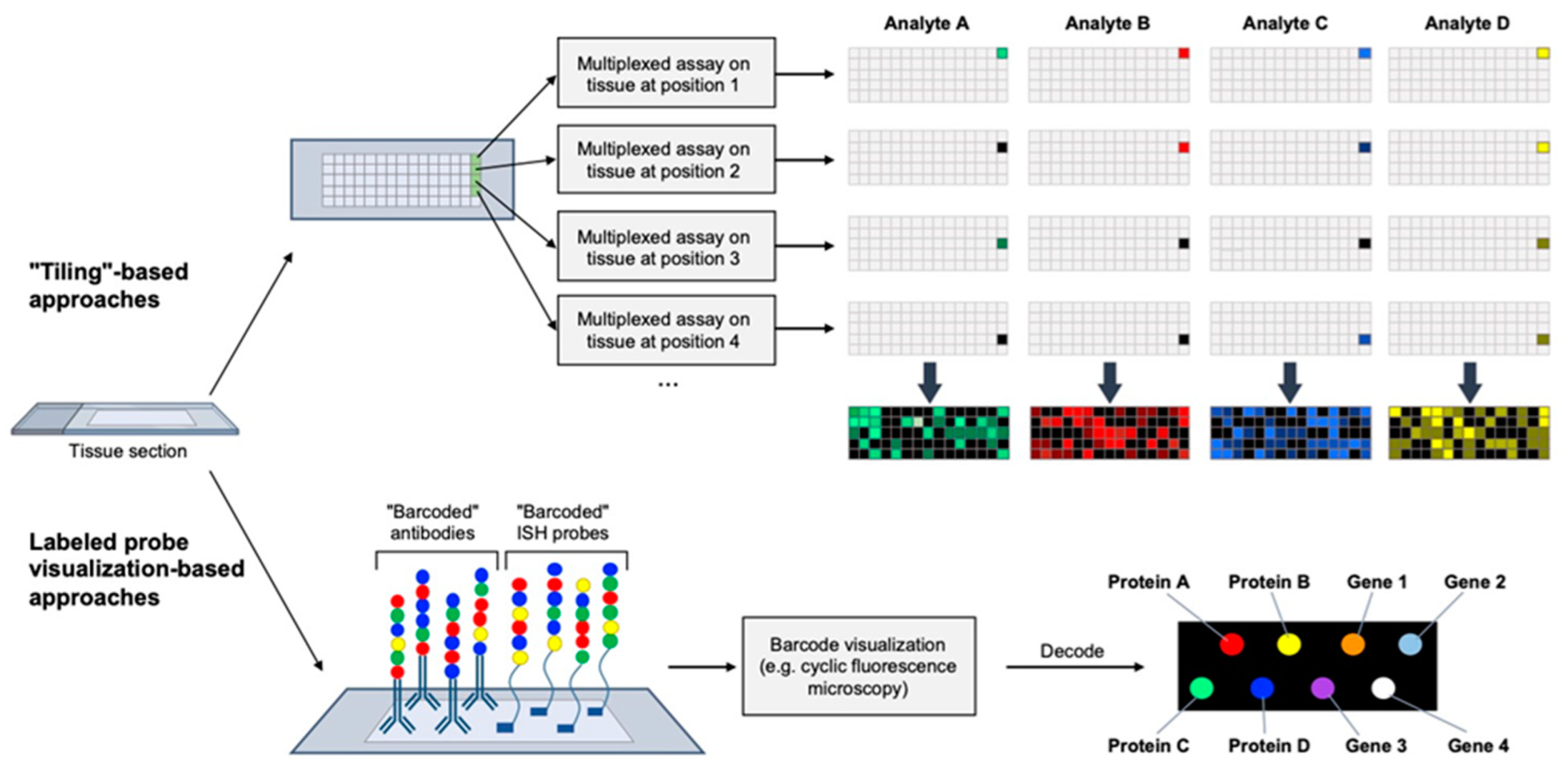
4. Spatial Multi-Omics
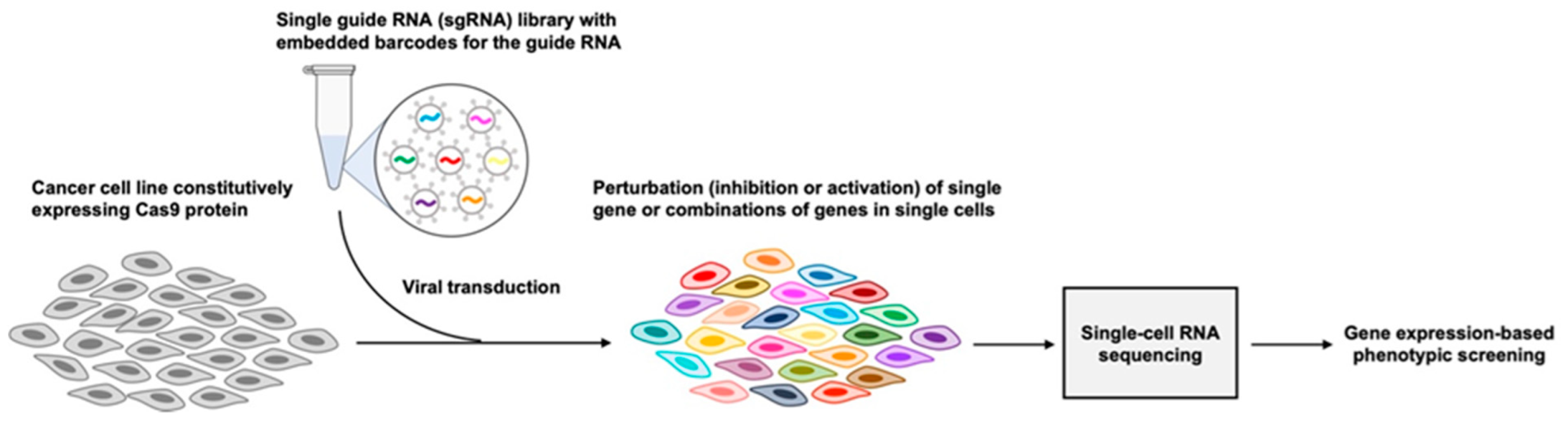
5. High-Throughput Functional Genomics
6. Need for Clinically and Pathologically Annotated Specimens
7. Artificial Intelligence/Machine Learning
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and Adolescent Cancer Statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Montella, A.; Tirelli, M.; Maiorino, T.; Cantalupo, S.; Iolascon, A. Genetic Predisposition to Solid Pediatric Cancers. Front. Oncol. 2020, 10, 590033. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, S.K.; Amatruda, J.F.; Bauer, S.; Collaud, S.; de Álava, E.; DuBois, S.G.; Hardes, J.; Hartmann, W.; Kovar, H.; Metzler, M.; et al. Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives. J. Clin. Med. Res. 2021, 10, 1685. [Google Scholar] [CrossRef] [PubMed]
- Spunt, S.L.; Million, L.; Chi, Y.-Y.; Anderson, J.; Tian, J.; Hibbitts, E.; Coffin, C.; McCarville, M.B.; Randall, R.L.; Parham, D.M.; et al. A Risk-Based Treatment Strategy for Non-Rhabdomyosarcoma Soft-Tissue Sarcomas in Patients Younger than 30 Years (ARST0332): A Children’s Oncology Group Prospective Study. Lancet Oncol. 2020, 21, 145–161. [Google Scholar] [CrossRef]
- Kinnaman, M.D.; Zhu, C.; Weiser, D.A.; Mohiuddin, S.; Hingorani, P.; Roth, M.; Gill, J.; Janeway, K.A.; Gorlick, R.; Lessnick, S.L.; et al. Survey of Paediatric Oncologists and Pathologists Regarding Their Views and Experiences with Variant Translocations in Ewing and Ewing-Like Sarcoma: A Report of the Children’s Oncology Group. Sarcoma 2020, 2020, 3498549. [Google Scholar] [CrossRef]
- Suurmeijer, A.J.; Dickson, B.C.; Swanson, D.; Zhang, L.; Sung, Y.-S.; Huang, H.-Y.; Fletcher, C.D.; Antonescu, C.R. The Histologic Spectrum of Soft Tissue Spindle Cell Tumors with NTRK3 Gene Rearrangements. Genes Chromosomes Cancer 2019, 58, 739–746. [Google Scholar] [CrossRef]
- Lei, L.; Stohr, B.A.; Berry, S.; Lockwood, C.M.; Davis, J.L.; Rudzinski, E.R.; Kunder, C.A. Recurrent EGFR Alterations in NTRK3 Fusion Negative Congenital Mesoblastic Nephroma. Pract. Lab. Med. 2020, 21, e00164. [Google Scholar] [CrossRef]
- Tsakiri, K.; Kotoula, V.; Lakis, S.; Müller, J.; Fostira, F.; Bobos, M.; Hytiroglou, P.; Fountzilas, G. Crizotinib Failure in a TPM4-ALK-Rearranged Inflammatory Myofibroblastic Tumor with an Emerging ALK Kinase Domain Mutation. JCO Precis. Oncol. 2017, 1, 1–7. [Google Scholar] [CrossRef]
- Lawlor, E.R.; Thiele, C.J. Epigenetic Changes in Pediatric Solid Tumors: Promising New Targets. Clin. Cancer Res. 2012, 18, 2768–2779. [Google Scholar] [CrossRef] [Green Version]
- Panditharatna, E.; Filbin, M.G. The Growing Role of Epigenetics in Childhood Cancers. Curr. Opin. Pediatr. 2020, 32, 67–75. [Google Scholar] [CrossRef]
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of Genome Folding into Topologically Associating Domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator Dysfunction and Oncogene Activation in IDH Mutant Gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of New Chromatin Domains Determines Pathogenicity of Genomic Duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers 2019, 5, 1. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Chi, Y.-Y.; Anderson, J.R.; Tian, J.; Arndt, C.A.S.; Bomgaars, L.; Donaldson, S.S.; Hayes-Jordan, A.; Mascarenhas, L.; McCarville, M.B.; et al. Addition of Vincristine and Irinotecan to Vincristine, Dactinomycin, and Cyclophosphamide Does Not Improve Outcome for Intermediate-Risk Rhabdomyosarcoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2018, 36, 2770–2777. [Google Scholar] [CrossRef]
- Weigel, B.J.; Lyden, E.; Anderson, J.R.; Meyer, W.H.; Parham, D.M.; Rodeberg, D.A.; Michalski, J.M.; Hawkins, D.S.; Arndt, C.A.S. Intensive Multiagent Therapy, Including Dose-Compressed Cycles of Ifosfamide/Etoposide and Vincristine/Doxorubicin/Cyclophosphamide, Irinotecan, and Radiation, in Patients with High-Risk Rhabdomyosarcoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2016, 34, 117–122. [Google Scholar] [CrossRef]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.A.; Parham, D.M.; Cripe, T.P.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Shorter-Duration Therapy Using Vincristine, Dactinomycin, and Lower-Dose Cyclophosphamide with or without Radiotherapy for Patients with Newly Diagnosed Low-Risk Rhabdomyosarcoma: A Report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J. Clin. Oncol. 2014, 32, 3547–3552. [Google Scholar]
- Pratt, C.B.; Pappo, A.S.; Gieser, P.; Jenkins, J.J.; Salzbergdagger, A.; Neff, J.; Rao, B.; Green, D.; Thomas, P.; Marcus, R.; et al. Role of Adjuvant Chemotherapy in the Treatment of Surgically Resected Pediatric Nonrhabdomyosarcomatous Soft Tissue Sarcomas: A Pediatric Oncology Group Study. J. Clin. Oncol. 1999, 17, 1219. [Google Scholar] [CrossRef]
- Parham, D.M.; Webber, B.L.; Jenkins, J.J., 3rd; Cantor, A.B.; Maurer, H.M. Nonrhabdomyosarcomatous Soft Tissue Sarcomas of Childhood: Formulation of a Simplified System for Grading. Mod. Pathol. 1995, 8, 705–710. [Google Scholar]
- Kim, S.Y.; Janeway, K.; Pappo, A. Pediatric and Wild-Type Gastrointestinal Stromal Tumor: New Therapeutic Approaches. Curr. Opin. Oncol. 2010, 22, 347–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, P.; Casanova, M.; Ferrari, A.; Fordham, A.; Trahair, T.; Venkatramani, R. Inflammatory Myofibroblastic Tumor: Molecular Landscape, Targeted Therapeutics, and Remaining Challenges. Curr. Probl. Cancer 2021, 45, 100768. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in Patients with TRK Fusion-Positive Solid Tumours: A Pooled Analysis of Three Phase 1/2 Clinical Trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Eaton, B.R.; Schwarz, R.; Vatner, R.; Yeh, B.; Claude, L.; Indelicato, D.J.; Laack, N. Osteosarcoma. Pediatr. Blood Cancer 2021, 68 (Suppl. 2), e28352. [Google Scholar] [CrossRef] [PubMed]
- Lilienthal, I.; Herold, N. Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies. Int. J. Mol. Sci. 2020, 21, 6885. [Google Scholar] [CrossRef]
- Kushlinskii, N.E.; Fridman, M.V.; Braga, E.A. Molecular Mechanisms and microRNAs in Osteosarcoma Pathogenesis. Biochemistry 2016, 81, 315–328. [Google Scholar] [CrossRef]
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex but Targetable Ecosystem. Cells 2020, 9, 976. [Google Scholar] [CrossRef] [Green Version]
- Raney, R.B.; Asmar, L.; Newton, W.A., Jr.; Bagwell, C.; Breneman, J.C.; Crist, W.; Gehan, E.A.; Webber, B.; Wharam, M.; Wiener, E.S.; et al. Ewing’s Sarcoma of Soft Tissues in Childhood: A Report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J. Clin. Oncol. 1997, 15, 574–582. [Google Scholar] [CrossRef]
- Leavey, P.J.; Laack, N.N.; Krailo, M.D.; Buxton, A.; Randall, R.L.; DuBois, S.G.; Reed, D.R.; Grier, H.E.; Hawkins, D.S.; Pawel, B.; et al. Phase III Trial Adding Vincristine-Topotecan-Cyclophosphamide to the Initial Treatment of Patients with Nonmetastatic Ewing Sarcoma: A Children’s Oncology Group Report. J. Clin. Oncol. 2021, 39, 4029–4038. [Google Scholar] [CrossRef]
- Cash, T.; McIlvaine, E.; Krailo, M.D.; Lessnick, S.L.; Lawlor, E.R.; Laack, N.; Sorger, J.; Marina, N.; Grier, H.E.; Granowetter, L.; et al. Comparison of Clinical Features and Outcomes in Patients with Extraskeletal versus Skeletal Localized Ewing Sarcoma: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2016, 63, 1771–1779. [Google Scholar] [CrossRef]
- Castex, M.-P.; Rubie, H.; Stevens, M.C.G.; Escribano, C.C.; de Gauzy, J.S.; Gomez-Brouchet, A.; Rey, A.; Delattre, O.; Oberlin, O. Extraosseous Localized Ewing Tumors: Improved Outcome with Anthracyclines--the French Society of Pediatric Oncology and International Society of Pediatric Oncology. J. Clin. Oncol. 2007, 25, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navin, N.E.; Chen, K. Genotyping Tumor Clones from Single-Cell Data. Nat. Methods 2016, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Suvà, M.L. Deciphering Human Tumor Biology by Single-Cell Expression Profiling. Annu. Rev. Cancer Biol. 2019, 3, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217.e12. [Google Scholar] [CrossRef] [Green Version]
- Leung, M.L.; Davis, A.; Gao, R.; Casasent, A.; Wang, Y.; Sei, E.; Vilar, E.; Maru, D.; Kopetz, S.; Navin, N.E. Single-Cell DNA Sequencing Reveals a Late-Dissemination Model in Metastatic Colorectal Cancer. Genome Res. 2017, 27, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Francis, J.M.; Zhang, C.-Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S.; et al. EGFR Variant Heterogeneity in Glioblastoma Resolved through Single-Nucleus Sequencing. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef] [Green Version]
- Gawad, C.; Koh, W.; Quake, S.R. Single-Cell Genome Sequencing: Current State of the Science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-Cell RNA-Seq Enables Comprehensive Tumour and Immune Cell Profiling in Primary Breast Cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Zhang, L.; Zhang, Y.; Li, Z.; Siemers, N.; Zhang, Z. Insights Gained from Single-Cell Analysis of Immune Cells in the Tumor Microenvironment. Annu. Rev. Immunol. 2021, 39, 583–609. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.; Carissimo, A.; Panariello, F.; Grimaldi, A.; Bouché, V.; Gambardella, G.; Cacchiarelli, D. Single-Cell RNA Sequencing Analysis: A Step-by-Step Overview. Methods Mol. Biol. 2021, 2284, 343–365. [Google Scholar]
- Andrews, T.S.; Kiselev, V.Y.; McCarthy, D.; Hemberg, M. Tutorial: Guidelines for the Computational Analysis of Single-Cell RNA Sequencing Data. Nat. Protoc. 2021, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Pomella, S.; Sayers, C.; Wu, X.S.; Song, Y.; Chiarella, A.M.; Bagchi, S.; Chou, H.-C.; Sinniah, R.S.; Walton, A.; et al. Histone Hyperacetylation Disrupts Core Gene Regulatory Architecture in Rhabdomyosarcoma. Nat. Genet. 2019, 51, 1714–1722. [Google Scholar] [CrossRef] [PubMed]
- Hovestadt, V.; Smith, K.S.; Bihannic, L.; Filbin, M.G.; Shaw, M.L.; Baumgartner, A.; DeWitt, J.C.; Groves, A.; Mayr, L.; Weisman, H.R.; et al. Resolving Medulloblastoma Cellular Architecture by Single-Cell Genomics. Nature 2019, 572, 74–79. [Google Scholar] [CrossRef]
- Andersson, N.; Bakker, B.; Karlsson, J.; Valind, A.; Holmquist Mengelbier, L.; Spierings, D.C.J.; Foijer, F.; Gisselsson, D. Extensive Clonal Branching Shapes the Evolutionary History of High-Risk Pediatric Cancers. Cancer Res. 2020, 80, 1512–1523. [Google Scholar] [CrossRef]
- Aynaud, M.-M.; Mirabeau, O.; Gruel, N.; Grossetête, S.; Boeva, V.; Durand, S.; Surdez, D.; Saulnier, O.; Zaïdi, S.; Gribkova, S.; et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep. 2020, 30, 1767–1779.e6. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.; Xia, T.; Ye, C.-J.; Zhan, Y.; Yang, R.; Liu, J.; Li, Y.; Chen, Z.-X.; Yao, W.; Li, K.; et al. Single-Cell Transcriptional Profiling Reveals the Heterogeneity in Embryonal Rhabdomyosarcoma. Medicine 2021, 100, e26775. [Google Scholar] [CrossRef]
- Wu, H.; Fu, R.; Zhang, Y.-H.; Liu, Z.; Chen, Z.; Xu, J.; Tian, Y.; Jin, W.; Wong, S.Z.H.; Wu, Q.-F. Single-Cell RNA Sequencing of Pediatric Ependymoma Unravels Subclonal Heterogeneity Associated with Patient Survival. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gojo, J.; Englinger, B.; Jiang, L.; Hübner, J.M.; Shaw, M.L.; Hack, O.A.; Madlener, S.; Kirchhofer, D.; Liu, I.; Pyrdol, J.; et al. Single-Cell RNA-Seq Reveals Cellular Hierarchies and Impaired Developmental Trajectories in Pediatric Ependymoma. Cancer Cell 2020, 38, 44–59.e9. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.-D.; Ye, C.-J.; Yao, X.-Y.; Luo, W.-Q.; Cheng, X.-M.; Miao, J.-J.; Wang, J.-F.; et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 2020, 38, 716–733.e6. [Google Scholar] [CrossRef] [PubMed]
- Khoogar, R.; Li, F.; Chen, Y.; Ignatius, M.; Lawlor, E.R.; Kitagawa, K.; Huang, T.H.-M.; Phelps, D.A.; Houghton, P.J. Single-Cell RNA Profiling Identifies Diverse Cellular Responses to EWSR1/FLI1 Downregulation in Ewing Sarcoma Cells. Cell. Oncol. 2022, 45, 19–40. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Neftel, C.; Shore, M.E.; Weisman, H.R.; Mathewson, N.D.; McBride, M.J.; Haas, B.; Izar, B.; Volorio, A.; Boulay, G.; et al. Opposing Immune and Genetic Mechanisms Shape Oncogenic Programs in Synovial Sarcoma. Nat. Med. 2021, 27, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.E.; Gorthi, A.; Bassani, N.; Lawrence, L.A.; Iskra, B.S.; Bishop, A.J.R. Reconstruction of Ewing Sarcoma Developmental Context from Mass-Scale Transcriptomics Reveals Characteristics of EWSR1-FLI1 Permissibility. Cancers 2020, 12, 948. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-Cell RNA Landscape of Intratumoral Heterogeneity and Immunosuppressive Microenvironment in Advanced Osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Patel, A.G.; Chen, X.; Huang, X.; Clay, M.R.; Komorova, N.; Krasin, M.J.; Pappo, A.; Tillman, H.; Orr, B.A.; McEvoy, J.; et al. The Myogenesis Program Drives Clonal Selection and Drug Resistance in Rhabdomyosarcoma. Dev. Cell 2022. [Google Scholar] [CrossRef]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and Temporal Diversity in Genomic Instability Processes Defines Lung Cancer Evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Yudushkin, I.A.; Schleifenbaum, A.; Kinkhabwala, A.; Neel, B.G.; Schultz, C.; Bastiaens, P.I.H. Live-Cell Imaging of Enzyme-Substrate Interaction Reveals Spatial Regulation of PTP1B. Science 2007, 315, 115–119. [Google Scholar] [CrossRef]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In Situ Sequencing for RNA Analysis in Preserved Tissue and Cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-Cell In Situ RNA Profiling by Sequential Hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.F.; Li, C.; Amamoto, R.; et al. Highly Multiplexed Subcellular RNA Sequencing in Situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA Imaging. Spatially Resolved, Highly Multiplexed RNA Profiling in Single Cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren, L.; Bosse, M.; Marquez, D.; Angoshtari, R.; Jain, S.; Varma, S.; Yang, S.-R.; Kurian, A.; Van Valen, D.; West, R.; et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 2018, 174, 1373–1387.e19. [Google Scholar] [CrossRef] [Green Version]
- Codeluppi, S.; Borm, L.E.; Zeisel, A.; La Manno, G.; van Lunteren, J.A.; Svensson, C.I.; Linnarsson, S. Spatial Organization of the Somatosensory Cortex Revealed by osmFISH. Nat. Methods 2018, 15, 932–935. [Google Scholar] [CrossRef]
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.; Black, S.; Nolan, G.P. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018, 174, 968–981.e15. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, J.R.; Bambah-Mukku, D.; Eichhorn, S.W.; Vaughn, E.; Shekhar, K.; Perez, J.D.; Rubinstein, N.D.; Hao, J.; Regev, A.; Dulac, C.; et al. Molecular, Spatial, and Functional Single-Cell Profiling of the Hypothalamic Preoptic Region. Science 2018, 362, eaau5324. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J.; et al. Three-Dimensional Intact-Tissue Sequencing of Single-Cell Transcriptional States. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.-H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.-C.; et al. Transcriptome-Scale Super-Resolved Imaging in Tissues by RNA seqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-Definition Spatial Transcriptomics for in Situ Tissue Profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Onozato, M.L.; Yapp, C.; Richardson, D.; Sundaresan, T.; Chahal, V.; Lee, J.; Sullivan, J.P.; Madden, M.W.; Shim, H.S.; Liebers, M.; et al. Highly Multiplexed Fluorescence in Situ Hybridization for in Situ Genomics. J. Mol. Diagn. 2019, 21, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Vickovic, S.; Lötstedt, B.; Klughammer, J.; Segerstolpe, Å.; Rozenblatt-Rosen, O.; Regev, A. SM-Omics: An Automated Platform for High-Throughput Spatial Multi-Omics. Nat. Commun. 2022, 13, 795. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681. [Google Scholar] [CrossRef]
- McMahon, N.P.; Jones, J.A.; Kwon, S.; Chin, K.; Nederlof, M.A.; Gray, J.W.; Gibbs, S.L. Oligonucleotide Conjugated Antibodies Permit Highly Multiplexed Immunofluorescence for Future Use in Clinical Histopathology. J. Biomed. Opt. 2020, 25, 056004. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Chiang, Z.D.; Morriss, J.W.; LaFave, L.M.; Murray, E.M.; Del Priore, I.; Meli, K.; Lareau, C.A.; Nadaf, N.M.; Li, J.; et al. Spatial Genomics Enables Multi-Modal Study of Clonal Heterogeneity in Tissues. Nature 2022, 601, 85–91. [Google Scholar] [CrossRef]
- Payne, A.C.; Chiang, Z.D.; Reginato, P.L.; Mangiameli, S.M.; Murray, E.M.; Yao, C.-C.; Markoulaki, S.; Earl, A.S.; Labade, A.S.; Jaenisch, R.; et al. In Situ Genome Sequencing Resolves DNA Sequence and Structure in Intact Biological Samples. Science 2021, 371, eaay3446. [Google Scholar] [CrossRef]
- Lundberg, E.; Borner, G.H.H. Spatial Proteomics: A Powerful Discovery Tool for Cell Biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-Spectrometry-Based Draft of the Human Proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Zhang, M.; Sheffield, T.; Zhan, X.; Li, Q.; Yang, D.M.; Wang, Y.; Wang, S.; Xie, Y.; Wang, T.; Xiao, G. Spatial Molecular Profiling: Platforms, Applications and Analysis Tools. Brief. Bioinform. 2021, 22, bbaa145. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Huang, R.; Jin, J.; Gao, J.; Liu, F.; Wei, Z.; Xu, X.; Chang, Z.; Lin, J.; Ta, N.; et al. A Comparative Integrated Multi-Omics Analysis Identifies CA2 as a Novel Target for Chordoma. Neuro. Oncol. 2021, 23, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- Turczyk, B.M.; Busby, M.; Martin, A.L.; Daugharthy, E.R.; Myung, D.; Terry, R.C.; Inverso, S.A.; Kohman, R.E.; Church, G.M. Spatial Sequencing: A Perspective. J. Biomol. Tech. 2020, 31, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Weinberg, R.A. Modelling the Molecular Circuitry of Cancer. Nat. Rev. Cancer 2002, 2, 331–341. [Google Scholar] [CrossRef]
- Weinberg, R. Point: Hypotheses First. Nature 2010, 464, 678. [Google Scholar] [CrossRef]
- Kile, B.T.; Hilton, D.J. The Art and Design of Genetic Screens: Mouse. Nat. Rev. Genet. 2005, 6, 557–567. [Google Scholar] [CrossRef]
- Grimm, S. The Art and Design of Genetic Screens: Mammalian Culture Cells. Nat. Rev. Genet. 2004, 5, 179–189. [Google Scholar] [CrossRef]
- Patton, E.E.; Zon, L.I. The Art and Design of Genetic Screens: Zebrafish. Nat. Rev. Genet. 2001, 2, 956–966. [Google Scholar] [CrossRef]
- Schneeberger, K. Using next-Generation Sequencing to Isolate Mutant Genes from Forward Genetic Screens. Nat. Rev. Genet. 2014, 15, 662–676. [Google Scholar] [CrossRef]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered Regularly Interspaced Short Palindrome Repeats (CRISPRs) Have Spacers of Extrachromosomal Origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef] [Green Version]
- Garneau, J.E.; Dupuis, M.-È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas Bacterial Immune System Cleaves Bacteriophage and Plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA Maturation by Trans-Encoded Small RNA and Host Factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA Ribonucleoprotein Complex Mediates Specific DNA Cleavage for Adaptive Immunity in Bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus Thermophilus CRISPR/Cas System Provides Immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in Sarcoma Genomics and New Therapeutic Targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Seebacher, N.A.; Hornicek, F.; Guo, Z.; Duan, Z. Advances in Sarcoma Gene Mutations and Therapeutic Targets. Cancer Treat. Rev. 2018, 62, 98–109. [Google Scholar] [CrossRef]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016, 167, 1853–1866.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nuñez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 2016, 167, 1867–1882.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Replogle, J.M.; Norman, T.M.; Xu, A.; Hussmann, J.A.; Chen, J.; Cogan, J.Z.; Meer, E.J.; Terry, J.M.; Riordan, D.P.; Srinivas, N.; et al. Combinatorial Single-Cell CRISPR Screens by Direct Guide RNA Capture and Targeted Sequencing. Nat. Biotechnol. 2020, 38, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Stolte, B.; Iniguez, A.B.; Dharia, N.V.; Robichaud, A.L.; Conway, A.S.; Morgan, A.M.; Alexe, G.; Schauer, N.J.; Liu, X.; Bird, G.H.; et al. Genome-Scale CRISPR-Cas9 Screen Identifies Druggable Dependencies in TP53 Wild-Type Ewing Sarcoma. J. Exp. Med. 2018, 215, 2137–2155. [Google Scholar] [CrossRef] [PubMed]
- Oberlick, E.M.; Rees, M.G.; Seashore-Ludlow, B.; Vazquez, F.; Nelson, G.M.; Dharia, N.V.; Weir, B.A.; Tsherniak, A.; Ghandi, M.; Krill-Burger, J.M.; et al. Small-Molecule and CRISPR Screening Converge to Reveal Receptor Tyrosine Kinase Dependencies in Pediatric Rhabdoid Tumors. Cell Rep. 2019, 28, 2331–2344.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, R.; Shen, B.; Li, N.; Zhou, H.; Wang, W.; Zhao, Y.; Huang, M.; Fang, P.; Wang, S.; et al. High-Throughput Functional Screening for next-Generation Cancer Immunotherapy Using Droplet-Based Microfluidics. Sci. Adv. 2021, 7, eabe3839. [Google Scholar] [CrossRef] [PubMed]
- Seong, B.K.A.; Dharia, N.V.; Lin, S.; Donovan, K.A.; Chong, S.; Robichaud, A.; Conway, A.; Hamze, A.; Ross, L.; Alexe, G.; et al. TRIM8 Modulates the EWS/FLI Oncoprotein to Promote Survival in Ewing Sarcoma. Cancer Cell 2021, 39, 1262–1278.e7. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.P.; Bailey, J.N.; Vleeshouwer-Neumann, T.; Chen, E.Y. CRISPR Screen Identifies the NCOR/HDAC3 Complex as a Major Suppressor of Differentiation in Rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 15090–15095. [Google Scholar] [CrossRef] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In Vivo CRISPR Screening Identifies Ptpn2 as a Cancer Immunotherapy Target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.; Hamilton, M.; Shima, Y.; Chambers, K.; Spinler, K.; Van Nostrand, E.L.; Yee, B.A.; Blue, S.M.; Chen, M.; Rizzeri, D.; et al. An In Vivo Genome-Wide CRISPR Screen Identifies the RNA-Binding Protein Staufen2 as a Key Regulator of Myeloid Leukemia. Nat. Cancer 2020, 1, 410–422. [Google Scholar] [CrossRef]
- Dong, M.B.; Wang, G.; Chow, R.D.; Ye, L.; Zhu, L.; Dai, X.; Park, J.J.; Kim, H.R.; Errami, Y.; Guzman, C.D.; et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 2019, 178, 1189–1204.e23. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional Genomics Reveal That the Serine Synthesis Pathway Is Essential in Breast Cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossi, D.; Cicalese, A.; Dellino, G.I.; Luzi, L.; Riva, L.; D’Alesio, C.; Diaferia, G.R.; Carugo, A.; Cavallaro, E.; Piccioni, R.; et al. In Vivo Genetic Screens of Patient-Derived Tumors Revealed Unexpected Frailty of the Transformed Phenotype. Cancer Discov. 2016, 6, 650–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-Wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [Green Version]
- Chow, R.D.; Guzman, C.D.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.A.; Zhang, S.; et al. AAV-Mediated Direct in Vivo CRISPR Screen Identifies Functional Suppressors in Glioblastoma. Nat. Neurosci. 2017, 20, 1329. [Google Scholar] [CrossRef] [Green Version]
- Bric, A.; Miething, C.; Bialucha, C.U.; Scuoppo, C.; Zender, L.; Krasnitz, A.; Xuan, Z.; Zuber, J.; Wigler, M.; Hicks, J.; et al. Functional Identification of Tumor-Suppressor Genes through an in Vivo RNA Interference Screen in a Mouse Lymphoma Model. Cancer Cell 2009, 16, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Carugo, A.; Genovese, G.; Seth, S.; Nezi, L.; Rose, J.L.; Bossi, D.; Cicalese, A.; Shah, P.K.; Viale, A.; Pettazzoni, P.F.; et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Braun, C.J.; Bruno, P.M.; Horlbeck, M.A.; Gilbert, L.A.; Weissman, J.S.; Hemann, M.T. Versatile In Vivo Regulation of Tumor Phenotypes by dCas9-Mediated Transcriptional Perturbation. Proc. Natl. Acad. Sci. USA 2016, 113, E3892–E3900. [Google Scholar] [CrossRef] [Green Version]
- Yeddula, N.; Xia, Y.; Ke, E.; Beumer, J.; Verma, I.M. Screening for Tumor Suppressors: Loss of Ephrin Receptor A2 Cooperates with Oncogenic KRas in Promoting Lung Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, E6476–E6485. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.; Santinha, A.J.; Platt, R.J. Moving from In Vitro to In Vivo CRISPR Screens. Gene Genome Ed. 2021, 2, 100008. [Google Scholar] [CrossRef]
- Borinstein, S.C.; Beeler, N.; Block, J.J.; Gorlick, R.; Grohar, P.; Jedlicka, P.; Krailo, M.; Morris, C.; Phillips, S.; Siegal, G.P.; et al. A Decade in Banking Ewing Sarcoma: A Report from the Children’s Oncology Group. Front. Oncol. 2013, 3, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, J.; Krailo, M.; Tello, T.; Marina, N.; Janeway, K.; Barkauskas, D.; Fan, T.M.; Gorlick, R.; Khanna, C.; COG Osteosarcoma Biology Group. A Summary of the Osteosarcoma Banking Efforts: A Report from the Children’s Oncology Group and the QuadW Foundation. Pediatr. Blood Cancer 2015, 62, 450–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volchenboum, S.L.; Andrade, J.; Huang, L.; Barkauskas, D.A.; Krailo, M.; Womer, R.B.; Ranft, A.; Potratz, J.; Dirksen, U.; Triche, T.J.; et al. Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group. J. Pathol. Clin. Res. 2015, 1, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.H.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.G.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent Clonal Selection Dominates Medulloblastoma at Recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep Learning: New Computational Modelling Techniques for Genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Libbrecht, M.W.; Noble, W.S. Machine Learning Applications in Genetics and Genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Zrimec, J.; Börlin, C.S.; Buric, F.; Muhammad, A.S.; Chen, R.; Siewers, V.; Verendel, V.; Nielsen, J.; Töpel, M.; Zelezniak, A. Deep Learning Suggests That Gene Expression Is Encoded in All Parts of a Co-Evolving Interacting Gene Regulatory Structure. Nat. Commun. 2020, 11, 6141. [Google Scholar] [CrossRef]
- Jiao, W.; Atwal, G.; Polak, P.; Karlic, R.; Cuppen, E.; PCAWG Tumor Subtypes and Clinical Translation Working Group; Danyi, A.; de Ridder, J.; van Herpen, C.; Lolkema, M.P.; et al. A Deep Learning System Accurately Classifies Primary and Metastatic Cancers Using Passenger Mutation Patterns. Nat. Commun. 2020, 11, 728. [Google Scholar] [CrossRef]
- He, H.; Yan, S.; Lyu, D.; Xu, M.; Ye, R.; Zheng, P.; Lu, X.; Wang, L.; Ren, B. Deep Learning for Biospectroscopy and Biospectral Imaging: State-of-the-Art and Perspectives. Anal. Chem. 2021, 93, 3653–3665. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and Mutation Prediction from Non–small Cell Lung Cancer Histopathology Images Using Deep Learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Shamai, G.; Binenbaum, Y.; Slossberg, R.; Duek, I.; Gil, Z.; Kimmel, R. Artificial Intelligence Algorithms to Assess Hormonal Status from Tissue Microarrays in Patients with Breast Cancer. JAMA Netw. Open 2019, 2, e197700. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Nomikou, S.; Coudray, N.; Jour, G.; Dawood, Z.; Hong, R.; Esteva, E.; Sakellaropoulos, T.; Donnelly, D.; Moran, U.; et al. Deep Learning and Pathomics Analyses Reveal Cell Nuclei as Important Features for Mutation Prediction of BRAF-Mutated Melanomas. J. Investig. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rajkomar, A.; Dean, J.; Kohane, I. Machine Learning in Medicine. N. Engl. J. Med. 2019, 380, 1347–1358. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Jayatunga, M.K.P.; Xie, W.; Ruder, L.; Schulze, U.; Meier, C. AI in Small-Molecule Drug Discovery: A Coming Wave? Nat. Rev. Drug Discov. 2022, 21, 175–176. [Google Scholar] [CrossRef]
- Xu, W.; Hao, D.; Hou, F.; Zhang, D.; Wang, H. Soft Tissue Sarcoma: Preoperative MRI-Based Radiomics and Machine Learning May Be Accurate Predictors of Histopathologic Grade. AJR Am. J. Roentgenol. 2020, 215, 963–969. [Google Scholar] [CrossRef]
- Blackledge, M.D.; Winfield, J.M.; Miah, A.; Strauss, D.; Thway, K.; Morgan, V.A.; Collins, D.J.; Koh, D.-M.; Leach, M.O.; Messiou, C. Supervised Machine-Learning Enables Segmentation and Evaluation of Heterogeneous Post-Treatment Changes in Multi-Parametric MRI of Soft-Tissue Sarcoma. Front. Oncol. 2019, 9, 941. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wang, J.; Sun, M.; Chen, X.; Xu, D.; Li, Z.-P.; Ma, J.; Feng, S.-T.; Gao, Z. Feasibility of Multi-Parametric Magnetic Resonance Imaging Combined with Machine Learning in the Assessment of Necrosis of Osteosarcoma after Neoadjuvant Chemotherapy: A Preliminary Study. BMC Cancer 2020, 20, 322. [Google Scholar] [CrossRef] [Green Version]
- Peeken, J.C.; Neumann, J.; Asadpour, R.; Leonhardt, Y.; Moreira, J.R.; Hippe, D.S.; Klymenko, O.; Foreman, S.C.; von Schacky, C.E.; Spraker, M.B.; et al. Prognostic Assessment in High-Grade Soft-Tissue Sarcoma Patients: A Comparison of Semantic Image Analysis and Radiomics. Cancers 2021, 13, 1929. [Google Scholar] [CrossRef]
- Tian, L.; Zhang, D.; Bao, S.; Nie, P.; Hao, D.; Liu, Y.; Zhang, J.; Wang, H. Radiomics-Based Machine-Learning Method for Prediction of Distant Metastasis from Soft-Tissue Sarcomas. Clin. Radiol. 2021, 76, 158.e19–158.e25. [Google Scholar] [CrossRef]
- Zhang, M.; Tong, E.; Hamrick, F.; Lee, E.H.; Tam, L.T.; Pendleton, C.; Smith, B.W.; Hug, N.F.; Biswal, S.; Seekins, J.; et al. Machine-Learning Approach to Differentiation of Benign and Malignant Peripheral Nerve Sheath Tumors: A Multicenter Study. Neurosurgery 2021, 89, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.; Zhuo, Y.; Lahoud, R.M.; Tian, F.; Ma, X.; Xing, F.; Moteabbed, M.; Liu, X.; Grogg, K.; Shusharina, N.; et al. Deep Learning-Based GTV Contouring Modeling Inter- and Intra- Observer Variability in Sarcomas. Radiother. Oncol. 2022, 167, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Ström, P.; Kartasalo, K.; Olsson, H.; Solorzano, L.; Delahunt, B.; Berney, D.M.; Bostwick, D.G.; Evans, A.J.; Grignon, D.J.; Humphrey, P.A.; et al. Artificial Intelligence for Diagnosis and Grading of Prostate Cancer in Biopsies: A Population-Based, Diagnostic Study. Lancet Oncol. 2020, 21, 222–232. [Google Scholar] [CrossRef]
- van Leenders, G.J.L.H.; van der Kwast, T.H.; Grignon, D.J.; Evans, A.J.; Kristiansen, G.; Kweldam, C.F.; Litjens, G.; McKenney, J.K.; Melamed, J.; Mottet, N.; et al. The 2019 International Society of Urological Pathology (ISUP) Consensus Conference on Grading of Prostatic Carcinoma. Am. J. Surg. Pathol. 2020, 44, e87–e99. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.F.; Nagpal, K.; Sayres, R.; Foote, D.J.; Wedin, B.D.; Pearce, A.; Cai, C.J.; Winter, S.R.; Symonds, M.; Yatziv, L.; et al. Evaluation of the Use of Combined Artificial Intelligence and Pathologist Assessment to Review and Grade Prostate Biopsies. JAMA Netw. Open 2020, 3, e2023267. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.-H.; Wang, F.; Berry, G.J.; Ré, C.; Altman, R.B.; Snyder, M.; Kohane, I.S. Classifying Non-Small Cell Lung Cancer Types and Transcriptomic Subtypes Using Convolutional Neural Networks. J. Am. Med. Inform. Assoc. 2020, 27, 757–769. [Google Scholar] [CrossRef] [PubMed]
- van IJzendoorn, D.G.P.; Szuhai, K.; Briaire-de Bruijn, I.H.; Kostine, M.; Kuijjer, M.L.; Bovée, J.V.M.G. Machine Learning Analysis of Gene Expression Data Reveals Novel Diagnostic and Prognostic Biomarkers and Identifies Therapeutic Targets for Soft Tissue Sarcomas. PLoS Comput. Biol. 2019, 15, e1006826. [Google Scholar] [CrossRef]
- Lietz, C.E.; Garbutt, C.; Barry, W.T.; Deshpande, V.; Chen, Y.-L.; Lozano-Calderon, S.A.; Wang, Y.; Lawney, B.; Ebb, D.; Cote, G.M.; et al. MicroRNA-mRNA Networks Define Translatable Molecular Outcome Phenotypes in Osteosarcoma. Sci. Rep. 2020, 10, 4409. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.O.; Lathara, M.; Shaw, C.Y.; Wogmon, O.; Jackson, J.M.; Clark, M.M.; Eshraghi, N.; Keenen, S.E.; Woods, A.D.; Purohit, R.; et al. Machine Learning for Rhabdomyosarcoma Histopathology. Mod. Pathol. 2022, 1–11. [Google Scholar] [CrossRef]
- Ren, E.-H.; Deng, Y.-J.; Yuan, W.-H.; Wu, Z.-L.; Zhang, G.-Z.; Xie, Q.-Q. An Immune-Related Gene Signature for Determining Ewing Sarcoma Prognosis Based on Machine Learning. J. Cancer Res. Clin. Oncol. 2021, 147, 153–165. [Google Scholar] [CrossRef]
- Chaber, R.; Arthur, C.J.; Łach, K.; Raciborska, A.; Michalak, E.; Bilska, K.; Drabko, K.; Depciuch, J.; Kaznowska, E.; Cebulski, J. Predicting Ewing Sarcoma Treatment Outcome Using Infrared Spectroscopy and Machine Learning. Molecules 2019, 24, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.J.; Berndl, M.; Michael Ando, D.; Barch, M.; Narayanaswamy, A.; Christiansen, E.; Hoyer, S.; Roat, C.; Hung, J.; Rueden, C.T.; et al. Assessing Microscope Image Focus Quality with Deep Learning. BMC Bioinform. 2018, 19, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannikopoulos, P.; Parham, D.M. Pediatric Sarcomas: The Next Generation of Molecular Studies. Cancers 2022, 14, 2515. https://doi.org/10.3390/cancers14102515
Giannikopoulos P, Parham DM. Pediatric Sarcomas: The Next Generation of Molecular Studies. Cancers. 2022; 14(10):2515. https://doi.org/10.3390/cancers14102515
Chicago/Turabian StyleGiannikopoulos, Petros, and David M. Parham. 2022. "Pediatric Sarcomas: The Next Generation of Molecular Studies" Cancers 14, no. 10: 2515. https://doi.org/10.3390/cancers14102515
APA StyleGiannikopoulos, P., & Parham, D. M. (2022). Pediatric Sarcomas: The Next Generation of Molecular Studies. Cancers, 14(10), 2515. https://doi.org/10.3390/cancers14102515