
Looking at Thyroid Cancer from the Tumor-Suppressor Genes Point of View

 ,
,  , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Major Classes of Tumor-Suppressor Genes
2.1. Gatekeepers
2.2. Caretakers
2.3. Landscapers
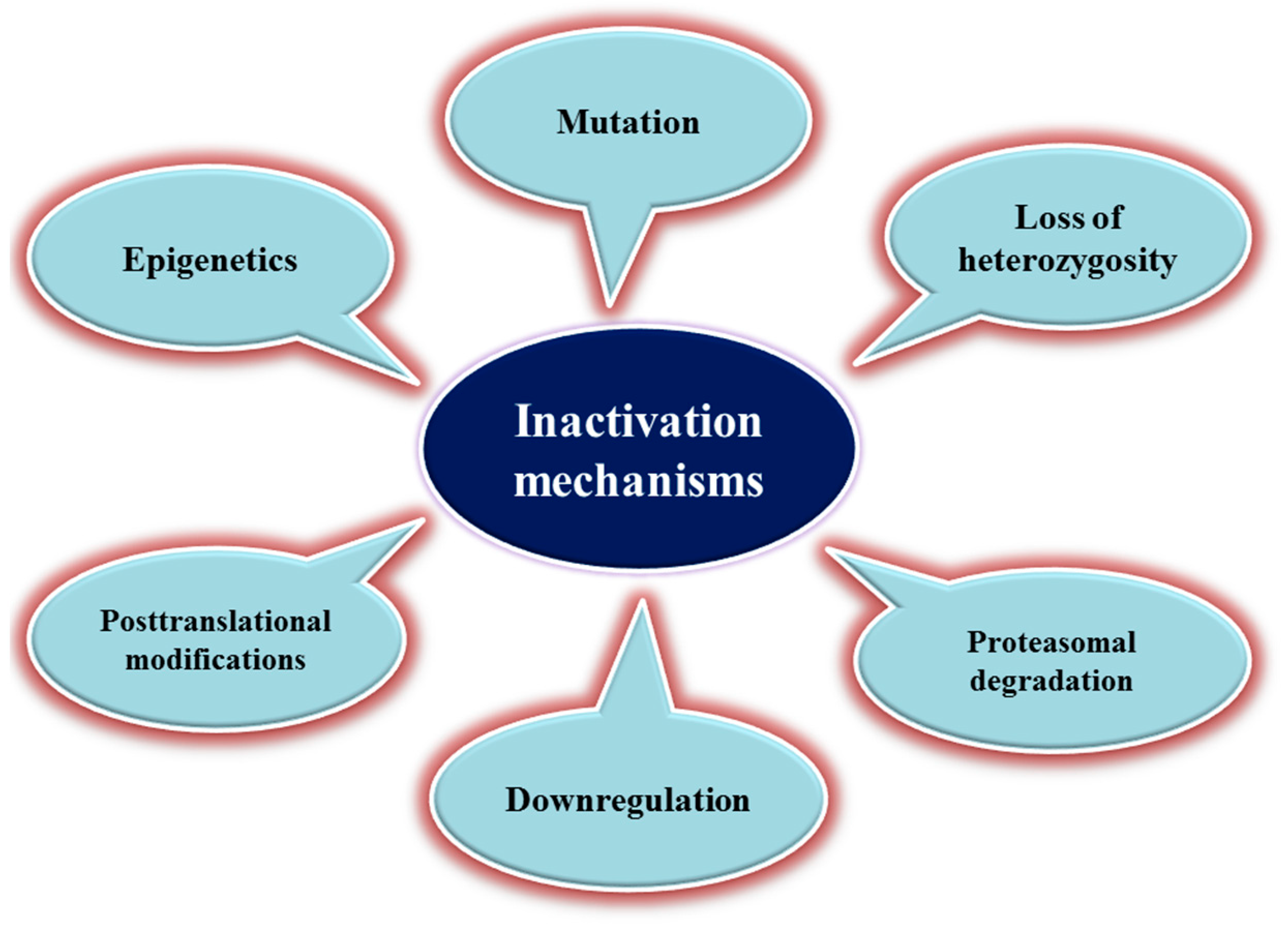
3. Mechanisms of TSG Inactivation
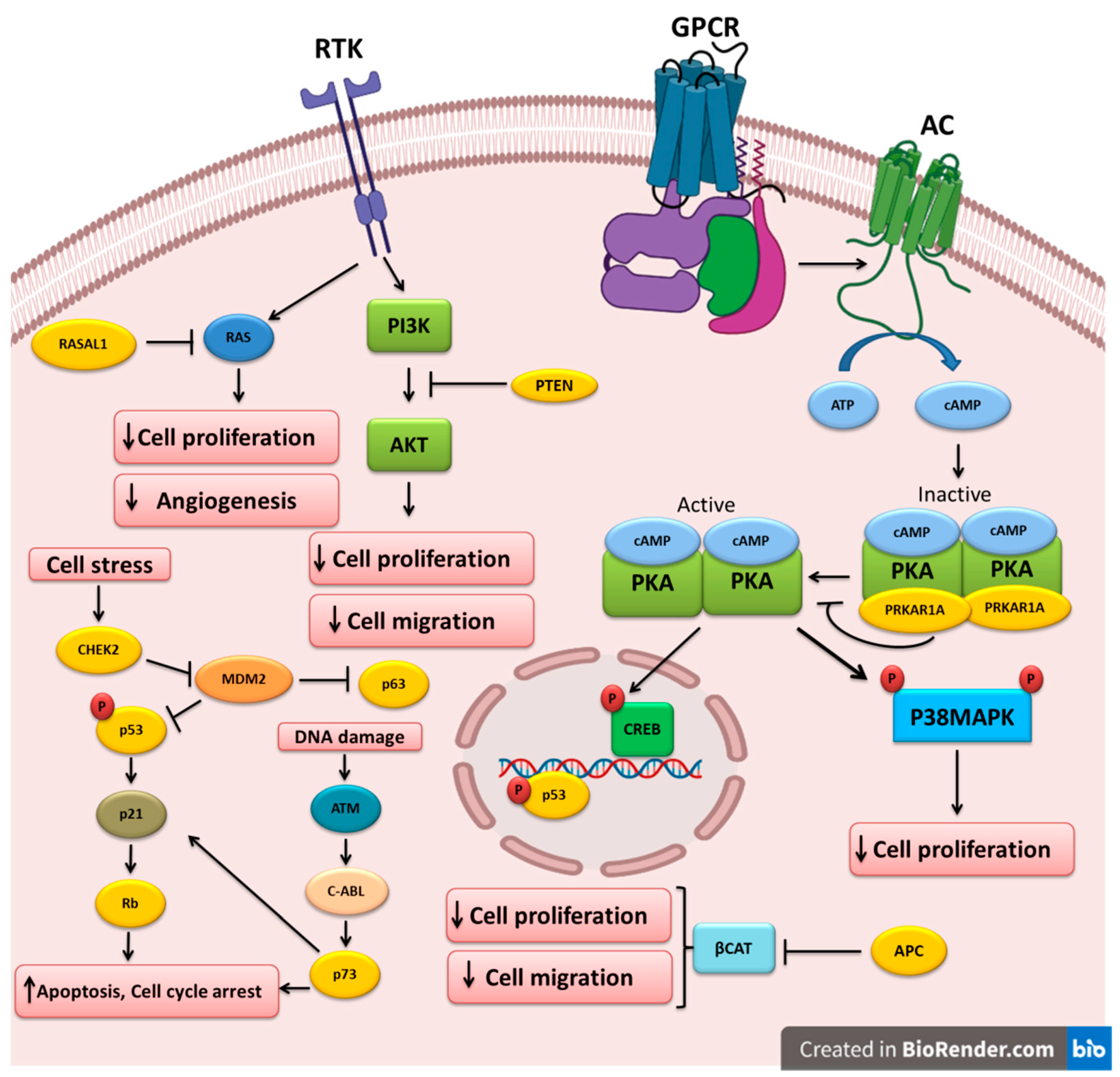
4. Altered Tumor-Suppressor Genes in Thyroid Cancer
4.1. TP53
4.2. PTEN
4.3. APC
4.4. RASAL1
4.5. TP63 and TP73
4.6. RB
4.7. PRKAR1A
4.8. CHEK2
5. Clinical Significance and Therapeutic Potential
6. Future Directions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rajabi, S.; Dehghan, M.H.; Dastmalchi, R.; Jalali Mashayekhi, F.; Salami, S.; Hedayati, M. The roles and role-players in thyroid cancer angiogenesis. Endocr. J. 2019, 66, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Vianna, D.M.; Curioni, O.A.; Franca, L.J.; Paiva, D.L.; Pompeu, B.F.; Dedivitis, R.A.; Rapoport, A. The histological rarity of thyroid cancer. Braz. J. Otorhinolaryngol. 2012, 78, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Howley, P.M.; Israel, M.A.; Gray, J.W.; Thompson, C. The Molecular Basis of Cancer, 4th ed.; Elsevier Inc.: New York, NY, USA, 2015; pp. 835–863. [Google Scholar]
- Lee, J.; Hwang, J.A.; Lee, E.K. Recent progress of genome study for anaplastic thyroid cancer. Genom. Inform. 2013, 11, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, S.; Hedayati, M. Medullary Thyroid Cancer: Clinical Characteristics and New Insights into Therapeutic Strategies Targeting Tyrosine Kinases. Mol. Diagn. Ther. 2017, 21, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, T.A.; Weil, M.M.; Story, M.D.; Strom, E.A.; Brock, W.A.; McNeese, M.D. Tumor suppressor genes and breast cancer. Radiat. Oncol. Investig. 1999, 7, 55–65. [Google Scholar] [CrossRef]
- Osborne, C.; Wilson, P.; Tripathy, D. Oncogenes and tumor suppressor genes in breast cancer: Potential diagnostic and therapeutic applications. Oncologist 2004, 9, 361–377. [Google Scholar] [CrossRef]
- Maziveyi, M.; Alahari, S.K. Breast Cancer Tumor Suppressors: A Special Emphasis on Novel Protein Nischarin. Cancer Res. 2015, 75, 4252–4259. [Google Scholar] [CrossRef]
- Oliveira, A.M.; Ross, J.S.; Fletcher, J.A. Tumor suppressor genes in breast cancer: The gatekeepers and the caretakers. Am. J. Clin. Pathol. 2005, 124, S16–S28. [Google Scholar]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Dardell, J. Proto-Oncogenes and Tumor-Suppressor Genes. In Molecular Cell Biology, 4th ed.; Freeman & Company: New York, NY, USA, 2000; pp. 845–867. [Google Scholar]
- Caldas, C.; Venkitaraman, A.R. Tumor Suppressor Genes. In Encyclopedia of Genetics, 3rd ed.; Miller, S.B.A.J.H., Ed.; Elsevier: Cambridge, UK, 2001; pp. 2081–2088. [Google Scholar]
- Ashworth, A.; Lord, C.J.; Reis-Filho, J.S. Genetic interactions in cancer progression and treatment. Cell 2011, 145, 30–38. [Google Scholar] [CrossRef]
- Macaluso, M.; Paggi, M.G.; Giordano, A. Genetic and epigenetic alterations as hallmarks of the intricate road to cancer. Oncogene 2003, 22, 6472–6478. [Google Scholar] [CrossRef]
- Frank, S.A. Somatic mutation: Early cancer steps depend on tissue architecture. Curr. Biol. 2003, 13, R261–R263. [Google Scholar] [CrossRef][Green Version]
- Van Heemst, D.; Den Reijer, P.; Westendorp, R. Ageing or cancer: A review: On the role of caretakers and gatekeepers. Eur. J. Cancer 2007, 43, 2144–2152. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Gatekeepers and caretakers. Nature 1997, 386, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Skotheim, J.M.; De Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Macleod, K.F. The RB tumor suppressor: A gatekeeper to hormone independence in prostate cancer? J. Clin. Investig. 2010, 120, 4179–4182. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Iwasa, Y.; Komarova, N.L.; Nowak, M.A. Local regulation of homeostasis favors chromosomal instability. Curr. Biol. 2003, 13, 581–584. [Google Scholar] [CrossRef][Green Version]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Apoptosis and APC in colorectal tumorigenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 7950–7954. [Google Scholar] [CrossRef]
- Macleod, K. Tumor suppressor genes. Curr. Opin. Genet. Dev. 2000, 10, 81–93. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Rubbi, C.P.; Milner, J. P53: Gatekeeper, caretaker or both? In 25 Years of p53 Research; Springer: Dordrecht, The Netherlands, 2007; pp. 233–253. [Google Scholar]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Vogelstein, B.; Sur, S.; Prives, C. p53: The most frequently altered gene in human cancers. Nat. Educ. 2010, 3, 6. [Google Scholar]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, M.; Montanari, M.; Giordano, A. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene 2006, 25, 5263–5267. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, W. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef]
- Kritikou, E. PTEN—A new guardian of the genome. Nat. Rev. Mol. Cell Biol. 2007, 8, 179–180. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Parada, L.F.; Silva, A.J.; Ratner, N. Neurofibromatosis type 1: Modeling CNS dysfunction. J. Neurosci. 2012, 32, 14087–14093. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Gutmann, D.H. Neurofibromatosis type 1: A multidisciplinary approach to care. Lancet Neurol. 2014, 13, 834–843. [Google Scholar] [CrossRef]
- Jafri, M.; Wake, N.C.; Ascher, D.B.; Pires, D.E.; Gentle, D.; Morris, M.R.; Rattenberry, E.; Simpson, M.A.; Trembath, R.C.; Weber, A. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell carcinoma. Cancer Discov. 2015, 5, 723–729. [Google Scholar] [CrossRef]
- Poi, M.J.; Knobloch, T.J.; Li, J. Deletion of RDINK4/ARF enhancer: A novel mutation to “inactivate” the INK4-ARF locus. DNA Repair 2017, 57, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Chen, H.; Collins, A.R.; Connell, M.; Damia, G.; Dasgupta, S.; Malhotra, M.; Meeker, A.K.; Amedei, A.; Amin, A. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 2015, 35, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gómez-González, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.K.; Schiffman, J.D.; Boddy, A.M. Evolution of Cancer Defense Mechanisms Across Species. In Ecology and Evolution of Cancer; Elsevier Inc.: New York, NY, USA, 2017. [Google Scholar]
- Levitt, N.C.; Hickson, I.D. Caretaker tumour suppressor genes that defend genome integrity. Trends Mol. Med. 2002, 8, 179–186. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Deininger, P. Genetic instability in cancer: Caretaker and gatekeeper genes. Ochsner J. 1999, 1, 206–209. [Google Scholar]
- Morris, L.G.; Chan, T.A. Therapeutic targeting of tumor suppressor genes. Cancer 2015, 121, 1357–1368. [Google Scholar] [CrossRef]
- Chen, J.-J.; Silver, D.; Cantor, S.; Livingston, D.M.; Scully, R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res. 1999, 59, 1752s–1756s. [Google Scholar]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.-S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef]
- Zheng, L.; Shang, L.; Boyer, T.G.; Wen-Hwa, L. Lessons learned from BRCA1 and BRCA2. Oncogene 2000, 19, 6159–6175. [Google Scholar] [CrossRef] [PubMed]
- O’donovan, P.J.; Livingston, D.M. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdottir, K.; Ashworth, A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006, 25, 5864–5874. [Google Scholar] [CrossRef]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer; GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1247/ (accessed on 3 February 2022).
- McCarthy, A.M.; Armstrong, K. The role of testing for BRCA1 and BRCA2 mutations in cancer prevention. JAMA Intern. Med. 2014, 174, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.-X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Landscaping the cancer terrain. Science 1998, 280, 1036–1037. [Google Scholar] [CrossRef]
- Woodford-Richens, K.; Williamson, J.; Bevan, S.; Young, J.; Leggett, B.; Frayling, I.; Thway, Y.; Hodgson, S.; Kim, J.C.; Iwama, T. Allelic loss at SMAD4 in polyps from juvenile polyposis patients and use of fluorescence in situ hybridization to demonstrate clonal origin of the epithelium. Cancer Res. 2000, 60, 2477–2482. [Google Scholar]
- Volmer, M.W.; Radacz, Y.; Hahn, S.A.; Klein-Scory, S.; Stühler, K.; Zapatka, M.; Schmiegel, W.; Meyer, H.E.; Schwarte-Waldhoff, I. Tumor suppressor Smad4 mediates downregulation of the anti-adhesive invasion-promoting matricellular protein SPARC: Landscaping activity of Smad4 as revealed by a “secretome” analysis. Proteomics 2004, 4, 1324–1334. [Google Scholar] [CrossRef]
- Yang, G.; Yang, X. Smad4-mediated TGF-β signaling in tumorigenesis. Int. J. Biol. Sci. 2010, 6, 1–8. [Google Scholar] [CrossRef]
- Naik, V.G.; Adhyaru, P.; Gudigenavar, A. Tumor suppressor genes in oral cancer. Clin. Cancer Investig. J. 2015, 4, 697. [Google Scholar] [CrossRef]
- Payne, S.R.; Kemp, C.J. Tumor suppressor genetics. Carcinogenesis 2005, 26, 2031–2045. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical st.tudy of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef]
- Komarova, N.L.; Sengupta, A.; Nowak, M.A. Mutation-selection networks of cancer initiation: Tumor suppressor genes and chromosomal instability. J. Theor. Biol. 2003, 223, 433–450. [Google Scholar] [CrossRef]
- Nowak, M.A.; Michor, F.; Komarova, N.L.; Iwasa, Y. Evolutionary dynamics of tumor suppressor gene inactivation. Proc. Natl. Acad. Sci. USA 2004, 101, 10635–10638. [Google Scholar] [CrossRef]
- Spatz, A.; Borg, C.; Feunteun, J. X-chromosome genetics and human cancer. Nat. Rev. Cancer 2004, 4, 617–629. [Google Scholar] [CrossRef]
- Santarosa, M.; Ashworth, A. Haploinsufficiency for tumour suppressor genes: When you don’t need to go all the way. Biochim. Biophys. Acta 2004, 1654, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.B.; Conway, K.; Wang, X.W.; Bhamra, R.K.; Lin, X.H.; Cohen, M.D.; Annab, L.; Barrett, J.C.; Costa, M. Senescence of nickel-transformed cells by an X chromosome: Possible epigenetic control. Science 1991, 251, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Wang, L.; Morrison, C.; Chang, X.; Zhang, H.; Li, W.; Liu, Y.; Wang, Y.; Liu, X.; Chan, M.W.; et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell 2007, 129, 1275–1286. [Google Scholar] [CrossRef]
- Rivera, M.N.; Kim, W.J.; Wells, J.; Driscoll, D.R.; Brannigan, B.W.; Han, M.; Kim, J.C.; Feinberg, A.P.; Gerald, W.L.; Vargas, S.O.; et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 2007, 315, 642–645. [Google Scholar] [CrossRef]
- Liu, R.; Kain, M.; Wang, L. Inactivation of X-linked tumor suppressor genes in human cancer. Future Oncol. 2012, 8, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Zheng, P. X-linked tumor suppressors: Perplexing inheritance, a unique therapeutic opportunity. Trends Genet. 2010, 26, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Is DNA methylation of tumour suppressor genes epigenetic? Elife 2014, 3, 2475. [Google Scholar] [CrossRef]
- Mott, J.L. MicroRNAs involved in tumor suppressor and oncogene pathways: Implications for hepatobiliary neoplasia. Hepatology 2009, 50, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperact.tivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, Z.; Zhou, S.F.; Lu, N. Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors. Drug Des. Dev. Ther. 2014, 8, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Farid, N.R. P53 mutations in thyroid carcinoma: Tidings from an old foe. J. Endocrinol. Investig. 2001, 24, 536–545. [Google Scholar] [CrossRef]
- Dralle, H.; Machens, A.; Basa, J.; Fatourechi, V.; Franceschi, S.; Hay, I.D.; Nikiforov, Y.E.; Pacini, F.; Pasieka, J.L.; Sherman, S.I. Follicular cell-derived thyroid cancer. Nat. Rev. Dis. Primers 2015, 1, 15077. [Google Scholar] [CrossRef]
- Pollina, L.; Pacini, F.; Fontanini, G.; Vignati, S.; Bevilacqua, G.; Basolo, F. bcl-2, p53 and proliferating cell nuclear antigen expression is related to the degree of differentiation in thyroid carcinomas. Br. J. Cancer 1996, 73, 139–143. [Google Scholar] [CrossRef]
- Donghi, R.; Longoni, A.; Pilotti, S.; Michieli, P.; Della Porta, G.; Pierotti, M.A. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J. Clin. Investig. 1993, 91, 1753–1760. [Google Scholar] [CrossRef]
- Park, K.Y.; Koh, J.M.; Kim, Y.I.; Park, H.J.; Gong, G.; Hong, S.J.; Ahn, I.M. Prevalences of Gs alpha, ras, p53 mutations and ret/PTC rearrangement in differentiated thyroid tumours in a Korean population. Clin. Endocrinol. 1998, 49, 317–323. [Google Scholar] [CrossRef]
- Soares, P.; Cameselle-Teijeiro, J.; Sobrinho-Simoes, M. Immunohistochemical detection of p53 in differentiated, poorly differentiated and undifferentiated carcinomas of the thyroid. Histopathology 1994, 24, 205–210. [Google Scholar] [CrossRef]
- Chen, D.; Li, M.; Luo, J.; Gu, W. Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function. J. Biol. Chem. 2003, 278, 13595–13598. [Google Scholar] [CrossRef]
- Prodosmo, A.; Giglio, S.; Moretti, S.; Mancini, F.; Barbi, F.; Avenia, N.; Di Conza, G.; Schunemann, H.J.; Pistola, L.; Ludovini, V.; et al. Analysis of human MDM4 variants in papillary thyroid carcinomas reveals new potential markers of cancer properties. J. Mol. Med. 2008, 86, 585–596. [Google Scholar] [CrossRef]
- Kurokawa, M.; Kim, J.; Geradts, J.; Matsuura, K.; Liu, L.; Ran, X.; Xia, W.; Ribar, T.J.; Henao, R.; Dewhirst, M.W.; et al. A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci. Signal. 2013, 6, ra32. [Google Scholar] [CrossRef]
- Ma, W.; Zhao, P.; Zang, L.; Zhang, K.; Liao, H.; Hu, Z. Tumour suppressive function of HUWE1 in thyroid cancer. J. Biosci. 2016, 41, 395–405. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, H.; Hu, X.; Xin, S.; Duan, Z. Abnormality of pl6/p38MAPK/p53/Wipl pathway in papillary thyroid cancer. Gland Surg. 2012, 1, 33–38. [Google Scholar]
- Zou, M.; Baitei, E.Y.; Al-Rijjal, R.A.; Parhar, R.S.; Al-Mohanna, F.A.; Kimura, S.; Pritchard, C.; Binessa, H.A.; Alzahrani, A.S.; Al-Khalaf, H.H.; et al. TSH overcomes Braf(V600E)-induced senescence to promote tumor progression via downregulation of p53 expression in papillary thyroid cancer. Oncogene 2016, 35, 1909–1918. [Google Scholar] [CrossRef]
- Xing, M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid 2010, 20, 697–706. [Google Scholar] [CrossRef]
- Eng, C. The role of PTEN, a phosphatase gene, in inherited and sporadic nonmedullary thyroid tumors. Recent Prog. Horm. Res. 1999, 54, 441–452. [Google Scholar]
- Ngeow, J.; Mester, J.; Rybicki, L.A.; Ni, Y.; Milas, M.; Eng, C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J. Clin. Endocrinol. Metab. 2011, 96, E2063–E2071. [Google Scholar] [CrossRef]
- Ngeow, J.; He, X.; Mester, J.L.; Lei, J.; Romigh, T.; Orloff, M.S.; Milas, M.; Eng, C. Utility of PTEN protein dosage in predicting for underlying germline PTEN mutations among patients presenting with thyroid cancer and Cowden-like phenotypes. J. Clin. Endocrinol. Metab. 2012, 97, E2320–E2327. [Google Scholar] [CrossRef]
- Goschzik, T.; Gessi, M.; Denkhaus, D.; Pietsch, T. PTEN mutations and activation of the PI3K/Akt/mTOR signaling pathway in papillary tumors of the pineal region. J. Neuropathol. Exp. Neurol. 2014, 73, 747–751. [Google Scholar] [CrossRef]
- Lotan, T.L.; Carvalho, F.L.; Peskoe, S.B.; Hicks, J.L.; Good, J.; Fedor, H.; Humphreys, E.; Han, M.; Platz, E.A.; Squire, J.A.; et al. PTEN loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy. Mod. Pathol. 2015, 28, 128–137. [Google Scholar] [CrossRef]
- Piras, G.; Monne, M.; Palmas, A.D.; Calvisi, A.; Asproni, R.; Vacca, F.; Pilo, L.; Gabbas, A.; Latte, G. Methylation analysis of the phosphates and tensin homologue on chromosome 10 gene (PTEN) in multiple myeloma. Clin. Epigenet. 2014, 6, 16. [Google Scholar] [CrossRef]
- Yang, Z.; Yuan, X.G.; Chen, J.; Luo, S.W.; Luo, Z.J.; Lu, N.H. Reduced expression of PTEN and increased PTEN phosphorylation at residue Ser380 in gastric cancer tissues: A novel mechanism of PTEN inactivation. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 72–79. [Google Scholar] [CrossRef]
- Dahia, P.L.; Marsh, D.J.; Zheng, Z.; Zedenius, J.; Komminoth, P.; Frisk, T.; Wallin, G.; Parsons, R.; Longy, M.; Larsson, C.; et al. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997, 57, 4710–4713. [Google Scholar]
- Smith, J.R.; Marqusee, E.; Webb, S.; Nose, V.; Fishman, S.J.; Shamberger, R.C.; Frates, M.C.; Huang, S.A. Thyroid nodules and cancer in children with PTEN hamartoma tumor syndrome. J. Clin. Endocrinol. Metab. 2011, 96, 34–37. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Nagy, R.; Ganapathi, S.; Comeras, I.; Peterson, C.; Orloff, M.; Porter, K.; Eng, C.; Ringel, M.D.; Kloos, R.T. Frequency of germline PTEN mutations in differentiated thyroid cancer. Thyroid 2011, 21, 505–510. [Google Scholar] [CrossRef]
- Gimm, O.; Perren, A.; Weng, L.P.; Marsh, D.J.; Yeh, J.J.; Ziebold, U.; Gil, E.; Hinze, R.; Delbridge, L.; Lees, J.A.; et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am. J. Pathol. 2000, 156, 1693–1700. [Google Scholar] [CrossRef]
- Halachmi, N.; Halachmi, S.; Evron, E.; Cairns, P.; Okami, K.; Saji, M.; Westra, W.H.; Zeiger, M.A.; Jen, J.; Sidransky, D. Somatic mutations of the PTEN tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosom. Cancer 1998, 23, 239–243. [Google Scholar] [CrossRef]
- Hsieh, M.C.; Lin, S.F.; Shin, S.J.; Liu, T.C.; Chang, J.G.; Lee, J.P. Mutation analysis of PTEN/MMAC 1 in sporadic thyroid tumors. Kaohsiung J. Med. Sci. 2000, 16, 9–12. [Google Scholar]
- Hou, P.; Ji, M.; Xing, M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer 2008, 113, 2440–2447. [Google Scholar] [CrossRef]
- Alvarez-Nunez, F.; Bussaglia, E.; Mauricio, D.; Ybarra, J.; Vilar, M.; Lerma, E.; de Leiva, A.; Matias-Guiu, X. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid 2006, 16, 17–23. [Google Scholar] [CrossRef]
- Pringle, D.R.; Vasko, V.V.; Yu, L.; Manchanda, P.K.; Lee, A.A.; Zhang, X.; Kirschner, J.M.; Parlow, A.F.; Saji, M.; Jarjoura, D.; et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. J. Clin. Endocrinol. Metab. 2014, 99, E804–E812. [Google Scholar] [CrossRef]
- Bruni, P.; Boccia, A.; Baldassarre, G.; Trapasso, F.; Santoro, M.; Chiappetta, G.; Fusco, A.; Viglietto, G. PTEN expression is reduced in a subset of sporadic thyroid carcinomas: Evidence that PTEN-growth suppressing activity in thyroid cancer cells mediated by p27kip1. Oncogene 2000, 19, 3146–3155. [Google Scholar] [CrossRef]
- Beg, S.; Siraj, A.K.; Jehan, Z.; Prabakaran, S.; Al-Sobhi, S.S.; Al-Dawish, M.; Al-Dayel, F.; Al-Kuraya, K.S. PTEN loss is associated with follicular variant of Middle Eastern papillary thyroid carcinoma. Br. J. Cancer 2015, 112, 1938–1943. [Google Scholar] [CrossRef]
- Yu, W.; Ni, Y.; Saji, M.; Ringel, M.D.; Jaini, R.; Eng, C. Cowden syndrome-associated germline succinate dehydrogenase complex subunit D (SDHD) variants cause PTEN-mediated down-regulation of autophagy in thyroid cancer cells. Hum. Mol. Genet. 2017, 26, 1365–1375. [Google Scholar] [CrossRef]
- Frisk, T.; Foukakis, T.; Dwight, T.; Lundberg, J.; Hoog, A.; Wallin, G.; Eng, C.; Zedenius, J.; Larsson, C. Silencing of the PTEN tumor-suppressor gene in anaplastic thyroid cancer. Genes Chromosom. Cancer 2002, 35, 74–80. [Google Scholar] [CrossRef]
- Groden, J.; Thliveris, A.; Samowitz, W.; Carlson, M.; Gelbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66, 589–600. [Google Scholar] [CrossRef]
- Beroud, C.; Soussi, T. APC gene: Database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1996, 24, 121–124. [Google Scholar] [CrossRef]
- Crail, H.W. Multiple primary malignancies arising in the rectum, brain, and thyroid; report of a case. US Nav. Med. Bull. 1949, 49, 123–128. [Google Scholar]
- Bulow, C.; Bulow, S. Is screening for thyroid carcinoma indicated in familial adenomatous polyposis? The Leeds Castle Polyposis Group. Int. J. Colorectal Dis. 1997, 12, 240–242. [Google Scholar]
- Cetta, F.; Montalto, G.; Gori, M.; Curia, M.C.; Cama, A.; Olschwang, S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J. Clin. Endocrinol. Metab. 2000, 85, 286–292. [Google Scholar]
- Groen, E.J.; Roos, A.; Muntinghe, F.L.; Enting, R.H.; de Vries, J.; Kleibeuker, J.H.; Witjes, M.J.; Links, T.P.; van Beek, A.P. Extra-intestinal manifestations of familial adenomatous polyposis. Ann. Surg. Oncol. 2008, 15, 2439–2450. [Google Scholar] [CrossRef]
- Martayan, A.; Sanchez-Mete, L.; Baldelli, R.; Falvo, E.; Barnabei, A.; Conti, L.; Giacomini, P.; Appetecchia, M.; Stigliano, V. Gene variants associated to malignant thyroid disease in familial adenomatous polyposis: A novel APC germline mutation. J. Endocrinol. Investig. 2010, 33, 603–606. [Google Scholar] [CrossRef]
- Zeki, K.; Spambalg, D.; Sharifi, N.; Gonsky, R.; Fagin, J.A. Mutations of the adenomatous polyposis coli gene in sporadic thyroid neoplasms. J. Clin. Endocrinol. Metab. 1994, 79, 1317–1321. [Google Scholar]
- Cetta, F.; Chiappetta, G.; Melillo, R.M.; Petracci, M.; Montalto, G.; Santoro, M.; Fusco, A. The ret/ptc1 oncogene is activated in familial adenomatous polyposis-associated thyroid papillary carcinomas. J. Clin. Endocrinol. Metab. 1998, 83, 1003–1006. [Google Scholar] [CrossRef]
- Uchino, S.; Noguchi, S.; Yamashita, H.; Yamashita, H.; Watanabe, S.; Ogawa, T.; Tsuno, A.; Murakami, A.; Miyauchi, A. Mutational analysis of the APC gene in cribriform-morula variant of papillary thyroid carcinoma. World J. Surg. 2006, 30, 775–779. [Google Scholar] [CrossRef]
- Cetta, F.; Olschwang, S.; Petracci, M.; Montalto, G.; Baldi, C.; Zuckermann, M.; Mariani Costantini, R.; Fusco, A. Genetic alterations in thyroid carcinoma associated with familial adenomatous polyposis: Clinical implications and suggestions for early detection. World J. Surg. 1998, 22, 1231–1236. [Google Scholar] [CrossRef]
- Truta, B.; Allen, B.A.; Conrad, P.G.; Kim, Y.S.; Berk, T.; Gallinger, S.; Bapat, B.; Terdiman, J.P.; Sleisenger, M.H. Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam. Cancer 2003, 2, 95–99. [Google Scholar] [CrossRef]
- Septer, S.; Slowik, V.; Morgan, R.; Dai, H.; Attard, T. Thyroid cancer complicating familial adenomatous polyposis: Mutation spectrum of at-risk individuals. Hered. Cancer Clin. Pract. 2013, 11, 13. [Google Scholar] [CrossRef]
- Han, S.H.; Ryu, J.S.; Kim, Y.J.; Cho, H.I.; Yang, Y.H.; Lee, K.R. Mutation analysis of the APC gene in unrelated Korean patients with FAP: Four novel mutations with unusual phenotype. Fam. Cancer 2011, 10, 21–26. [Google Scholar] [CrossRef]
- Zhang, X.; Li, M.; Zuo, K.; Li, D.; Ye, M.; Ding, L.; Cai, H.; Fu, D.; Fan, Y.; Lv, Z. Upregulated miR-155 in papillary thyroid carcinoma promotes tumor growth by targeting APC and activating Wnt/beta-catenin signaling. J. Clin. Endocrinol. Metab. 2013, 98, E1305–E1313. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Albert, I.; Porfiri, E.; Munemitsu, S.; Polakis, P. Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res. 1997, 57, 4624–4630. [Google Scholar]
- Kumamoto, K.; Ishida, H.; Ohsawa, T.; Ishibashi, K.; Ushiama, M.; Yoshida, T.; Iwama, T. Germline and somatic mutations of the APC gene in papillary thyroid carcinoma associated with familial adenomatous polyposis: Analysis of three cases and a review of the literature. Oncol. Lett. 2015, 10, 2239–2243. [Google Scholar] [CrossRef]
- Xing, M. RASAL1 in thyroid cancer: Promise from a new friend. J. Clin. Endocrinol. Metab. 2014, 99, 3619–3621. [Google Scholar] [CrossRef]
- Liu, D.; Yang, C.; Bojdani, E.; Murugan, A.K.; Xing, M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J. Natl. Cancer Inst. Monogr. 2013, 105, 1617–1627. [Google Scholar] [CrossRef]
- Ngeow, J.; Ni, Y.; Tohme, R.; Song Chen, F.; Bebek, G.; Eng, C. Germline alterations in RASAL1 in Cowden syndrome patients presenting with follicular thyroid cancer and in individuals with apparently sporadic epithelial thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1316–E1321. [Google Scholar] [CrossRef]
- Lyssikatos, C.; Quezado, M.M.; Faucz, F.R.; Angelousi, A.; Nasiri-Ansari, N.; Stratakis, C.A.; Kassi, E. A rare case of medullary thyroid cancer, mesothelioma and meningioma, due to APC and RASAL1 mutations. In Proceedings of the 19th European Congress of Endocrinology, Lisbon, Portugal, 20–23 May 2017. [Google Scholar] [CrossRef]
- Angelousi, A.; Settas, N.; Faucz, F.R.; Lyssikatos, C.; Quezado, M.; Nasiri-Ansari, N.; Stratakis, C.A.; Kassi, E. Medullary thyroid cancer, leukemia, mesothelioma and meningioma associated with germline APC and RASAL1 variants: A new syndrome? Hormones 2017, 16, 423–428. [Google Scholar]
- Jeon, M.J.; Chun, S.M.; Kim, D.; Kwon, H.; Jang, E.K.; Kim, T.Y.; Kim, W.B.; Shong, Y.K.; Jang, S.J.; Song, D.E.; et al. Genomic Alterations of Anaplastic Thyroid Carcinoma Detected by Targeted Massive Parallel Sequencing in a BRAF(V600E) Mutation-Prevalent Area. Thyroid 2016, 26, 683–690. [Google Scholar] [CrossRef]
- Preto, A.; Reis-Filho, J.S.; Ricardo, S.; Soares, P. P63 expression in papillary and anaplastic carcinomas of the thyroid gland: Lack of an oncogenetic role in tumorigenesis and progression. Pathol. Res. Pract. 2002, 198, 449–454. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Preto, A.; Soares, P.; Ricardo, S.; Cameselle-Teijeiro, J.; Sobrinho-Simoes, M. p63 expression in solid cell nests of the thyroid: Further evidence for a stem cell origin. Mod. Pathol. 2003, 16, 43–48. [Google Scholar] [CrossRef]
- Bonzanini, M.; Amadori, P.L.; Sagramoso, C.; Dalla Palma, P. Expression of cytokeratin 19 and protein p63 in fine needle aspiration biopsy of papillary thyroid carcinoma. Acta Cytol. 2008, 52, 541–548. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Mandarino, A.; Mazzon, E.; Vella, V.; Gangemi, P.; Vancheri, C.; Vigneri, P.; Aloisi, A.; Vigneri, R.; Frasca, F. The p53-homologue p63 may promote thyroid cancer progression. Endocr. Relat. Cancer 2005, 12, 953–971. [Google Scholar] [CrossRef]
- Ferru, A.; Denis, S.; Guilhot, J.; Gibelin, H.; Tourani, J.M.; Kraimps, J.L.; Larsen, C.J.; Karayan-Tapon, L. Expression of TAp73 and DeltaNp73 isoform transcripts in thyroid tumours. Eur. J. Surg. Oncol. 2006, 32, 228–230. [Google Scholar] [CrossRef]
- Frasca, F.; Vella, V.; Aloisi, A.; Mandarino, A.; Mazzon, E.; Vigneri, R.; Vigneri, P. p73 tumor-suppressor activity is impaired in human thyroid cancer. Cancer Res. 2003, 63, 5829–5837. [Google Scholar]
- Ito, Y.; Uramoto, H.; Funa, K.; Yoshida, H.; Jikuzono, T.; Asahi, S.; Higashiyama, T.; Tomoda, C.; Takamura, Y.; Miya, A.; et al. Delta Np73 expression in thyroid neoplasms originating from follicular cells. Pathology 2006, 38, 205–209. [Google Scholar] [CrossRef]
- Puppin, C.; Fabbro, D.; Dima, M.; Di Loreto, C.; Puxeddu, E.; Filetti, S.; Russo, D.; Damante, G. High periostin expression correlates with aggressiveness in papillary thyroid carcinomas. J. Endocrinol. 2008, 197, 401–408. [Google Scholar] [CrossRef]
- Puppin, C.; Passon, N.; Frasca, F.; Vigneri, R.; Tomay, F.; Tomaciello, S.; Damante, G. In thyroid cancer cell lines expression of periostin gene is controlled by p73 and is not related to epigenetic marks of active transcription. Cell. Oncol. 2011, 34, 131–140. [Google Scholar] [CrossRef]
- Vella, V.; Puppin, C.; Damante, G.; Vigneri, R.; Sanfilippo, M.; Vigneri, P.; Tell, G.; Frasca, F. DeltaNp73alpha inhibits PTEN expression in thyroid cancer cells. Int. J. Cancer 2009, 124, 2539–2548. [Google Scholar] [CrossRef]
- Figge, J.; Bakst, G.; Weisheit, D.; Solis, O.; Ross, J.S. Image analysis quantitation of immunoreactive retinoblastoma protein in human thyroid neoplasms with a streptavidin-biotin-peroxidase staining technique. Am. J. Pathol. 1991, 139, 1213–1219. [Google Scholar]
- Zou, M.; Shi, Y.; Farid, N.R.; Al-Sedairy, S.T. Inverse association between cyclin D1 overexpression and retinoblastoma gene mutation in thyroid carcinomas. Endocrine 1998, 8, 61–64. [Google Scholar] [CrossRef]
- Holm, R.; Nesland, J.M. Retinoblastoma and p53 tumour suppressor gene protein expression in carcinomas of the thyroid gland. J. Pathol. 1994, 172, 267–272. [Google Scholar] [CrossRef]
- Van Veelen, W.; van Gasteren, C.J.; Acton, D.S.; Franklin, D.S.; Berger, R.; Lips, C.J.; Hoppener, J.W. Synergistic effect of oncogenic RET and loss of p18 on medullary thyroid carcinoma development. Cancer Res. 2008, 68, 1329–1337. [Google Scholar] [CrossRef]
- Cote, G.J.; Grubbs, E.G.; Hofmann, M.C. Thyroid C-Cell Biology and Oncogenic Transformation. Recent Results Cancer Res. 2015, 204, 1–39. [Google Scholar]
- Pozo, K.; Hillmann, A.; Augustyn, A.; Plattner, F.; Hai, T.; Singh, T.; Ramezani, S.; Sun, X.; Pfragner, R.; Minna, J.D.; et al. Differential expression of cell cycle regulators in CDK5-dependent medullary thyroid carcinoma tumorigenesis. Oncotarget 2015, 6, 12080–12093. [Google Scholar] [CrossRef]
- Song, H.; Lin, C.; Yao, E.; Zhang, K.; Li, X.; Wu, Q.; Chuang, P.T. Selective Ablation of Tumor Suppressors in Parafollicular C Cells Elicits Medullary Thyroid Carcinoma. J. Biol. Chem. 2017, 292, 3888–3899. [Google Scholar] [CrossRef]
- Valenciaga, A.; Grubbs, E.G.; Porter, K.; Wakely, P.E., Jr.; Williams, M.D.; Cote, G.J.; Vasko, V.V.; Saji, M.; Ringel, M.D. Reduced Retinoblastoma Protein Expression Is Associated with Decreased Patient Survival in Medullary Thyroid Cancer. Thyroid 2017, 27, 1523–1533. [Google Scholar] [CrossRef]
- Brzezinski, J.; Migodzinski, A.; Toczek, A.; Tazbir, J.; Dedecjus, M. Patterns of cyclin E, retinoblastoma protein, and p21Cip1/WAF1 immunostaining in the oncogenesis of papillary thyroid carcinoma. Clin. Cancer Res. 2005, 11, 1037–1043. [Google Scholar]
- Skalhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000, 5, D678–D693. [Google Scholar]
- Gu, T.; Zhang, Z.; Wang, J.; Guo, J.; Shen, W.H.; Yin, Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011, 71, 2821–2825. [Google Scholar] [CrossRef]
- Massimi, M.; Cardarelli, S.; Galli, F.; Giardi, M.F.; Ragusa, F.; Panera, N.; Cinque, B.; Cifone, M.G.; Biagioni, S.; Giorgi, M. Increase of Intracellular Cyclic AMP by PDE4 Inhibitors Affects HepG2 Cell Cycle Progression and Survival. J. Cell. Biochem. 2017, 118, 1401–1411. [Google Scholar] [CrossRef]
- Yasuda, M.T.; Sakakibara, H.; Shimoi, K. Estrogen- and stress-induced DNA damage in breast cancer and chemoprevention with dietary flavonoid. Genes Environ. 2017, 39, 10. [Google Scholar] [CrossRef]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- Sandrini, F.; Matyakhina, L.; Sarlis, N.J.; Kirschner, L.S.; Farmakidis, C.; Gimm, O.; Stratakis, C.A. Regulatory subunit type I-alpha of protein kinase A (PRKAR1A): A tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes Cancer 2002, 35, 182–192. [Google Scholar] [CrossRef]
- Pringle, D.R.; Yin, Z.; Lee, A.A.; Manchanda, P.K.; Yu, L.; Parlow, A.F.; Jarjoura, D.; La Perle, K.M.; Kirschner, L.S. Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr. Relat. Cancer 2012, 19, 435–446. [Google Scholar] [CrossRef][Green Version]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Cybulski, C.; Huzarski, T.; Gorski, B.; Masojc, B.; Mierzejewski, M.; Debniak, T.; Gliniewicz, B.; Matyjasik, J.; Zlowocka, E.; Kurzawski, G.; et al. A novel founder CHEK2 mutation is associated with increased prostate cancer risk. Cancer Res. 2004, 64, 2677–2679. [Google Scholar] [CrossRef]
- Kilpivaara, O.; Vahteristo, P.; Falck, J.; Syrjakoski, K.; Eerola, H.; Easton, D.; Bartkova, J.; Lukas, J.; Heikkila, P.; Aittomaki, K.; et al. CHEK2 variant I157T may be associated with increased breast cancer risk. Int. J. Cancer 2004, 111, 543–547. [Google Scholar] [CrossRef]
- Cybulski, C.; Gorski, B.; Huzarski, T.; Masojc, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef]
- Wojcicka, A.; Czetwertynska, M.; Swierniak, M.; Dlugosinska, J.; Maciag, M.; Czajka, A.; Dymecka, K.; Kubiak, A.; Kot, A.; Ploski, R.; et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer 2014, 53, 516–523. [Google Scholar] [CrossRef]
- Fayaz, S.; Fard-Esfahani, P.; Torbati, P.M. Lack of CHEK2 gene mutations in differentiated thyroid carcinoma patients using high resolution melting analysis. Asian Pac. J. Cancer Prev. 2014, 15, 5019–5022. [Google Scholar] [CrossRef][Green Version]
- Kaczmarek-Rys, M.; Ziemnicka, K.; Hryhorowicz, S.T.; Gorczak, K.; Hoppe-Golebiewska, J.; Skrzypczak-Zielinska, M.; Tomys, M.; Golab, M.; Szkudlarek, M.; Budny, B.; et al. The c.470 T > C CHEK2 missense variant increases the risk of differentiated thyroid carcinoma in the Great Poland population. Hered. Cancer Clin. Pract 2015, 13, 8. [Google Scholar] [CrossRef]
- Swierniak, M.; Pfeifer, A.; Stokowy, T.; Rusinek, D.; Chekan, M.; Lange, D.; Krajewska, J.; Oczko-Wojciechowska, M.; Czarniecka, A.; Jarzab, M.; et al. Somatic mutation profiling of follicular thyroid cancer by next generation sequencing. Mol. Cell. Endocrinol. 2016, 433, 130–137. [Google Scholar] [CrossRef]
- Spitzweg, C.; Morris, J.C. Gene therapy for thyroid cancer: Current status and future prospects. Thyroid 2004, 14, 424–434. [Google Scholar] [CrossRef]
- Levy, H.C.; Hulvey, D.; Adamson-Small, L.; Jn-Simon, N.; Prima, V.; Rivkees, S.; Hobbs, J.A. Improved cell-specificity of adeno-associated viral vectors for medullary thyroid carcinoma using calcitonin gene regulatory elements. PLoS ONE 2020, 15, e0228005. [Google Scholar] [CrossRef]
- Moretti, F.; Farsetti, A.; Soddu, S.; Misiti, S.; Crescenzi, M.; Filetti, S.; Andreoli, M.; Sacchi, A.; Pontecorvi, A. p53 re-expression inhibits proliferation and restores differentiation of human thyroid anaplastic carcinoma cells. Oncogene 1997, 14, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Fagin, J.A.; Tang, S.H.; Zeki, K.; Di Lauro, R.; Fusco, A.; Gonsky, R. Reexpression of thyroid peroxidase in a derivative of an undifferentiated thyroid carcinoma cell line by introduction of wild-type p53. Cancer Res. 1996, 56, 765–771. [Google Scholar] [PubMed]
- Zeki, K.; Tanaka, Y.; Morimoto, I.; Nishimura, Y.; Kimura, A.; Yamashita, U.; Eto, S. Induction of expression of MHC-class-II antigen on human thyroid carcinoma by wild-type p53. Int. J. Cancer 1998, 75, 391–395. [Google Scholar] [CrossRef]
- Marcello, M.A.; Morari, E.C.; Cunha, L.L.; De Nadai Silva, A.C.; Carraro, D.M.; Carvalho, A.L.; Soares, F.A.; Vassallo, J.; Ward, L.S. P53 and expression of immunological markers may identify early stage thyroid tumors. Clin. Dev. Immunol. 2013, 2013, 846584. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, Y.; Shigematsu, K.; Namba, H.; Zeki, K.; Yamashita, S.; Niwa, M. Inhibition of angiogenesis and tumorigenesis, and induction of dormancy by p53 in a p53-null thyroid carcinoma cell line in vivo. Anticancer Res. 2000, 20, 2723–2728. [Google Scholar]
- Liu, L.; Li, D.; Chen, Z.; Yang, J.; Ma, Y.; Cai, H.; Shan, C.; Lv, Z.; Zhang, X. Wild-Type P53 Induces Sodium/Iodide Symporter Expression Allowing Radioiodide Therapy in Anaplastic Thyroid Cancer. Cell. Physiol. Biochem. 2017, 43, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, M.; Nagayama, Y.; Akino, K.; Yasuda, M.; Yamamoto, T.; Yang, T.T.; Ohtsuru, A.; Namba, H.; Yamashita, S.; Ayabe, H.; et al. Therapeutic usefulness of wild-type p53 gene introduction in a p53-null anaplastic thyroid carcinoma cell line. J. Clin. Endocrinol. Metab. 1998, 83, 3668–3672. [Google Scholar] [CrossRef]
- Imanishi, R.; Ohtsuru, A.; Iwamatsu, M.; Iioka, T.; Namba, H.; Seto, S.; Yano, K.; Yamashita, S. A histone deacetylase inhibitor enhances killing of undifferentiated thyroid carcinoma cells by p53 gene therapy. J. Clin. Endocrinol. Metab. 2002, 87, 4821–4824. [Google Scholar] [CrossRef][Green Version]
- Moretti, F.; Nanni, S.; Farsetti, A.; Narducci, M.; Crescenzi, M.; Giuliacci, S.; Sacchi, A.; Pontecorvi, A. Effects of exogenous p53 transduction in thyroid tumor cells with different p53 status. J. Clin. Endocrinol. Metab. 2000, 85, 302–308. [Google Scholar] [CrossRef]
- Nagayama, Y.; Yokoi, H.; Takeda, K.; Hasegawa, M.; Nishihara, E.; Namba, H.; Yamashita, S.; Niwa, M. Adenovirus-mediated tumor suppressor p53 gene therapy for anaplastic thyroid carcinoma in vitro and in vivo. J. Clin. Endocrinol. Metab. 2000, 85, 4081–4086. [Google Scholar] [CrossRef]
- Kim, S.B.; Ahn, I.M.; Park, H.J.; Park, J.S.; Cho, H.J.; Gong, G.; Suh, C.; Lee, J.S.; Kim, W.K.; Kim, S.H. Growth inhibition and chemosensitivity of poorly differentiated human thyroid cancer cell line (NPA) transfected with p53 gene. Head Neck 2001, 23, 223–229. [Google Scholar] [CrossRef]
- DeGroot, L.J.; Zhang, R. Viral mediated gene therapy for the management of metastatic thyroid carcinoma. Endocr. Metab. Immune Disord. Drug Targets 2004, 4, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Chahal, M.S.; Tang, X.; Bruce, J.E.; Pommier, Y.; Daoud, S.S. Proteomic identification of heat shock protein 90 as a candidate target for p53 mutation reactivation by PRIMA-1 in breast cancer cells. Breast Cancer Res. 2005, 7, R765–R774. [Google Scholar] [CrossRef]
- Qiang, W.; Jin, T.; Yang, Q.; Liu, W.; Liu, S.; Ji, M.; He, N.; Chen, C.; Shi, B.; Hou, P. PRIMA-1 selectively induces global DNA demethylation in p53 mutant-type thyroid cancer cells. J. Biomed. Nanotechnol. 2014, 10, 1249–1258. [Google Scholar] [CrossRef]
- Messina, R.L.; Sanfilippo, M.; Vella, V.; Pandini, G.; Vigneri, P.; Nicolosi, M.L.; Giani, F.; Vigneri, R.; Frasca, F. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int. J. Cancer 2012, 130, 2259–2270. [Google Scholar] [CrossRef]
- Li, Z.; Xu, X.; Li, Y.; Zou, K.; Zhang, Z.; Xu, X.; Liao, Y.; Zhao, X.; Jiang, W.; Yu, W.; et al. Synergistic Antitumor Effect of BKM120 with Prima-1Met Via Inhibiting PI3K/AKT/mTOR and CPSF4/hTERT Signaling and Reactivating Mutant P53. Cell. Physiol. Biochem. 2018, 45, 1772–1786. [Google Scholar] [CrossRef]
- Garufi, A.; D’Orazi, V.; Crispini, A.; D’Orazi, G. Zn(II)-curc targets p53 in thyroid cancer cells. Int. J. Oncol. 2015, 47, 1241–1248. [Google Scholar] [CrossRef]
- Chen, H.; Luo, D.; Zhang, L.; Lin, X.; Luo, Q.; Yi, H.; Wang, J.; Yan, X.; Li, B.; Chen, Y.; et al. Restoration of p53 using the novel MDM2-p53 antagonist APG115 suppresses dedifferentiated papillary thyroid cancer cells. Oncotarget 2017, 8, 43008–43022. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, B.; Huang, Z.; Zhao, D.W.; Zeng, Q. Shikonin Inhibites Migration and Invasion of Thyroid Cancer Cells by Downregulating DNMT1. Med. Sci. Monit. 2018, 24, 661–670. [Google Scholar] [CrossRef]
- Biswas, R.; Mondal, A.; Ahn, J.C. Deregulation of EGFR/PI3K and activation of PTEN by photodynamic therapy combined with carboplatin in human anaplastic thyroid cancer cells and xenograft tumors in nude mice. J. Photochem. Photobiol. B 2015, 148, 118–127. [Google Scholar] [CrossRef]
- Weng, L.P.; Gimm, O.; Kum, J.B.; Smith, W.M.; Zhou, X.P.; Wynford-Thomas, D.; Leone, G.; Eng, C. Transient ectopic expression of PTEN in thyroid cancer cell lines induces cell cycle arrest and cell type-dependent cell death. Hum. Mol. Genet. 2001, 10, 251–258. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Vella, V.; Pandini, G.; Sanfilippo, M.; Pezzino, V.; Vigneri, R.; Frasca, F. TAp73 alpha increases p53 tumor suppressor activity in thyroid cancer cells via the inhibition of Mdm2-mediated degradation. Mol. Cancer Res. 2008, 6, 64–77. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Major Classes | Examples | Main Functions |
|---|---|---|
Gatekeepers
| APC TP53, PTEN, and TP63 PTEN PTEN | The regulation of cell proliferation/division, tissue growth, and apoptosis |
| Caretakers | ATM, XRCC3, XPC, ERCC5, ATR, and BRCA1 | The maintenance of genomic stability and DNA repair mechanisms |
| Landscapers | SMAD4, CDH1, NF1, RB, and APC | The regulation of extracellular matrix proteins, cell-surface markers, adhesion molecules, and growth factors |
| Tumor Suppressor Gene | Normal Function of Protein Product | Type of Alterations | Affected Thyroid Tumors | Mutation Frequency |
|---|---|---|---|---|
| TP53 | Cell-cycle regulation | Point mutations, negative regulation by MDM family members, and ubiquitination. Dual function: oncogene and TSG | ATC (50–80%), PTC | 40% in PTC, 60% in ATC |
| PTEN | Cell division regulation | Point mutations, deletion, promoter hypermethylation, LOH, ubiquitination, and post-translational modifications | FTC, DTC, ATC, and PTC | 65–85% |
| APC | The regulation of cell division, adhesion, and migration | Nonsense and missense mutations, frameshift mutations, polymorphisms, and epigenetic regulation | ATC, MTC, PTC, FTC, and CMVPTC | 87% in FAP-associated PTC |
| RASAL1 | The stimulation of the GTPase activity of RAS | Missense and nonsense mutations, promoter hypermethylation | PTC, ATC, FTC, and MTC | 17% in ATCs, 5% in FTCs, and 3% in PTCs |
| TP63 | The regulation of cell proliferation and differentiation, the transactivating effect, or dominant negative activity on p53 target genes | Downregulation, dual function: oncogene and TSG | PTC and FTC | |
| TP73 | Involved in cellular responses to stress and development | Downregulation and upregulation, dual function: oncogene and TSG | Follicular adenoma, FTC, and PTC | |
| RB | The control of DNA replication and cell division during cell damage | Mutations, deletions, downregulation, enhanced phosphorylation, and the loss of expression | MTC, PTC | 1.8% in MTC |
| PRKAR1A | Promoting cell growth and division | Downregulation, LOH | FTC, ATC | LOH of the PRKAR1A(CA)n locus in 37.5% of cases |
| CHEK2 | Cell-cycle control | Mutations, polymorphisms | PTC, FTC, and DTC | 15.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajabi, S.; Alix-Panabières, C.; Alaei, A.S.; Abooshahab, R.; Shakib, H.; Ashrafi, M.R. Looking at Thyroid Cancer from the Tumor-Suppressor Genes Point of View. Cancers 2022, 14, 2461. https://doi.org/10.3390/cancers14102461
Rajabi S, Alix-Panabières C, Alaei AS, Abooshahab R, Shakib H, Ashrafi MR. Looking at Thyroid Cancer from the Tumor-Suppressor Genes Point of View. Cancers. 2022; 14(10):2461. https://doi.org/10.3390/cancers14102461
Chicago/Turabian StyleRajabi, Sadegh, Catherine Alix-Panabières, Arshia Sharbatdar Alaei, Raziyeh Abooshahab, Heewa Shakib, and Mohammad Reza Ashrafi. 2022. "Looking at Thyroid Cancer from the Tumor-Suppressor Genes Point of View" Cancers 14, no. 10: 2461. https://doi.org/10.3390/cancers14102461
APA StyleRajabi, S., Alix-Panabières, C., Alaei, A. S., Abooshahab, R., Shakib, H., & Ashrafi, M. R. (2022). Looking at Thyroid Cancer from the Tumor-Suppressor Genes Point of View. Cancers, 14(10), 2461. https://doi.org/10.3390/cancers14102461