
Distinct Gene Expression Profiles of Matched Primary and Metastatic Triple-Negative Breast Cancers

 , , , ,
, , , , 

Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Patients and Patient Samples
2.2. RNA Isolation and Sequencing
2.3. RNA-Seq Data Processing
2.4. Cell Type Enrichment Analysis
3. Results
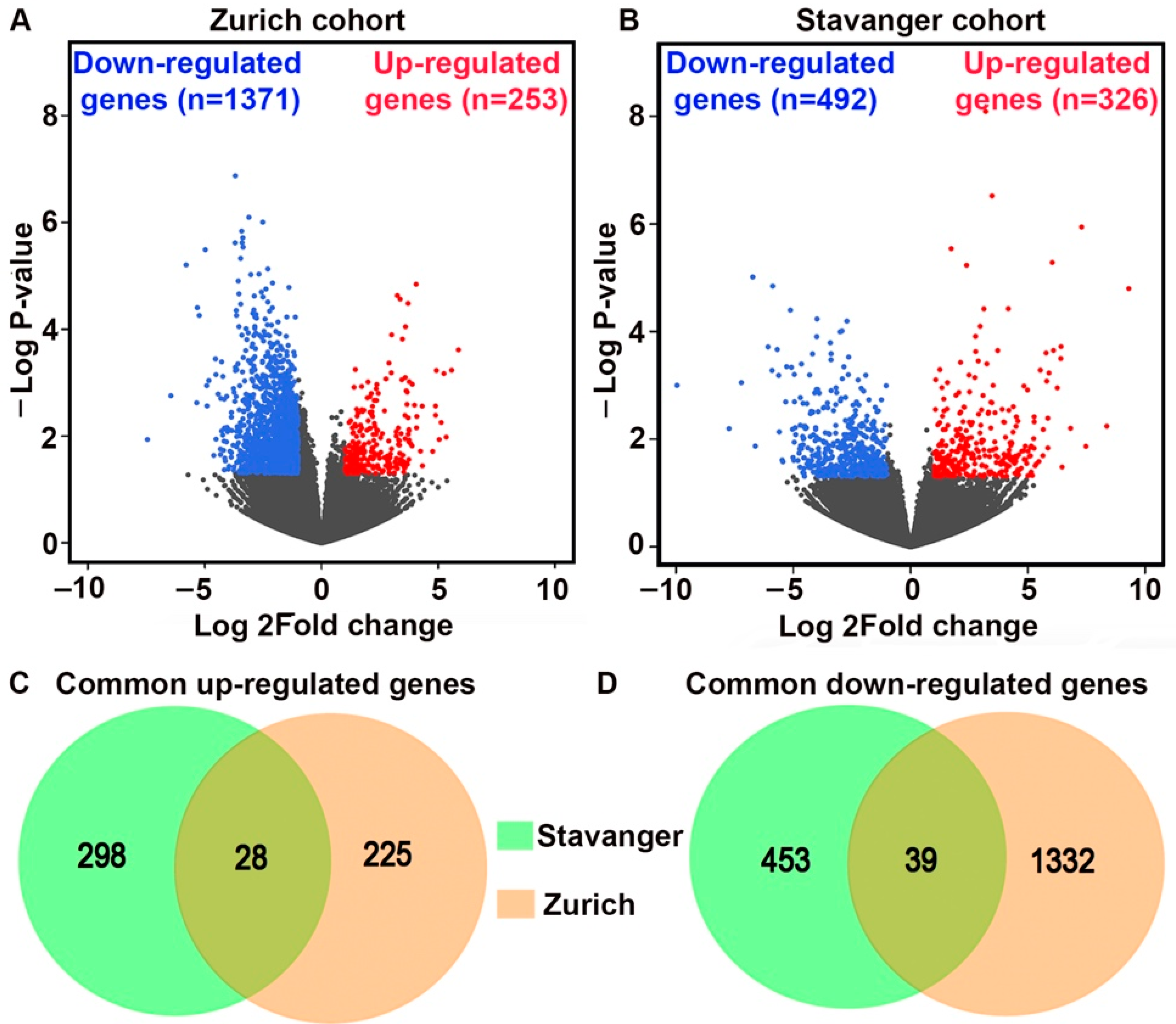
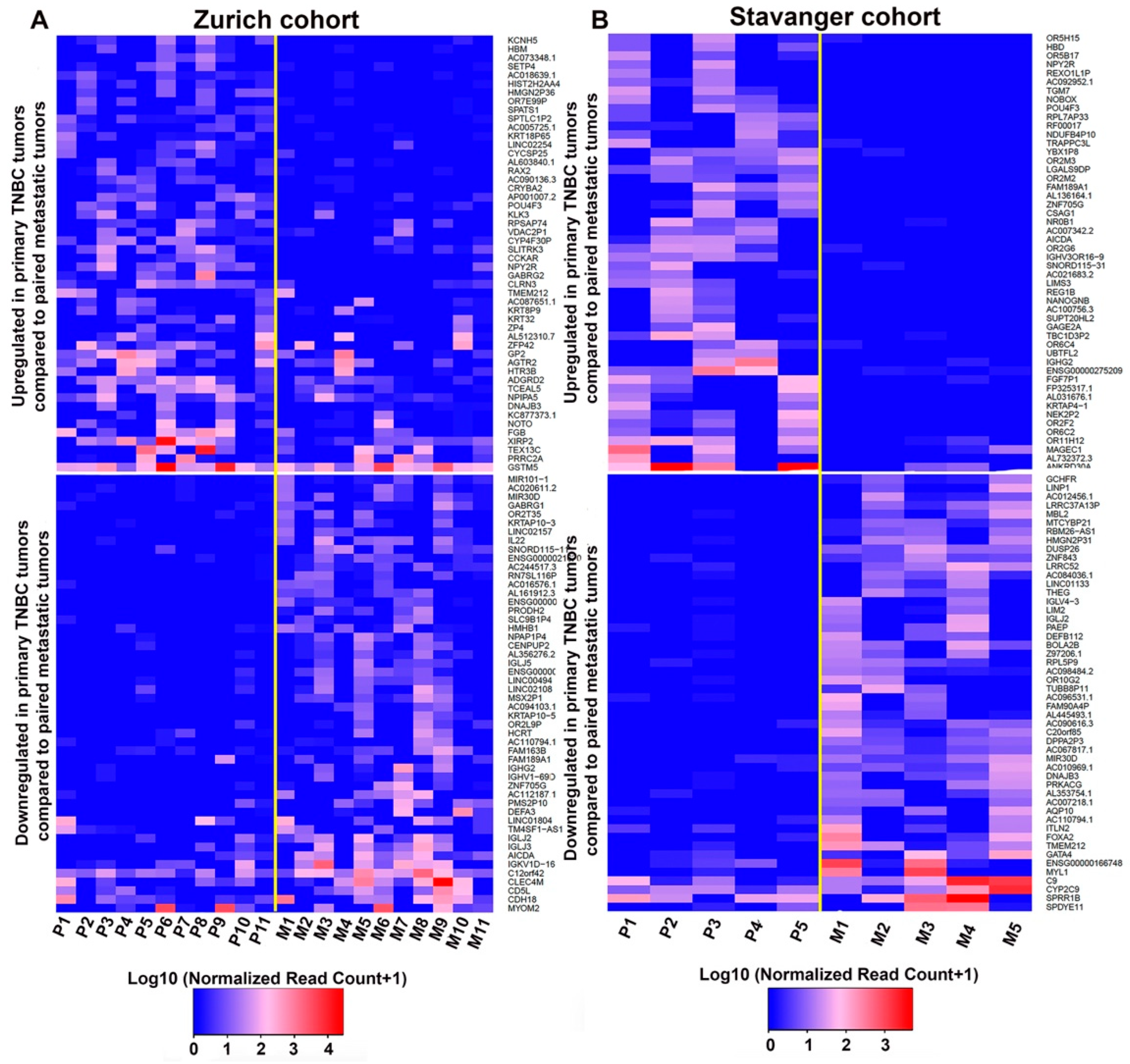
3.1. Identification of DEGs between Primary TNBC Tumors and Matched Metastatic Lesions
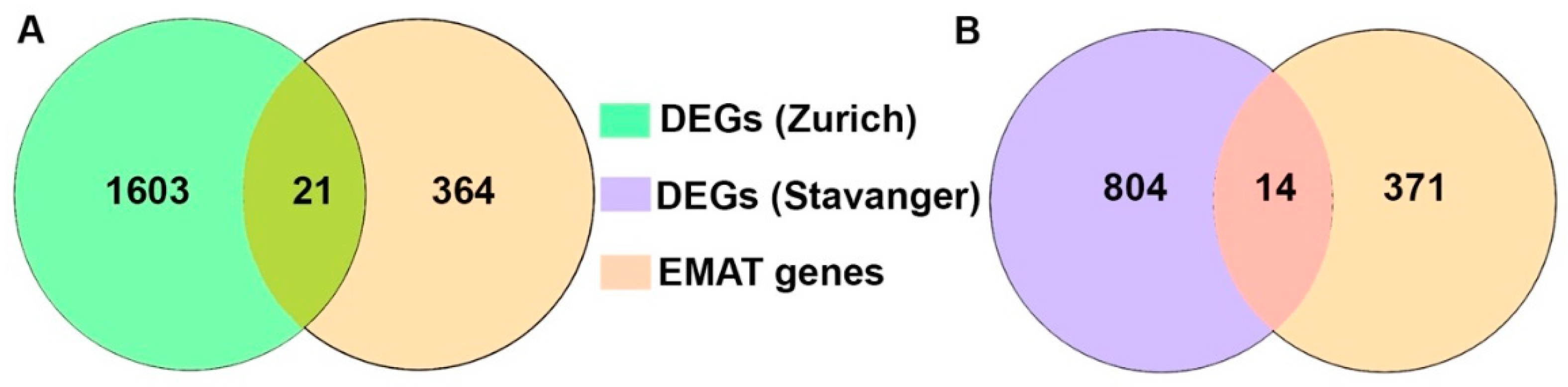
3.2. Analysis of EMAT-Related Gene Signature
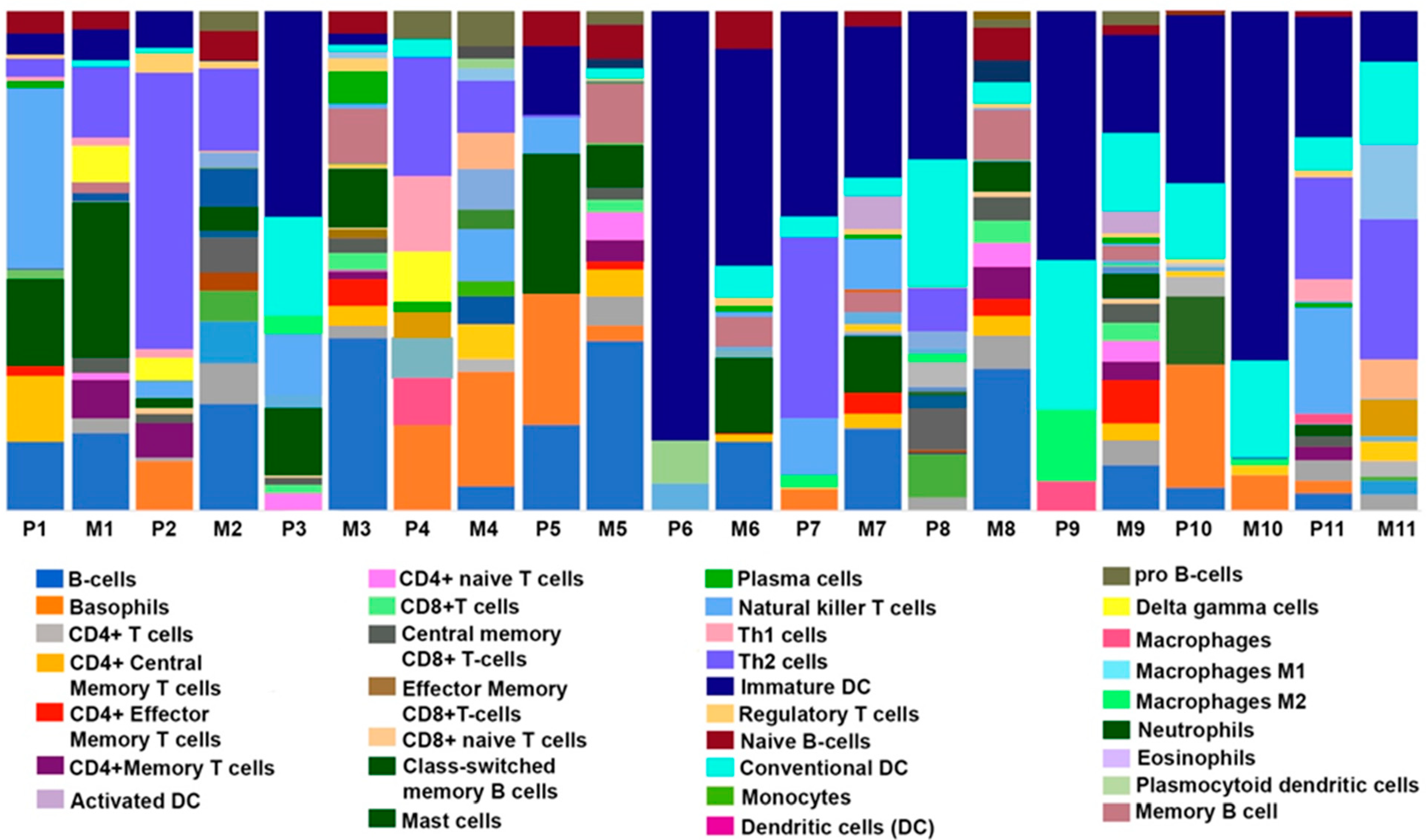
3.3. Comparison of Immune, Stromal, and Microenvironment Scores in Paired Primary and Metastatic TNBC Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Jitariu, A.A.; Cimpean, A.M.; Ribatti, D.; Raica, M. Triple negative breast cancer: The kiss of death. Oncotarget 2017, 8, 46652–46662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9 (Suppl. S2), S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Siddharth, S.; Sharma, D. Triple Negative Breast Cancer: A Mountain Yet to Be Scaled Despite the Triumphs. Cancers 2021, 13, 3697. [Google Scholar] [CrossRef]
- Montel, V.; Huang, T.Y.; Mose, E.; Pestonjamasp, K.; Tarin, D. Expression profiling of primary tumors and matched lymphatic and lung metastases in a xenogeneic breast cancer model. Am. J. Pathol. 2005, 166, 1565–1579. [Google Scholar] [CrossRef] [Green Version]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef]
- Gupta, G.P.; Minn, A.J.; Kang, Y.; Siegel, P.M.; Serganova, I.; Cordon-Cardo, C.; Olshen, A.B.; Gerald, W.L.; Massague, J. Identifying site-specific metastasis genes and functions. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool fro High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 May 2022).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveros, J.C. An Interactive Tool for Comapring Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 12 May 2022).
- Emad, A.; Ray, T.; Jensen, T.W.; Parat, M.; Natrajan, R.; Sinha, S.; Ray, P.S. Superior breast cancer metastasis risk stratification using an epithelial-mesenchymal-amoeboid transition gene signature. Breast Cancer Res. 2020, 22, 74. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruohola, J.K.; Viitanen, T.P.; Valve, E.M.; Seppanen, J.A.; Loponen, N.T.; Keskitalo, J.J.; Lakkakorpi, P.T.; Harkonen, P.L. Enhanced invasion and tumor growth of fibroblast growth factor 8b-overexpressing MCF-7 human breast cancer cells. Cancer Res. 2001, 61, 4229–4237. [Google Scholar] [PubMed]
- Yu, Z.; Lou, L.; Zhao, Y. Fibroblast growth factor 18 promotes the growth, migration and invasion of MDAMB231 cells. Oncol. Rep. 2018, 40, 704–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; Maguire, T.M.; Hill, A.; McDermott, E.; O’Higgins, N. Metalloproteinases: Role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000, 2, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Jansson, S.; Aaltonen, K.; Bendahl, P.O.; Falck, A.K.; Karlsson, M.; Pietras, K.; Ryden, L. The PDGF pathway in breast cancer is linked to tumour aggressiveness, triple-negative subtype and early recurrence. Breast Cancer Res. Treat. 2018, 169, 231–241. [Google Scholar] [CrossRef] [Green Version]
- van de Vijver, M.J.; He, Y.D.; van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- van ’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, T.; Niikura, N.; Ogiya, R.; Yasojima, H.; Watanabe, K.I.; Kanbayashi, C.; Tsuneizumi, M.; Matsui, A.; Fujisawa, T.; Iwasa, T.; et al. Distinct gene expression profiles between primary breast cancers and brain metastases from pair-matched samples. Sci. Rep. 2019, 9, 13343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, Z.; Wu, L.; Ye, J.; Tao, W. Cellular heterogeneity map of diverse immune and stromal phenotypes within breast tumor microenvironment. PeerJ 2020, 8, e9478. [Google Scholar] [CrossRef] [PubMed]
- Tauro, M.; Lynch, C.C. Cutting to the Chase: How Matrix Metalloproteinase-2 Activity Controls Breast-Cancer-to-Bone Metastasis. Cancers 2018, 10, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, A.; Janosky, M.; Harrison, B.; Zhong, J.; Moussai, D.; Siyah, P.; Schatz-Siemers, N.; Zeng, J.; Adams, S.; Mignatti, P. Inhibition of Breast Cancer Metastasis by Presurgical Treatment with an Oral Matrix Metalloproteinase Inhibitor: A Preclinical Proof-of-Principle Study. Mol. Cancer Ther. 2016, 15, 2370–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aukes, K.; Forsman, C.; Brady, N.J.; Astleford, K.; Blixt, N.; Sachdev, D.; Jensen, E.D.; Mansky, K.C.; Schwertfeger, K.L. Breast cancer cell-derived fibroblast growth factors enhance osteoclast activity and contribute to the formation of metastatic lesions. PLoS ONE 2017, 12, e0185736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, I.; Milanezi, F.; Martins, A.; Reis, R.M.; Schmitt, F. Overexpression of platelet-derived growth factor receptor alpha in breast cancer is associated with tumour progression. Breast Cancer Res. 2005, 7, R788–R795. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Brocker, E.B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003, 160, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Zhao, K.; Bao, K.; Li, J. Radiation increases COL1A1, COL3A1, and COL1A2 expression in breast cancer. Open Med. 2022, 17, 329–340. [Google Scholar] [CrossRef]
- Liu, S.; Gonzalez-Prieto, R.; Zhang, M.; Geurink, P.P.; Kooij, R.; Iyengar, P.V.; van Dinther, M.; Bos, E.; Zhang, X.; Le Devedec, S.E.; et al. Deubiquitinase Activity Profiling Identifies UCHL1 as a Candidate Oncoprotein That Promotes TGFbeta-Induced Breast Cancer Metastasis. Clin. Cancer Res. 2020, 26, 1460–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; He, J.; Yang, C.; Orange, M.; Ren, X.; Blair, N.; Tan, T.; Yang, J.M.; Zhu, H. UCH-L1 promotes invasion of breast cancer cells through activating Akt signaling pathway. J. Cell Biochem. 2018, 119, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.K.; Gao, B.; Dadmanesh, F.; Carlson, K.; Qu, Y.; Deng, N.; Cui, X.; Giuliano, A.E. Gene expression comparison between primary triple-negative breast cancer and paired axillary and sentinel lymph node metastasis. Breast J. 2020, 26, 904–910. [Google Scholar] [CrossRef]
- Glas, A.M.; Floore, A.; Delahaye, L.J.; Witteveen, A.T.; Pover, R.C.; Bakx, N.; Lahti-Domenici, J.S.; Bruinsma, T.J.; Warmoes, M.O.; Bernards, R.; et al. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genom. 2006, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Roepman, P.; Van’t Veer, L.J.; Bernards, R.; de Snoo, F.; Glas, A.M. Biological functions of the genes in the mammaprint breast cancer profile reflect the hallmarks of cancer. Biomark. Insights 2010, 5, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seystahl, K.; Papachristodoulou, A.; Burghardt, I.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P.; Weller, M. Biological Role and Therapeutic Targeting of TGF-beta3 in Glioblastoma. Mol. Cancer Ther. 2017, 16, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naber, H.P.; Wiercinska, E.; Pardali, E.; van Laar, T.; Nirmala, E.; Sundqvist, A.; van Dam, H.; van der Horst, G.; van der Pluijm, G.; Heckmann, B.; et al. BMP-7 inhibits TGF-beta-induced invasion of breast cancer cells through inhibition of integrin beta(3) expression. Cell Oncol. 2012, 35, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Kohrt, H.E.; Nouri, N.; Nowels, K.; Johnson, D.; Holmes, S.; Lee, P.P. Profile of immune cells in axillary lymph nodes predicts disease-free survival in breast cancer. PLoS Med. 2005, 2, e284. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Na, K.J.; Choi, H. Differences in Tumor Immune Microenvironment in Metastatic Sites of Breast Cancer. Front. Oncol. 2021, 11, 649004. [Google Scholar] [CrossRef]
- Rye, I.H.; Huse, K.; Josefsson, S.E.; Kildal, W.; Danielsen, H.E.; Schlichting, E.; Garred, O.; Riis, M.L.; Lingjaerde, O.C.; Myklebust, J.H.; et al. Breast cancer metastasis: Immune profiling of lymph nodes reveals exhaustion of effector T cells and immunosuppression. Mol. Oncol. 2022, 16, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, R.E.; Seebach, J.; Field, L.A.; Heckman, C.; Kane, J.; Hooke, J.A.; Love, B.; Shriver, C.D. A gene expression signature that defines breast cancer metastases. Clin. Exp. Metastasis. 2009, 26, 205–213. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics | Metastasis (Lymph Node or Other) | Metastasis-Free | Metastasis (Distant Organ) |
|---|---|---|---|
| Zurich cohort (n = 31) | Stavanger Cohort (n = 5) | ||
| Patient Age, n (%) | |||
| 20–29 | 0 (0.00) | 1 (5.55) | 0 (0.00) |
| 30–39 | 2 (15.38) | 1 (5.55) | 1 (20.00) |
| 40–49 | 1 (7.69) | 4 (22.22) | 1 (20.00) |
| 50–59 | 5 (38.46) | 6 (33.33) | 1 (20.00) |
| 60–69 | 3 (23.07) | 1 (5.55) | 2 (40.00) |
| 70+ | 2 (15.38) | 5 (27.78) | 0 (0.00) |
| Tumor Grade, n (%) | |||
| I | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| II | 1 (7.69) | 2 (11.11) | 0 (0.00) |
| III | 12 (92.31) | 16 (88.89) | 5 (100.00) |
| Missing | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Histological Type, n (%) | |||
| NST (ductal) | 10 (76.92) | 17 (94.44) | 5 (100.00) |
| NST (with secretory differentiation) | 1 (7.69) | 0 (0.00) | 0 (0.00) |
| Apocrine | 1 (7.69) | 1 (5.56) | 0 (0.00) |
| Metaplastic | 1 (7.69) | 0 (0.00) | 0 (0.00) |
| Missing | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Survival Status, n (%) | |||
| Alive | 5 (38.46) | 13 (72.22) | 0 (0.00) |
| Dead | 8 (61.54) | 5 (27.78) | 5 (100.00) |
| Missing | 0 (0.00) | 0 (0.00) | 0 (0.00) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, J.; Chandrashekar, D.S.; Varga, Z.; Sobottka, B.; Janssen, E.; Kowalski, J.; Kiraz, U.; Varambally, S.; Aneja, R. Distinct Gene Expression Profiles of Matched Primary and Metastatic Triple-Negative Breast Cancers. Cancers 2022, 14, 2447. https://doi.org/10.3390/cancers14102447
Kaur J, Chandrashekar DS, Varga Z, Sobottka B, Janssen E, Kowalski J, Kiraz U, Varambally S, Aneja R. Distinct Gene Expression Profiles of Matched Primary and Metastatic Triple-Negative Breast Cancers. Cancers. 2022; 14(10):2447. https://doi.org/10.3390/cancers14102447
Chicago/Turabian StyleKaur, Jaspreet, Darshan S. Chandrashekar, Zsuzsanna Varga, Bettina Sobottka, Emiel Janssen, Jeanne Kowalski, Umay Kiraz, Sooryanarayana Varambally, and Ritu Aneja. 2022. "Distinct Gene Expression Profiles of Matched Primary and Metastatic Triple-Negative Breast Cancers" Cancers 14, no. 10: 2447. https://doi.org/10.3390/cancers14102447
APA StyleKaur, J., Chandrashekar, D. S., Varga, Z., Sobottka, B., Janssen, E., Kowalski, J., Kiraz, U., Varambally, S., & Aneja, R. (2022). Distinct Gene Expression Profiles of Matched Primary and Metastatic Triple-Negative Breast Cancers. Cancers, 14(10), 2447. https://doi.org/10.3390/cancers14102447