
Clinical Impact of Pathogenic Variants in DNA Damage Repair Genes beyond BRCA1 and BRCA2 in Breast and Ovarian Cancer Patients

 , , , , , add
Show full author list
, , , , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials & Methods
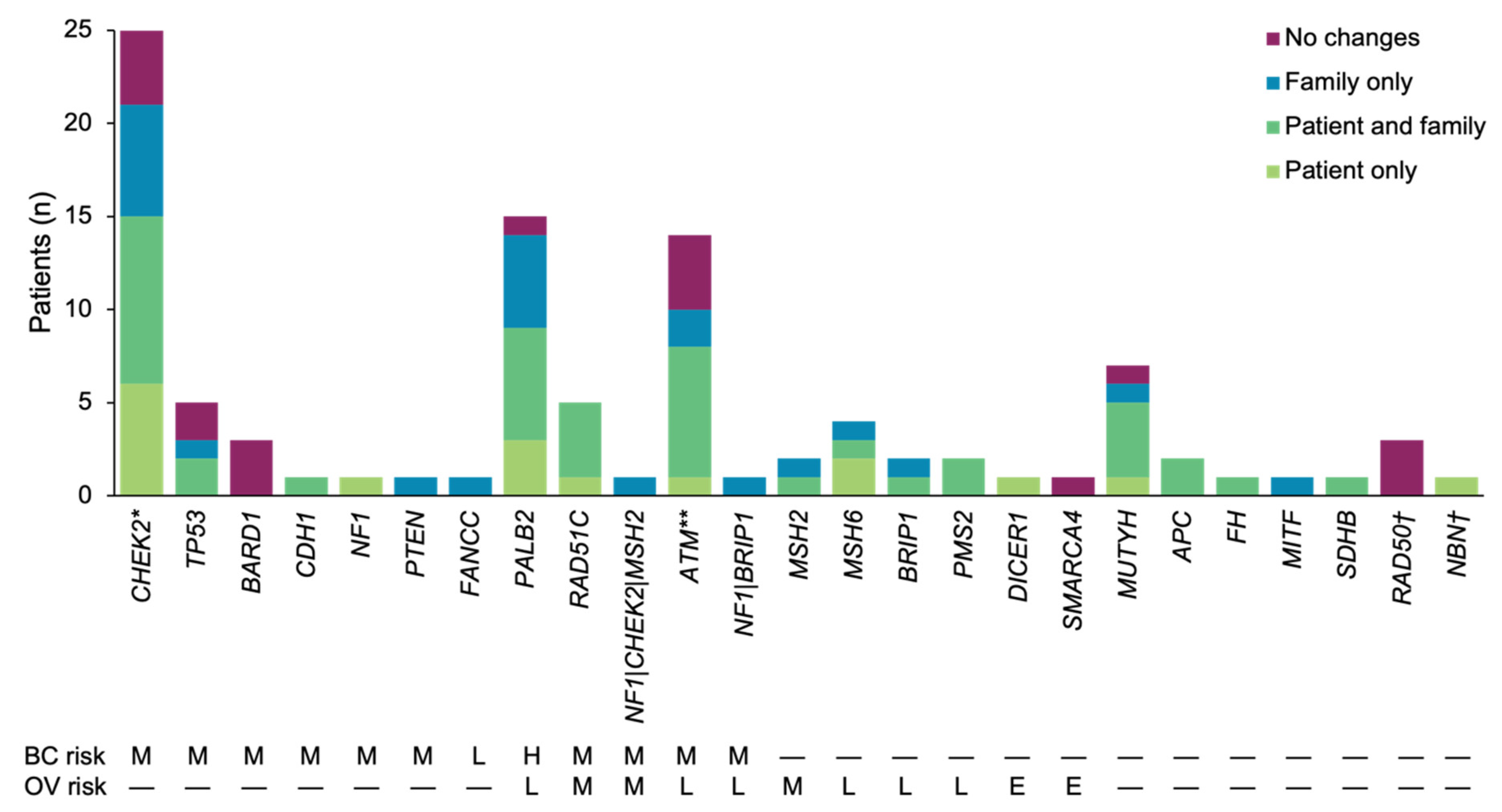
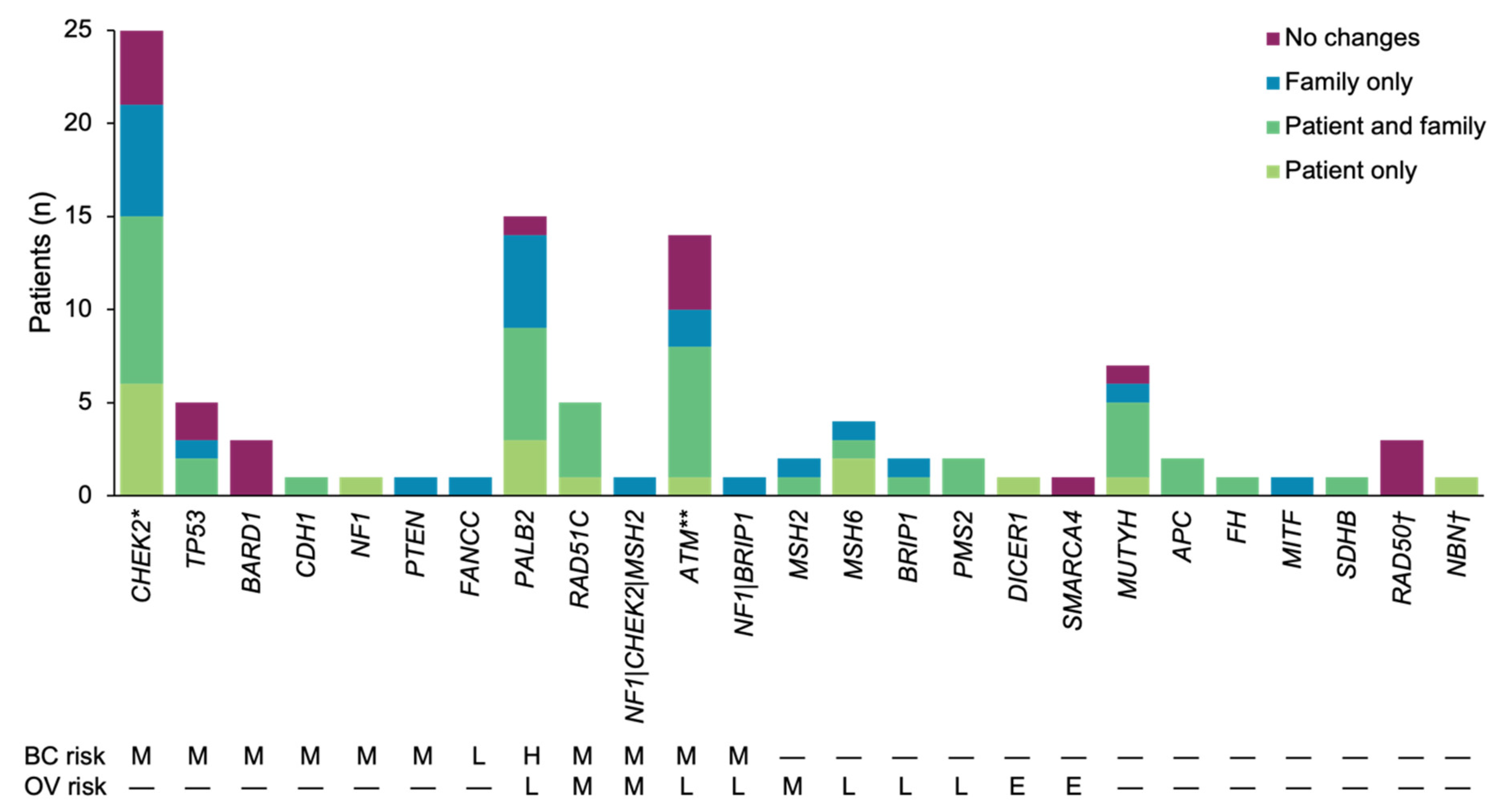
3. Results
3.1. Patient Population
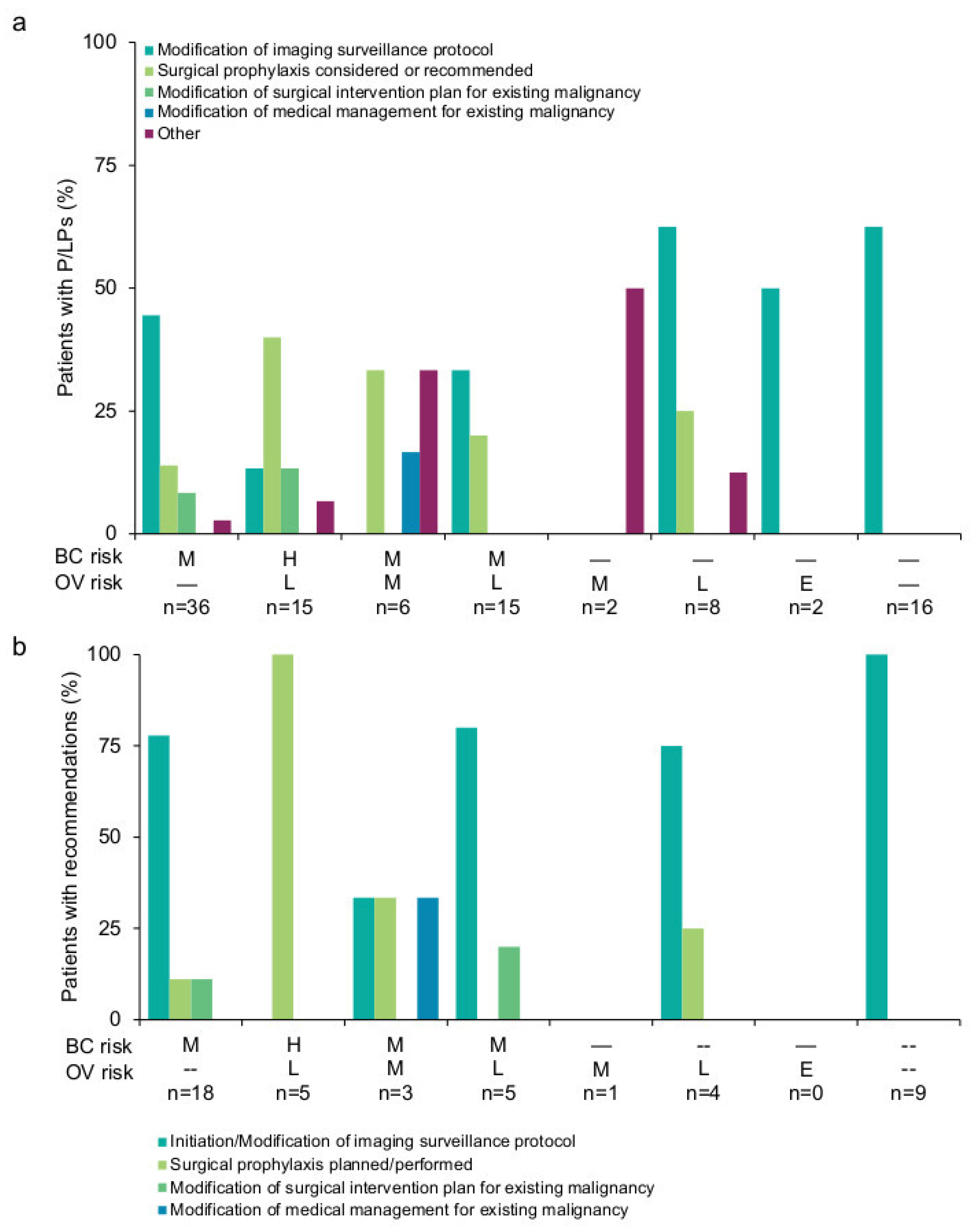
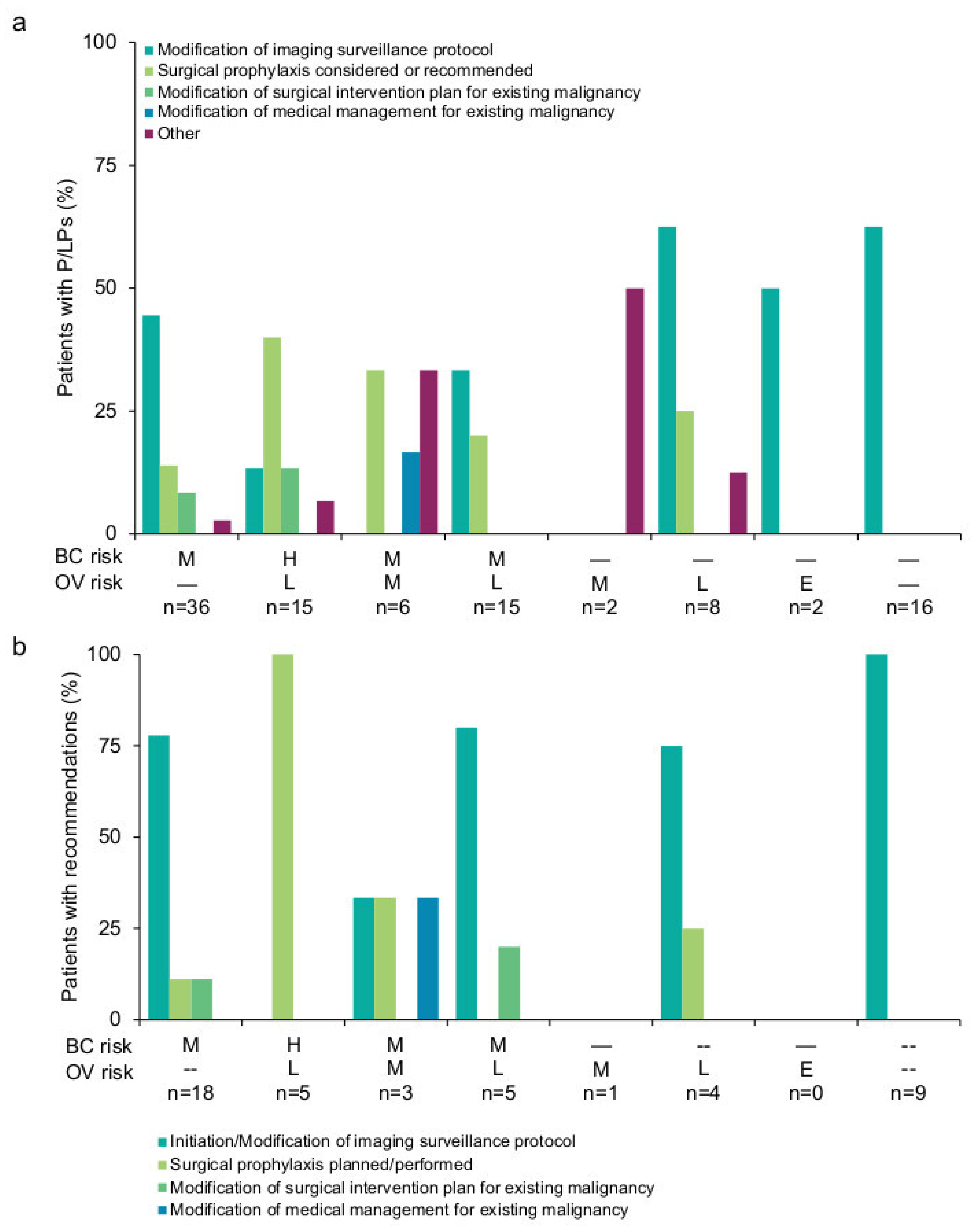
3.2. Results-Based Management Recommendations for Patients
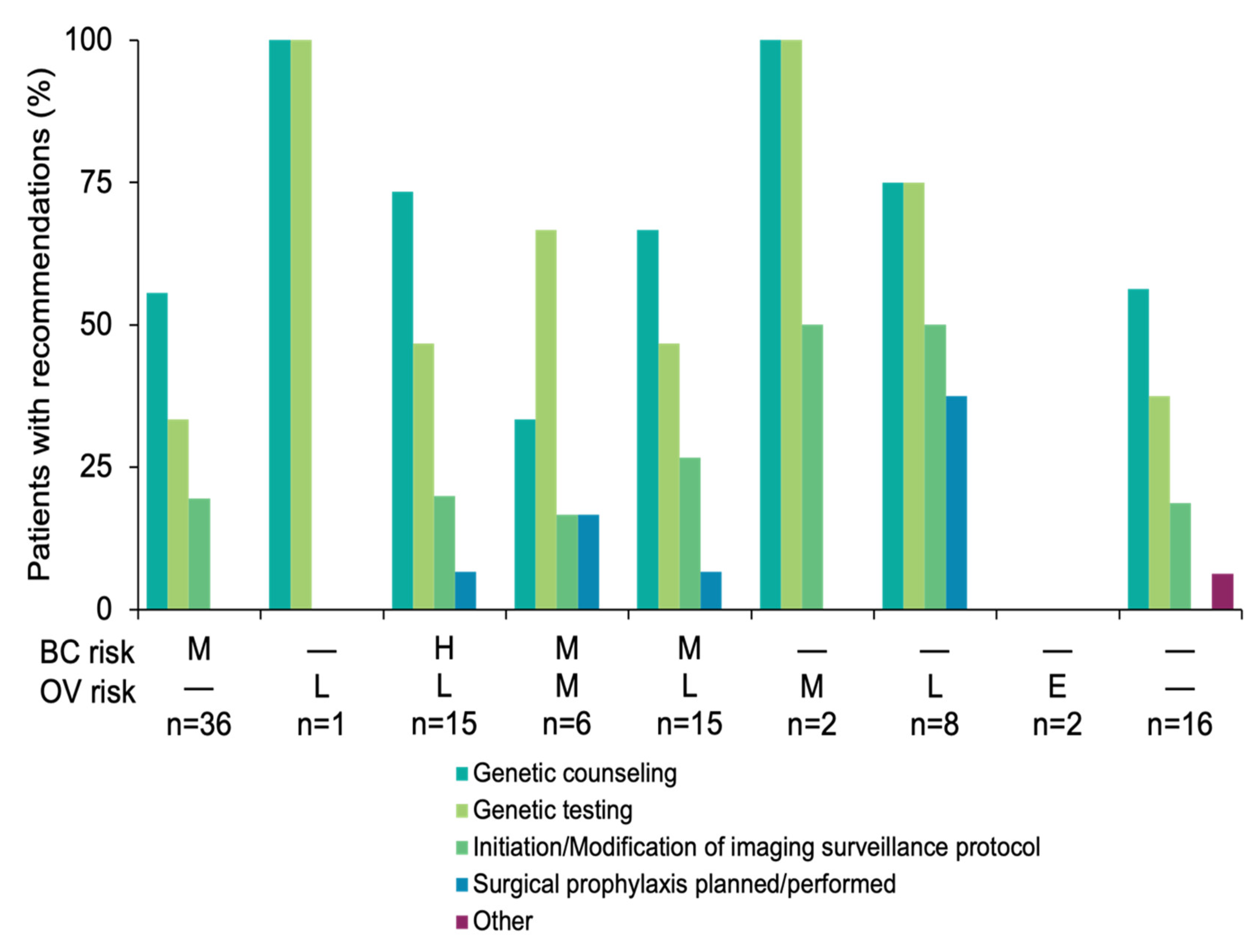
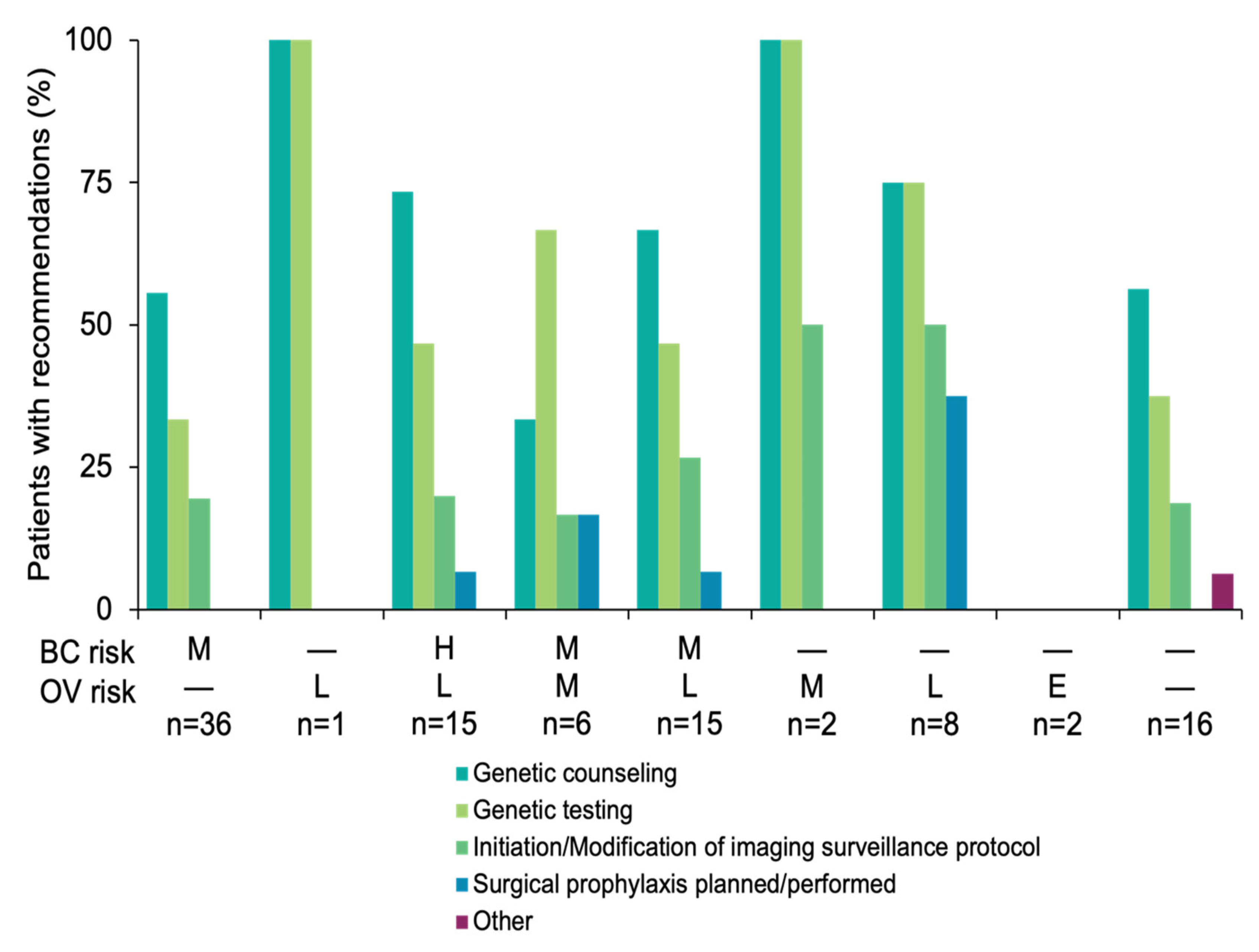
3.3. Results-Based Management Recommendations for Family Members
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Warner, E.; Plewes, D.B.; Hill, K.A.; Causer, P.A.; Zubovits, J.T.; Jong, R.A.; Cutrara, M.R.; DeBoer, G.; Yaffe, M.J.; Messner, S.J.; et al. Surveillance of BRCA1 and BRCA2 Mutation Carriers with Magnetic Resonance Imaging, Ultrasound, Mammography, and Clinical Breast Examination. JAMA 2004, 292, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Domchek, S.M.; Friebel, T.M.; Singer, C.F.; Evans, D.G.; Lynch, H.T.; Isaacs, C.; Garber, J.E.; Neuhausen, S.L.; Matloff, E.; Eeles, R.; et al. Association of Risk-Reducing Surgery in BRCA1 or BRCA2 Mutation Carriers with Cancer Risk and Mortality. JAMA 2010, 304, 967–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of Mutations in Individuals with Breast Cancer Referred for BRCA1 and BRCA2 Testing Using next-Generation Sequencing with a 25-Gene Panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.-R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients with Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurian, A.W.; Hughes, E.; Handorf, E.A.; Gutin, A.; Allen, B.; Hartman, A.-R.; Hall, M.J. Breast and Ovarian Cancer Penetrance Estimates Derived from Germline Multiple-Gene Sequencing Results in Women. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.M.M.; McIntosh, S.A.; Savage, K.I. Homologous Recombination Deficiency in Breast Cancer: Implications for Risk, Cancer Development, and Therapy. Genes Chromosomes Cancer 2021, 60, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.M.; Eccles, D.M.; Romero, I.L.; Al-Mulla, F.; Balmaña, J.; Biancolella, M.; Bslok, R.; Caligo, M.A.; Calvello, M.; Capone, G.L.; et al. Genetic Testing and Clinical Management Practices for Variants in Non-BRCA1/2 Breast (and Breast/Ovarian) Cancer Susceptibility Genes: An International Survey by the Evidence-Based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) Clinical Working Group. JCO Precis. Oncol. 2018, 2, 1. [Google Scholar] [CrossRef]
- Kurian, A.W.; Ward, K.C.; Howlader, N.; Deapen, D.; Hamilton, A.S.; Mariotto, A.; Miller, D.; Penberthy, L.S.; Katz, S.J. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J. Clin. Oncol. 2019, 37, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- LaDuca, H.; Polley, E.C.; Yussuf, A.; Hoang, L.; Gutierrez, S.; Hart, S.N.; Yadav, S.; Hu, C.; Na, J.; Goldgar, D.E.; et al. A Clinical Guide to Hereditary Cancer Panel Testing: Evaluation of Gene-Specific Cancer Associations and Sensitivity of Genetic Testing Criteria in a Cohort of 165,000 High-Risk Patients. Genet. Med. 2020, 22, 407–415. [Google Scholar] [CrossRef] [Green Version]
- National Comprehensive Cancer Network. National Comprehensive Cancer Network Genetic/Familial High-Risk Assessment: Breast, Ovarian and Pancreatic (version 1.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 27 March 2020).
- Lincoln, S.E.; Kobayashi, Y.; Anderson, M.J.; Yang, S.; Desmond, A.J.; Mills, M.A.; Nilsen, G.B.; Jacobs, K.B.; Monzon, F.A.; Kurian, A.W.; et al. A Systematic Comparison of Traditional and Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Genes in More than 1000 Patients. J. Mol. Diagn. 2015, 17, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Truty, R.; Paul, J.; Kennemer, M.; Lincoln, S.E.; Olivares, E.; Nussbaum, R.L.; Aradhya, S. Prevalence and Properties of Intragenic Copy-Number Variation in Mendelian Disease Genes. Genet. Med. 2019, 21, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoln, S.E.; Hambuch, T.; Zook, J.M.; Bristow, S.L.; Hatchell, K.; Truty, R.; Kennemer, M.; Shirts, B.H.; Fellowes, A.; Chowdhury, S.; et al. One in Seven Pathogenic Variants Can be Challenging to Detect by NGS: An Analysis of 450,000 Patients with Implications for Clinical Sensitivity and Genetic Test Implementation. Genet. Med. 2021, 23, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A Comprehensive Refinement of the ACMG-AMP Variant Classification Criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Else, T.; Greenberg, S.; Fishbein, L. Hereditary Paraganglioma-Pheocytoma Syndromes. In GeneReviews®; Adam, M.P., Ardingner, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2008. [Google Scholar]
- National Comprehensive Cancer Network. National Comprehensive Cancer Network Genetc/Familial High-Risk Assessment: Colorectal (version 3.2019). Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdef (accessed on 27 March 2020).
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of At-Risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Menko, F.H.; Maher, E.R.; Schmidt, L.S.; Middelton, L.A.; Aittomäki, K.; Tomlinson, I.; Richard, S.; Linehan, W.M. Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC): Renal Cancer Risk, Surveillance and Treatment. Fam. Cancer 2014, 13, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Pithukpakorn, M.; Toro, J.R. Hereditary Leiomyomatosis and Renal Cell Cancer. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2006. [Google Scholar]
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Endocrine Society Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Lefebvre, M.; Foulkes, W.D. Pheochromocytoma and Paraganglioma Syndromes: Genetics and Management Update. Curr. Oncol. 2014, 21, e8–e17. [Google Scholar] [CrossRef]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.-P. Paraganglioma and Phaeochromocytoma: From Genetics to Personalized Medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef]
- Cancer of the Breast (Female)—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 5 January 2022).
- Cancer of the Ovary—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 5 January 2022).
- Easton, D.F.; Pharoah, P.D.P.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, N.; Domchek, S.M.; Stadler, Z.; Nathanson, K.L.; Couch, F.; Garber, J.E.; Offit, K.; Robson, M.E. Counselling Framework for Moderate-Penetrance Cancer-Susceptibility Mutations. Nat. Rev. Clin. Oncol. 2016, 13, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; Ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer Risks by Gene, Age, and Gender in 6350 Carriers of Pathogenic Mismatch Repair Variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Association Consortium; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar]
- Phillips, K.-A.; Jenkins, M.A.; Lindeman, G.J.; McLachlan, S.A.; McKinley, J.M.; Weideman, P.C.; Hopper, J.L.; Friedlander, M.L. kConFab Investigators Risk-Reducing Surgery, Screening and Chemoprevention Practices of BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Clin. Genet. 2006, 70, 198–206. [Google Scholar] [CrossRef]
- Padamsee, T.J.; Wills, C.E.; Yee, L.D.; Paskett, E.D. Decision Making for Breast Cancer Prevention among Women at Elevated Risk. Breast Cancer Res. 2017, 19, 34. [Google Scholar] [CrossRef] [Green Version]
- Paquet, L.; Simmonds, L.; Yang, C.; Verma, S. An Exploratory Study of Patients’ Views about Being at High-Risk for Breast Cancer and Risk Management Beliefs and Intentions, before and after Risk Counselling: Preliminary Evidence of the Influence of Beliefs on Post-Counselling Prevention Intentions. Patient Educ. Couns. 2017, 100, 575–582. [Google Scholar] [CrossRef]
- Hao, J.; Hassen, D.; Manickam, K.; Murray, M.F.; Hartzel, D.N.; Hu, Y.; Liu, K.; Rahm, A.K.; Williams, M.S.; Lazzeri, A.; et al. Healthcare Utilization and Costs after Receiving a Positive BRCA1/2 Result from a Genomic Screening Program. J. Pers. Med. 2020, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Heikkinen, K.; Karppinen, S.-M.; Soini, Y.; Mäkinen, M.; Winqvist, R. Mutation Screening of Mre11 Complex Genes: Indication of RAD50 Involvement in Breast and Ovarian Cancer Susceptibility. J. Med. Genet. 2003, 40, e131. [Google Scholar] [CrossRef] [Green Version]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Manahan, E.R.; Kuerer, H.M.; Sebastian, M.; Hughes, K.S.; Boughey, J.C.; Euhus, D.M.; Boolbol, S.K.; Taylor, W.A. Consensus Guidelines on Genetic Testing for Hereditary Breast Cancer from the American Society of Breast Surgeons. Ann. Surg. Oncol. 2019, 26, 3025–3031. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Characteristic, n (%) | Patients (n = 101) |
|---|---|
| Ethnicity a | |
| White/Caucasian | 89 (88.1) |
| Black/African American | 4 (4.0) |
| Ashkenazi Jewish | 3 (3.0) |
| Other | 4 (4.0) |
| Unknown | 1 (1.0) |
| Age at testing a | |
| 20–29 years | 3 (3.0) |
| 30–39 years | 5 (5.0) |
| 40–49 years | 25 (24.8) |
| 50–59 years | 31 (30.7) |
| 60–69 years | 27 (26.7) |
| 70–79 years | 6 (5.9) |
| ≥80 years | 4 (4.0) |
| Personal Br/Ov Ca history a,b | |
| Breast cancer | 84 (83.2) |
| Ovarian cancer | 14 (13.9) |
| Breast and ovarian cancers | 3 (3.0) |
| Other personal cancer history (in addition to Br/Ov Ca) | |
| Yes | 19 (18.8) |
| No | 77 (76.2) |
| Not reported | 5 (5.0) |
| Family Br/Ov Ca history a | |
| Breast cancer | 44 (43.6) |
| Ovarian cancer | 4 (4.0) |
| Breast and ovarian cancers | 15 (14.9) |
| No | 17 (16.8) |
| Not reported | 22 (21.8) |
| Family non-Br/Ov Ca history | |
| Yes | 62 (60.4) |
| No | 38 (37.6) |
| Not reported | 2 (2.0) |
| Previous genetic testing a | |
| Patient only | 4 (4.0) |
| Patient and relative(s) | 2 (2.0) |
| Relative(s) only | 2 (2.0) |
| None | 93 (91.1) |
| Number of genes on ordered test | |
| 1–5 genes | 4 (4.0) |
| 6–15 genes | 16 (15.8) |
| 16–50 genes | 76 (75.2) |
| 51+ genes | 5 (5.0) |
| Reason for No Results-Based Recommendations | Number (Percentage) of Patients |
|---|---|
| Patient already had advanced/metastatic cancer (or died before results available) | 13 (30.2) |
| Gene/Variant/Finding with no clear recommendations available a | 9 (20.9) |
| Patient had previous interventions before genetic testing due to personal or family history of cancer | 4 (9.3) |
| Recommendations for patient’s current cancer diagnosis superseded recommendations for genetic testing result or no further recommendations were applicable at this point in time given the patient’s current cancer diagnosis | 6 (14.0) |
| Patient preference: patient did not follow recommendations or followed alternative recommendations | 3 (7.0) |
| No information provided | 8 (18.6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinel, W.; Champine, M.; Hampel, H.; Jeter, J.; Sweet, K.; Pilarski, R.; Pearlman, R.; Shane, K.; Brock, P.; Westman, J.A.; et al. Clinical Impact of Pathogenic Variants in DNA Damage Repair Genes beyond BRCA1 and BRCA2 in Breast and Ovarian Cancer Patients. Cancers 2022, 14, 2426. https://doi.org/10.3390/cancers14102426
Espinel W, Champine M, Hampel H, Jeter J, Sweet K, Pilarski R, Pearlman R, Shane K, Brock P, Westman JA, et al. Clinical Impact of Pathogenic Variants in DNA Damage Repair Genes beyond BRCA1 and BRCA2 in Breast and Ovarian Cancer Patients. Cancers. 2022; 14(10):2426. https://doi.org/10.3390/cancers14102426
Chicago/Turabian StyleEspinel, Whitney, Marjan Champine, Heather Hampel, Joanne Jeter, Kevin Sweet, Robert Pilarski, Rachel Pearlman, Kate Shane, Pamela Brock, Judith A. Westman, and et al. 2022. "Clinical Impact of Pathogenic Variants in DNA Damage Repair Genes beyond BRCA1 and BRCA2 in Breast and Ovarian Cancer Patients" Cancers 14, no. 10: 2426. https://doi.org/10.3390/cancers14102426
APA StyleEspinel, W., Champine, M., Hampel, H., Jeter, J., Sweet, K., Pilarski, R., Pearlman, R., Shane, K., Brock, P., Westman, J. A., Kipnis, L., Sotelo, J., Chittenden, A., Culver, S., Stopfer, J. E., Schneider, K. A., Sacca, R., Koeller, D. R., Gaonkar, S., ... Esplin, E. D. (2022). Clinical Impact of Pathogenic Variants in DNA Damage Repair Genes beyond BRCA1 and BRCA2 in Breast and Ovarian Cancer Patients. Cancers, 14(10), 2426. https://doi.org/10.3390/cancers14102426