
Current and Future Therapies for Pancreatic Ductal Adenocarcinoma


Abstract
Simple Summary
Abstract
1. Introduction
2. Current Approved Therapies
Chemotherapy
3. Targeted Therapeutic Approaches
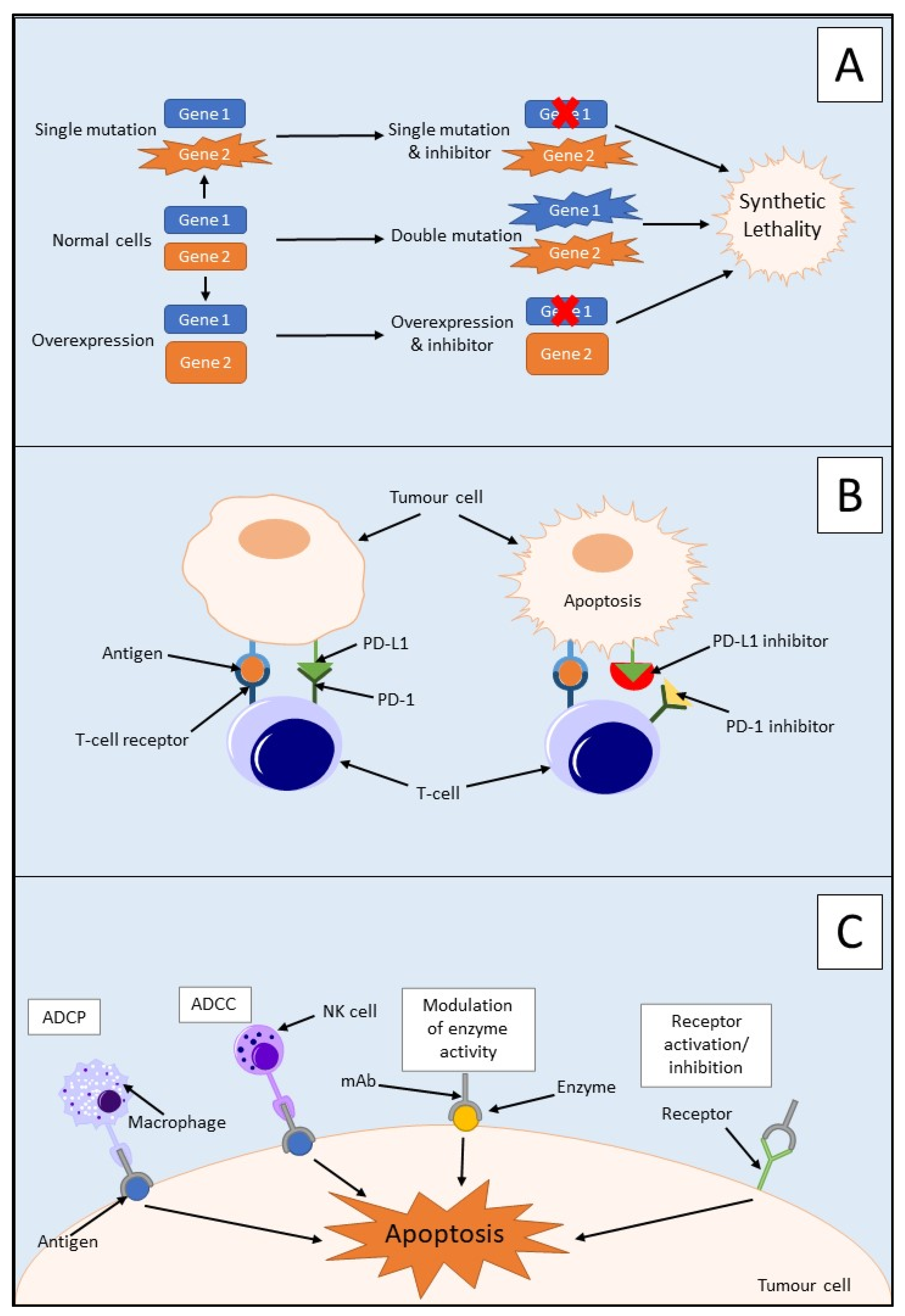
3.1. Synthetic Lethality
3.2. Immunotherapy
3.2.1. Immune Checkpoint Inhibitors
3.2.2. Monoclonal Antibodies
3.2.3. Adoptive Cell Transfer
3.2.4. Therapeutic Vaccination
3.3. Ongoing Clinical Trials
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef]
- Maron, R.; Schechter, B.; Nataraj, N.B.; Ghosh, S.; Romaniello, D.; Marrocco, I.; Noronha, A.; Carvalho, S.; Yarden, Y.; Sela, M. Inhibition of a pancreatic cancer model by cooperative pairs of clinically approved and experimental antibodies. Biochem. Biophys. Res. Commun. 2019, 513, 219–225. [Google Scholar] [CrossRef] [PubMed]
- NCRI/ICS. Diagnosing Cancer in an Emergency: Patterns of Emergency Presentation of Cancer in Ireland 2002–2015; Irish Cancer Society: Dublin, Ireland; National Cancer Registry: Cork, Ireland, 2018. [Google Scholar]
- Zhang, Y.; Yang, C.; Cheng, H.; Fan, Z.; Huang, Q.; Lu, Y.; Fan, K.; Luo, G.; Jin, K.; Wang, Z.; et al. Novel agents for pancreatic ductal adenocarcinoma: Emerging therapeutics and future directions. J. Hematol. Oncol. 2018, 11, 14. [Google Scholar] [CrossRef]
- NICE. Pancreatic Cancer in Adults: Diagnosis and Management; NICE: London, UK, 2018. [Google Scholar]
- Su, Y.H.; Hsu, T.W.; Chen, H.A.; Su, C.M.; Huang, M.T.; Chuang, T.H.; Leo Su, J.; Hsieh, C.L.; Chiu, C.F. ERK-mediated transcriptional activation of Dicer is involved in gemcitabine resistance of pancreatic cancer. J. Cell. Physiol. 2021, 236, 4420–4434. [Google Scholar] [CrossRef]
- Ahmad-Nielsen, S.A.; Bruun Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Frequency of mismatch repair deficiency in pancreatic ductal adenocarcinoma. Pathol. Res. Pract. 2020, 216, 152985. [Google Scholar] [CrossRef]
- Liang, C.; Shi, S.; Meng, Q.; Liang, D.; Ji, S.; Zhang, B.; Qin, Y.; Xu, J.; Ni, Q.; Yu, X. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: Where we are and where we are going. Exp. Mol. Med. 2017, 49, e406. [Google Scholar] [CrossRef]
- Peretti, U.; Zanon, S.; Michele, R. The personalized medicine for pancreatic ductal adenocarcinoma patients: The oncologist perspective. Endosc. Ultrasound 2017, 6, S66–S68. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Du, Y.; Zhao, X.; Wang, C. Tumor microenvironment and metabolic remodeling in gemcitabine-based chemoresistance of pancreatic cancer. Cancer Lett. 2021, 521, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Bardeesy, N. Pancreatic Cancer Metabolism: Breaking It Down to Build It Back Up. Cancer Discov. 2015, 5, 1247–1261. [Google Scholar] [CrossRef]
- Karamitopoulou, E. The Tumor Microenvironment of Pancreatic Cancer. Cancers 2020, 12, 3076. [Google Scholar] [CrossRef]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Vicent, S.; Ponz-Sarvisé, M. PDAC as an Immune Evasive Disease: Can 3D Model Systems Aid to Tackle This Clinical Problem? Front. Cell Dev. Biol. 2021, 9, 787249. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Golan, T.; Atias, D.; Barshack, I.; Avivi, C.; Goldstein, R.S.; Berger, R. Ascites-derived pancreatic ductal adenocarcinoma primary cell cultures as a platform for personalised medicine. Br. J. Cancer 2014, 110, 2269–2276. [Google Scholar] [CrossRef][Green Version]
- Salinas-Miranda, E.; Deniffel, D.; Dong, X.; Healy, G.M.; Khalvati, F.; O’Kane, G.M.; Knox, J.; Bathe, O.F.; Baracos, V.E.; Gallinger, S.; et al. Prognostic value of early changes in CT-measured body composition in patients receiving chemotherapy for unresectable pancreatic cancer. Eur. Radiol. 2021, 31, 8662–8670. [Google Scholar] [CrossRef]
- Hamed, S.S.; Straubinger, R.M.; Jusko, W.J. Pharmacodynamic modeling of cell cycle and apoptotic effects of gemcitabine on pancreatic adenocarcinoma cells. Cancer Chemother. Pharmacol. 2013, 72, 553–563. [Google Scholar] [CrossRef]
- Gelibter, A.; Malaguti, P.; Di Cosimo, S.; Bria, E.; Ruggeri, E.M.; Carlini, P.; Carboni, F.; Ettorre, G.M.; Pellicciotta, M.; Giannarelli, D.; et al. Fixed dose-rate gemcitabine infusion as first-line treatment for advanced-stage carcinoma of the pancreas and biliary tree. Cancer 2005, 104, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Schellens, J.H. Capecitabine. Oncologist 2007, 12, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Köhne, C.H.; Peters, G.J. UFT: Mechanism of drug action. Oncology 2000, 14, 13–18. [Google Scholar] [PubMed]
- Shen, Z.T.; Zhou, H.; Li, A.M.; Ji, X.Q.; Jiang, C.C.; Yuan, X.; Li, B.; Zhu, X.X.; Huang, G.C. Clinical outcomes and prognostic factors of stereotactic body radiation therapy combined with gemcitabine plus capecitabine for locally advanced unresectable pancreatic cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Brachmann, C.; Liu, X.; Pierce, D.W.; Dey, J.; Kerwin, W.S.; Li, Y.; Zhou, S.; Hou, S.; Carleton, M.; et al. Albumin-bound nanoparticle (nab) paclitaxel exhibits enhanced paclitaxel tissue distribution and tumor penetration. Cancer Chemother. Pharmacol. 2015, 76, 699–712. [Google Scholar] [CrossRef] [PubMed]
- De Vita, F.; Ventriglia, J.; Febbraro, A.; Laterza, M.M.; Fabozzi, A.; Savastano, B.; Petrillo, A.; Diana, A.; Giordano, G.; Troiani, T.; et al. NAB-paclitaxel and gemcitabine in metastatic pancreatic ductal adenocarcinoma (PDAC): From clinical trials to clinical practice. BMC Cancer 2016, 16, 709. [Google Scholar] [CrossRef]
- Deyme, L.; Barbolosi, D.; Gattacceca, F. Population pharmacokinetics of FOLFIRINOX: A review of studies and parameters. Cancer Chemother. Pharmacol. 2019, 83, 27–42. [Google Scholar] [CrossRef]
- Khushman, M.; Dempsey, N.; Maldonado, J.C.; Loaiza-Bonilla, A.; Velez, M.; Carcas, L.; Dammrich, D.; Hurtado-Cordovi, J.; Parajuli, R.; Pollack, T.; et al. Full dose neoadjuvant FOLFIRINOX is associated with prolonged survival in patients with locally advanced pancreatic adenocarcinoma. Pancreatology 2015, 15, 667–673. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Guo, M. Synthetic lethality strategies: Beyond BRCA1/2 mutations in pancreatic cancer. Cancer Sci. 2020, 111, 3111–3121. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Stoof, J.; Harrold, E.; Mariottino, S.; Lowery, M.A.; Walsh, N. DNA Damage Repair Deficiency in Pancreatic Ductal Adenocarcinoma: Preclinical Models and Clinical Perspectives. Front. Cell Dev. Biol. 2021, 9, 749490. [Google Scholar] [CrossRef]
- Wei, X.; Yang, J.; Adair, S.J.; Ozturk, H.; Kuscu, C.; Lee, K.Y.; Kane, W.J.; O’Hara, P.E.; Liu, D.; Demirlenk, Y.M.; et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28068–28079. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Q.; Wang, M.F.; Chen, H.L.; Shang, D.; Das, J.K.; Song, J. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol. Cancer 2020, 19, 32. [Google Scholar] [CrossRef]
- Dean-Colomb, W.; Esteva, F.J. Her2-positive breast cancer: Herceptin and beyond. Eur. J. Cancer 2008, 44, 2806–2812. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Faghfuri, E.; Faramarzi, M.A.; Nikfar, S.; Abdollahi, M. Nivolumab and pembrolizumab as immune-modulating monoclonal antibodies targeting the PD-1 receptor to treat melanoma. Expert Rev. Anticancer Ther. 2015, 15, 981–993. [Google Scholar] [CrossRef]
- Panni, R.Z.; Herndon, J.M.; Zuo, C.; Hegde, S.; Hogg, G.D.; Knolhoff, B.L.; Breden, M.A.; Li, X.; Krisnawan, V.E.; Khan, S.Q.; et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci. Transl. Med. 2019, 11, aau9240. [Google Scholar] [CrossRef] [PubMed]
- Olaoba, O.T.; Ligali, F.C.; Alabi, Z.O.; Akinyemi, A.O.; Ayinde, K.S. Of immune checkpoint maladies and remedies: The throwing of jabs in the oncogenic ring of PDAC. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188483. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Song, X.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017, 407, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, X.; Wei, X.; Jiang, H.; Lan, C.; Yang, S.; Wang, H.; Yang, Y.; Tian, C.; Xu, Z.; et al. PD-L1 is a direct target of cancer-FOXP3 in pancreatic ductal adenocarcinoma (PDAC), and combined immunotherapy with antibodies against PD-L1 and CCL5 is effective in the treatment of PDAC. Signal Transduct. Target. Ther. 2020, 5, 38. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, C.K.; Lin, C.H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.e1059. [Google Scholar] [CrossRef]
- Geng, L.; Huang, D.; Liu, J.; Qian, Y.; Deng, J.; Li, D.; Hu, Z.; Zhang, J.; Jiang, G.; Zheng, S. B7-H1 up-regulated expression in human pancreatic carcinoma tissue associates with tumor progression. J. Cancer Res. Clin. Oncol. 2008, 134, 1021–1027. [Google Scholar] [CrossRef]
- Wang, X.; Lang, M.; Zhao, T.; Feng, X.; Zheng, C.; Huang, C.; Hao, J.; Dong, J.; Luo, L.; Li, X.; et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3. Oncogene 2017, 36, 3048–3058. [Google Scholar] [CrossRef]
- Zheng, L. Study of Pembrolizumab with or without Defa Actinib Following Chemotherapy as a Neoadjuvant and Adjuvant Treatment for Resectable Pancreatic Ductal Adenocarcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03727880?recrs=ab&cond=pdac&draw=2&rank=1 (accessed on 11 August 2021).
- Bengsch, F.; Knoblock, D.M.; Liu, A.; Mcallister, F.; Beatty, G.L. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017, 66, 1609–1617. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Ware, M.B.; Mcquinn, C.; Zaidi, M.Y.; Knochelmann, H.; Mace, T.A.; Chen, Z.; Zhang, C.; Farren, M.R.; Ruggieri, A.N.; Bowers, J.; et al. Dual blockade of IL-6 and CTLA-4 regresses pancreatic tumors in a CD4+ T cell-dependent manner. Cold Spring Harb. Lab. 2020. preprint. [Google Scholar]
- Michaels, A.D.; Newhook, T.E.; Adair, S.J.; Morioka, S.; Goudreau, B.J.; Nagdas, S.; Mullen, M.G.; Persily, J.B.; Bullock, T.N.J.; Slingluff, C.L.; et al. CD47 Blockade as an Adjuvant Immunotherapy for Resectable Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Lu, Z.; Xu, J. Targeting cluster of differentiation 47 improves the efficacy of anti-cytotoxic T-lymphocyte associated protein 4 treatment via antigen presentation enhancement in pancreatic ductal adenocarcinoma. Exp. Ther. Med. 2020, 20, 3301–3309. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Yang, J.; Shangguan, J.; Eresen, A.; Li, Y.; Ma, Q.; Yaghmai, V.; Velichko, Y.; Hu, C.; Zhang, Z. Natural killer cell-based adoptive transfer immunotherapy for pancreatic ductal adenocarcinoma in a. Am. J. Cancer Res. 2019, 9, 1757–1765. [Google Scholar] [PubMed]
- Furukawa, K.; Tanemura, M.; Miyoshi, E.; Eguchi, H.; Nagano, H.; Matsunami, K.; Nagaoka, S.; Yamada, D.; Asaoka, T.; Noda, T.; et al. A practical approach to pancreatic cancer immunotherapy using resected tumor lysate vaccines processed to express α-gal epitopes. PLoS ONE 2017, 12, e0184901. [Google Scholar] [CrossRef]
- Ouyang, X.; Liu, Y.; Zhou, Y.; Guo, J.; Wei, T.T.; Liu, C.; Lee, B.; Chen, B.; Zhang, A.; Casey, K.M.; et al. Antitumor effects of iPSC-based cancer vaccine in pancreatic cancer. Stem Cell Rep. 2021, 16, 1468–1477. [Google Scholar] [CrossRef]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Lim, K.H. BMS-813160 with Nivolumab and Gemcitabine and Nab-Paclitaxel in Borderline Resectable and Locally Advanced Pancreatic Ductal Adenocarcinoma (PDAC). Available online: https://clinicaltrials.gov/ct2/show/NCT03496662?recrs=ab&cond=pdac&draw=2&rank=4 (accessed on 11 August 2021).
- NCI. CCR2/CCR5 Antagonist BMS-813160. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/ccr2-ccr5-antagonist-bms-813160 (accessed on 21 September 2021).
- O’Hara, M. Pilot Study of Mature Dendritic Cell Vaccination against Mutated KRAS in Patients with Resectable Pancreatic Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03549000?recrs=ab&cond=pdac&draw=3&rank=14 (accessed on 11 August 2021).
- Zaidi, N. Pooled Mutant KRAS-Targeted Long Peptide Vaccine Combined with Nivolumab and Ipilimumab for Patients with Resected MMR-p Colorectal and Pancreatic Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04117087?recrs=ab&cond=pdac&draw=4&rank=21 (accessed on 11 August 2021).
- Zheng, L. Trial of Neoadjuvant and Adjuvant Nivolumab and BMS-813160 with or without GVAX for Locally Advanced Pancreatic Ductal Adenocarcinomas. Available online: https://clinicaltrials.gov/ct2/show/NCT03767582?recrs=ab&cond=pdac&draw=2&rank=2 (accessed on 11 August 2021).



{kind=link}
{kind=link}
| Treatment | Target | Stage | Phase | Reference |
|---|---|---|---|---|
| Pembrolizumab Defactinib | PD-1 monoclonal antibody Focal adhesion kinase inhibitor | Resectable PDAC | Recruiting, Phase 2 | [50] |
| Nivolumab BMS-813160 GVAX | PD-1 monoclonal antibody CCR2/CCR5 dual antagonist Whole tumour cell vaccine | Locally advanced PDAC | Recruiting, Phase 1/2 | [64] |
| BMS-813160 Nivolumab Gemcitabine Nab-paclitaxel | CCR2/CCR5 dual antagonist PD-1 monoclonal antibody Chemotherapy Chemotherapy | Borderline resectable/locally advanced PDAC | Recruiting, Phase 1/2 | [60] |
| mDC3/8-KRAS vaccine | Mutant KRAS | Resectable PDAC | Recruiting, Phase 1 | [62] |
| KRAS peptide vaccine Nivolumab Ipilimumab | KRAS peptide vaccine PD-1 monoclonal antibody CTLA-4 monoclonal antibody | Resected PDAC | Recruiting, Phase 1 | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sally, Á.; McGowan, R.; Finn, K.; Moran, B.M. Current and Future Therapies for Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 2417. https://doi.org/10.3390/cancers14102417
Sally Á, McGowan R, Finn K, Moran BM. Current and Future Therapies for Pancreatic Ductal Adenocarcinoma. Cancers. 2022; 14(10):2417. https://doi.org/10.3390/cancers14102417
Chicago/Turabian StyleSally, Áine, Ryan McGowan, Karen Finn, and Brian Michael Moran. 2022. "Current and Future Therapies for Pancreatic Ductal Adenocarcinoma" Cancers 14, no. 10: 2417. https://doi.org/10.3390/cancers14102417
APA StyleSally, Á., McGowan, R., Finn, K., & Moran, B. M. (2022). Current and Future Therapies for Pancreatic Ductal Adenocarcinoma. Cancers, 14(10), 2417. https://doi.org/10.3390/cancers14102417