
EGFR R521K Polymorphism Is Not a Major Determinant of Clinical Cetuximab Resistance in Head and Neck Cancer

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Cultures
2.3. Genetic Characterization of HNSCC Cell Lines and Patient Samples
2.4. Cell Proliferation Assay
2.5. Flow Cytometry
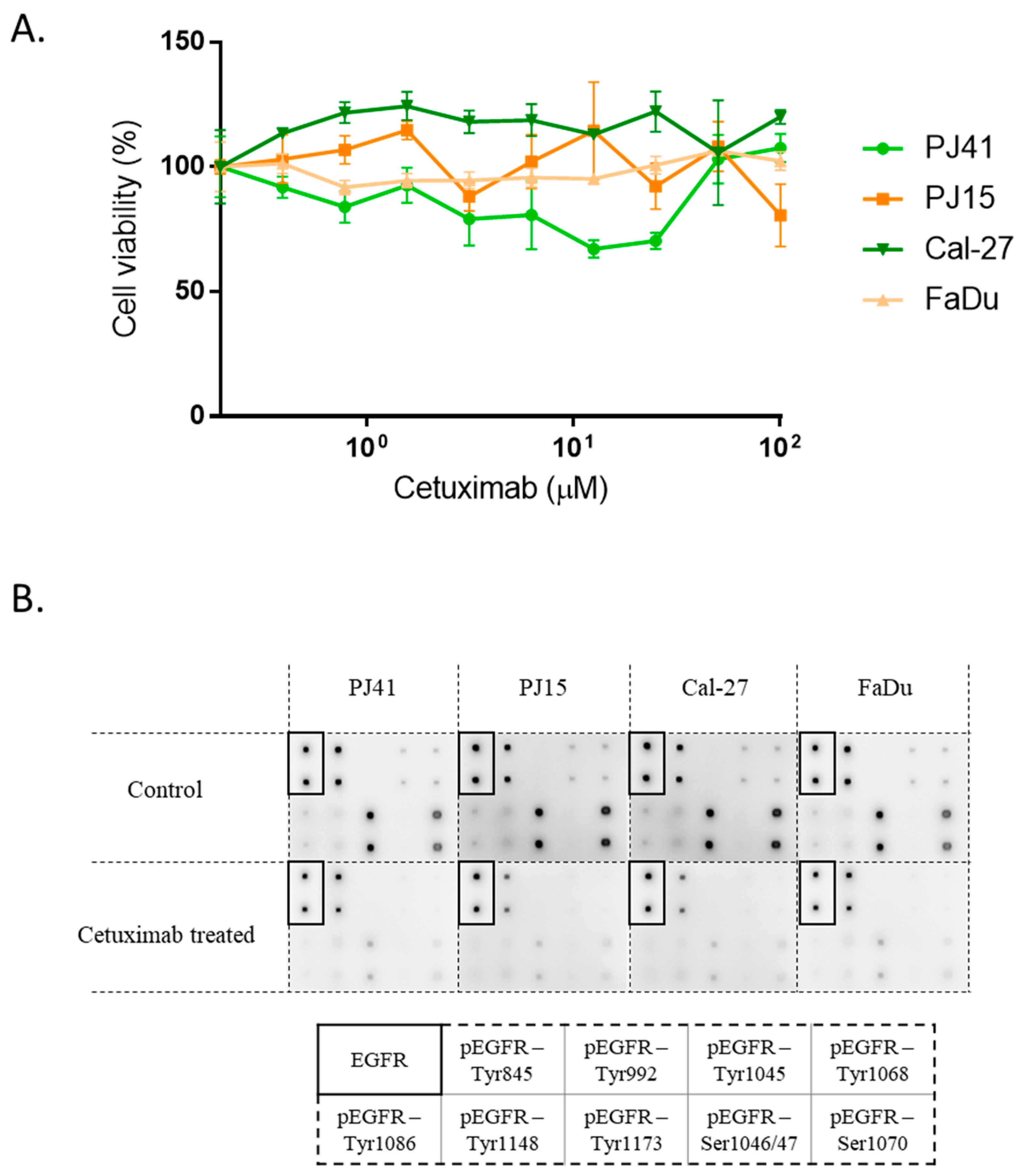
2.6. EGFR Phosphorylation Assay
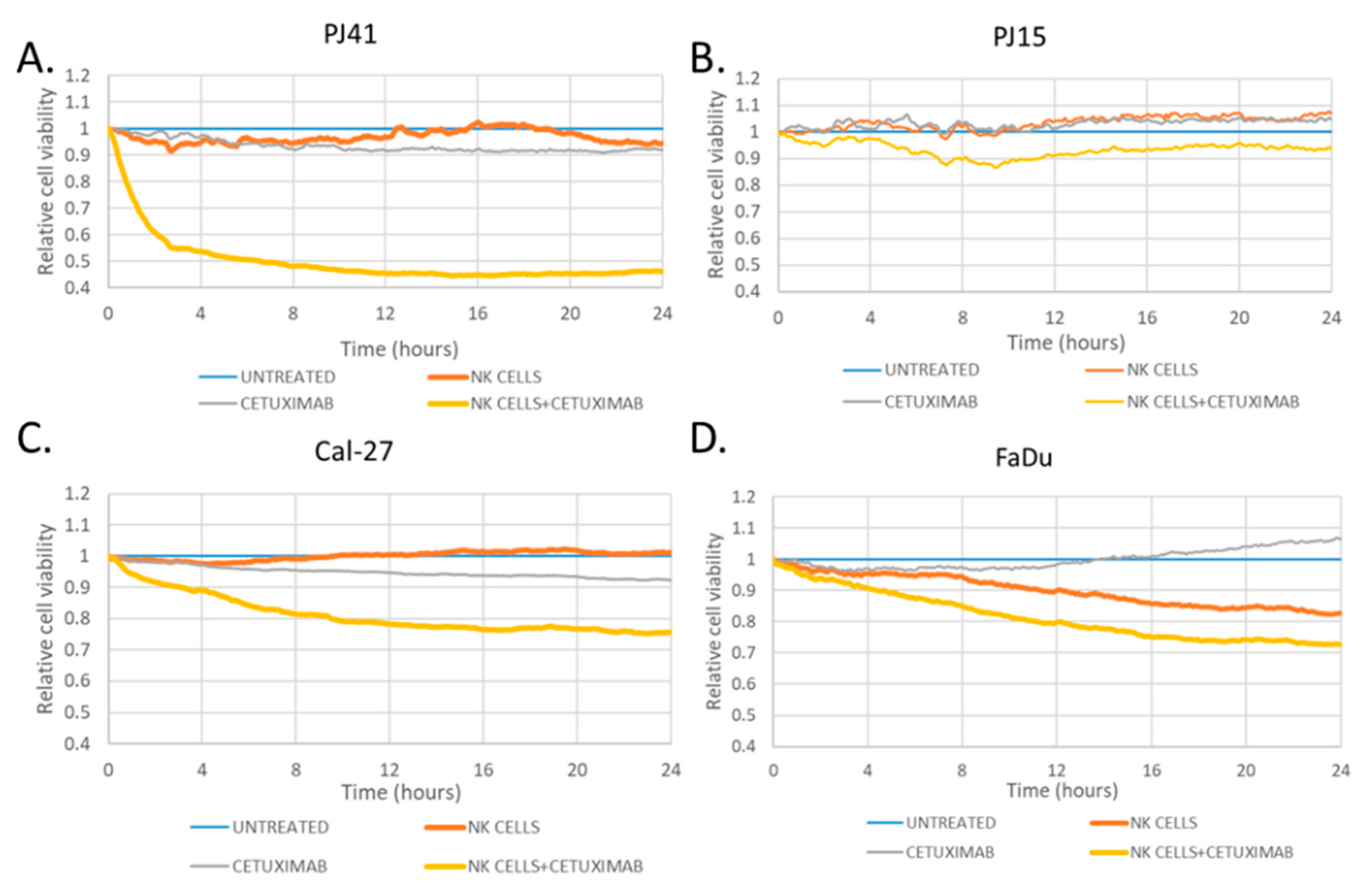
2.7. Antibody-Dependent Cellular Cytotoxicity Assay
2.8. Animal Experiments
2.9. Clinical Data Analysis
2.10. Immunohistochemistry
2.11. Statistical Analysis
3. Results
3.1. EGFRvIII and R521K Genotypes
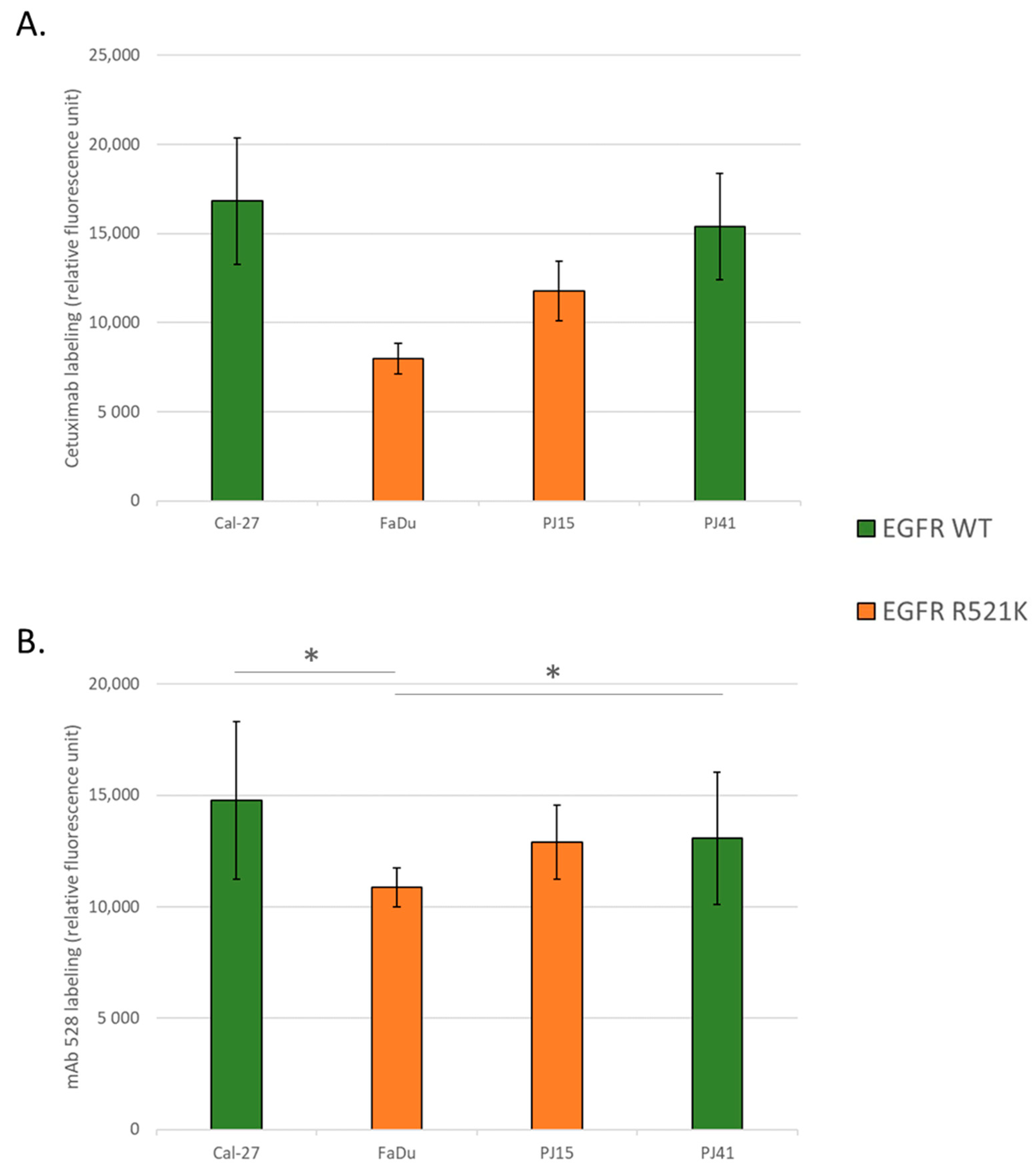
3.2. EGFR Expression Variances among the Used HNSCC Cell Lines
3.3. Cetuximab Sensitivity of HNSCC Cells in Vitro
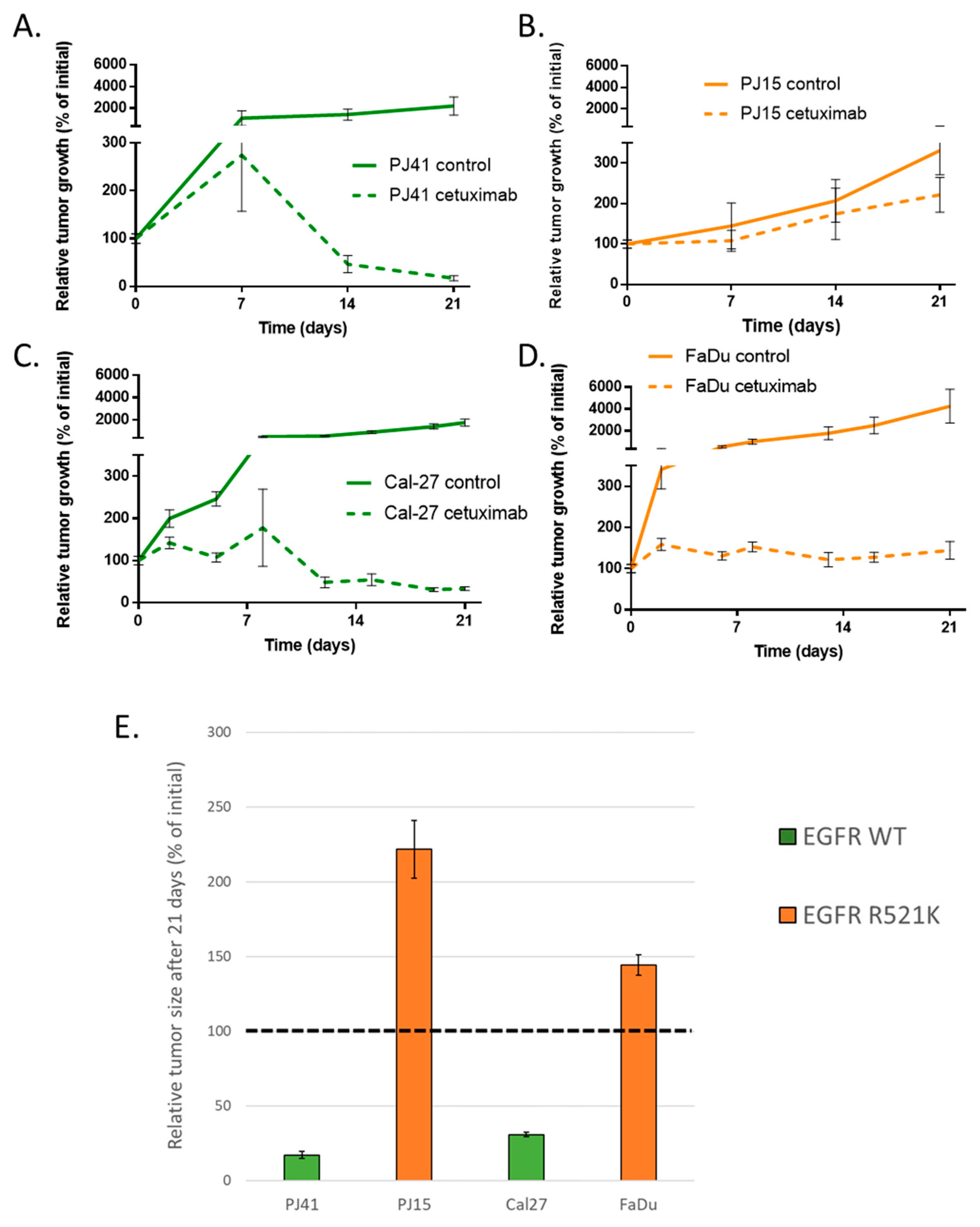
3.4. Human HNSCC Cell Line-Derived Xenograft Growth
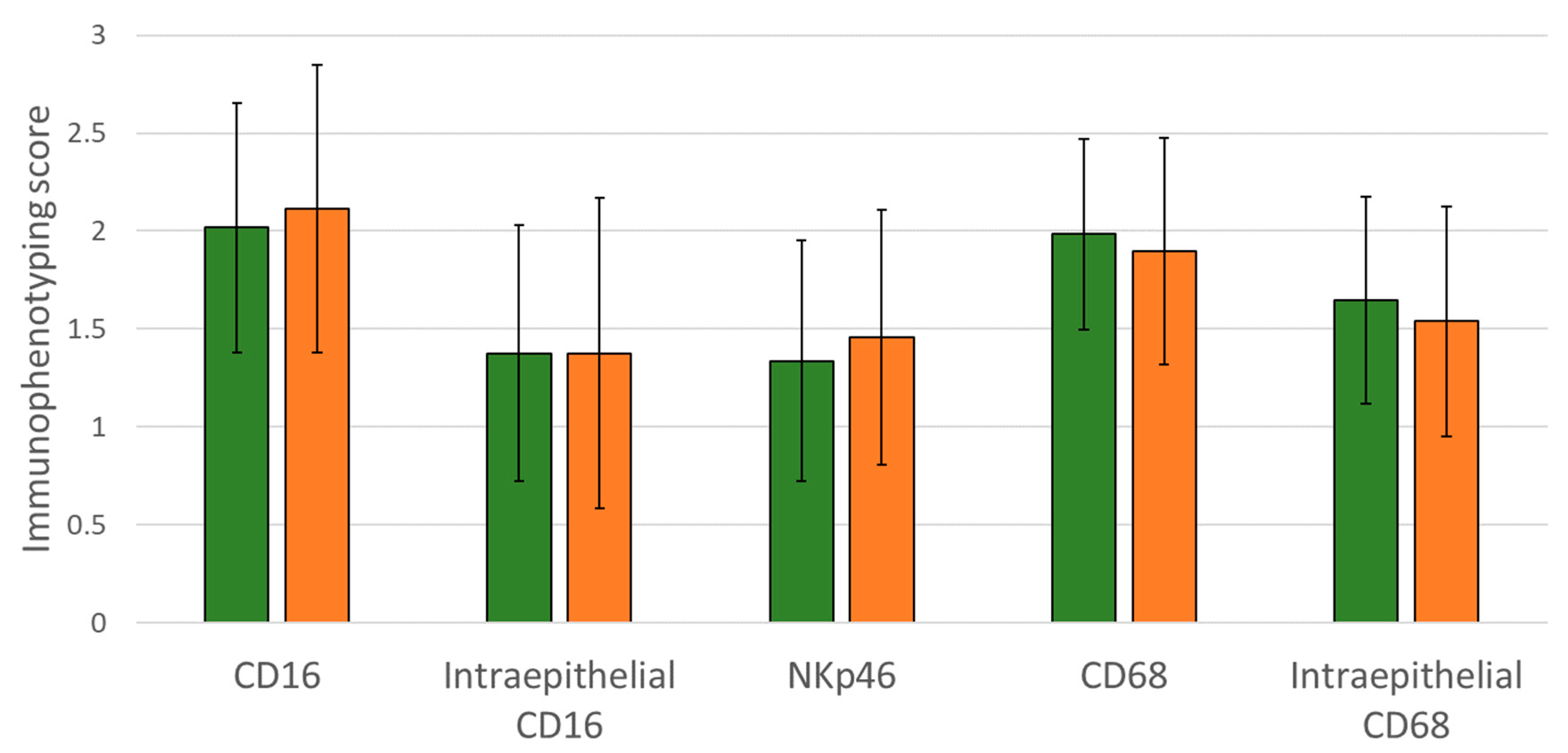
3.5. Immunophenotype in Wild-Type and EGFR R521 Tumor Samples and Its Correlation with Clinical Cetuximab Efficacy
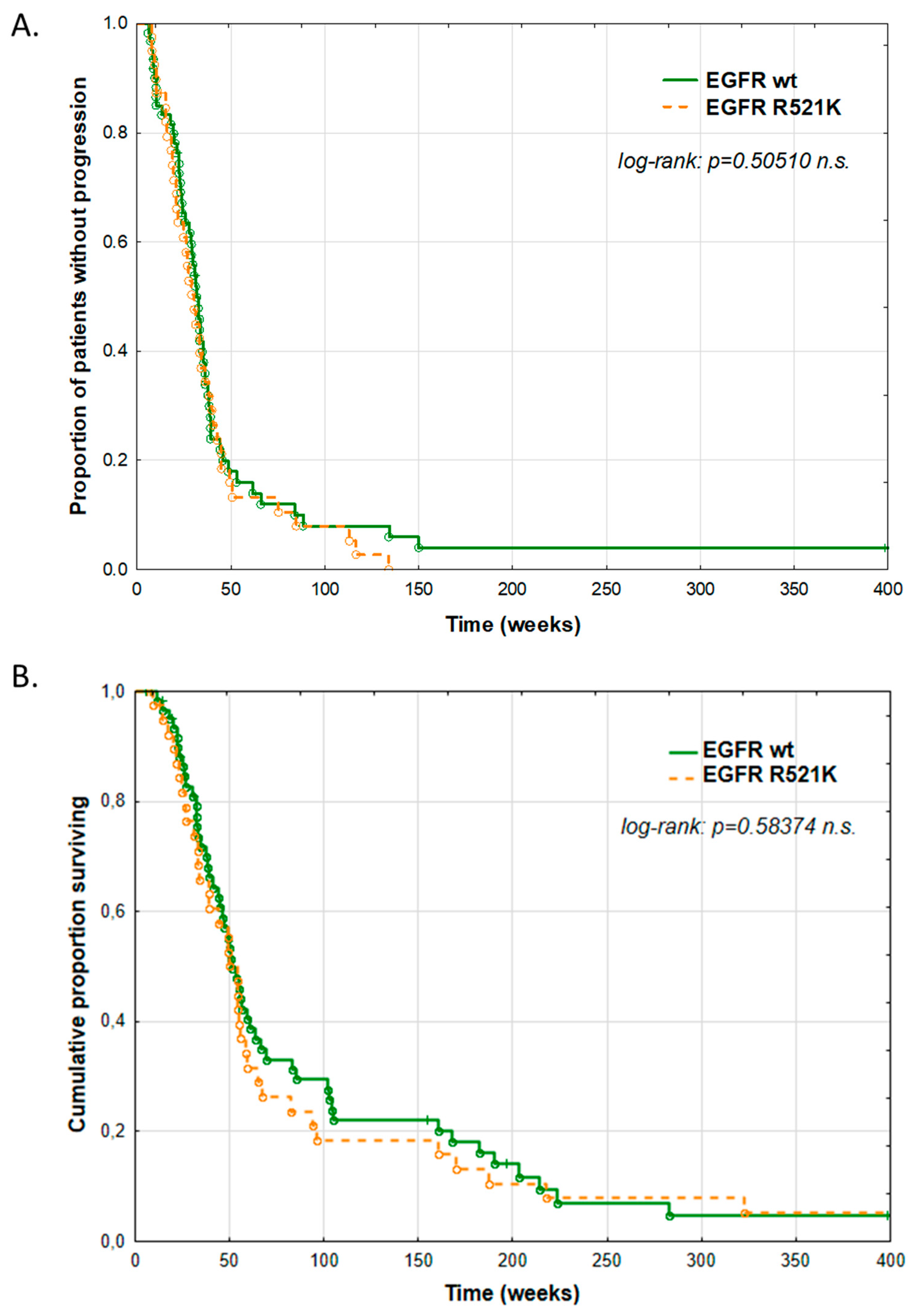
3.6. Clinical Impact of EGFR R521K Status on Cetuximab Therapy Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | epidermal growth factor receptor |
| ADCC | antibody-dependent cellular cytotoxicity |
| NK | natural killer |
| 5-FU | 5-fluorouracil |
| HNSCC | head and neck squamous cell carcinoma |
| PD-L1 | programmed death-ligand 1 |
| EPF | Erbitux-platinum-5-FU combination therapy |
| EMT | epithelial–mesenchymal transition |
| PFS | progression-free survival |
| OS | overall survival |
| HPV | human papillomavirus |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Oosting, S.F.; Haddad, R.I. Best Practice in Systemic Therapy for Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Mehra, R. Pembrolizumab for the Treatment of Head and Neck Squamous Cell Cancer. Expert Opin. Biol. Ther. 2019, 19, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G.J.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. Available online: https://www.nejm.org/doi/10.1056/nejmoa1602252 (accessed on 22 January 2021).
- Keytruda. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/keytruda (accessed on 7 September 2020).
- Opdivo. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/opdivo (accessed on 7 September 2020).
- Geoffrois, L.; Martin, L.; De Raucourt, D.; Sun, X.S.; Tao, Y.; Maingon, P.; Buffet, J.; Pointreau, Y.; Sire, C.; Tuchais, C.; et al. Induction Chemotherapy Followed by Cetuximab Radiotherapy Is Not Superior to Concurrent Chemoradiotherapy for Head and Neck Carcinomas: Results of the GORTEC 2007-02 Phase III Randomized Trial. J. Clin. Oncol. 2018, 36, 3077–3083. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.-R.; Cupissol, D.; et al. Platinum-Based Chemotherapy plus Cetuximab in Head and Neck Cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical Update on Head and Neck Cancer: Molecular Biology and Ongoing Challenges. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Aboud-Pirak, E.; Hurwitz, E.; Pirak, M.E.; Bellot, F.; Schlessinger, J.; Sela, M. Efficacy of Antibodies to Epidermal Growth Factor Receptor against KB Carcinoma in Vitro and in Nude Mice. J. Natl. Cancer Inst. 1988, 80, 1605–1611. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Mazzarella, L.; Guida, A.; Curigliano, G. Cetuximab for Treating Non-Small Cell Lung Cancer. Expert Opin. Biol. Ther. 2018, 18, 483–493. [Google Scholar] [CrossRef]
- Wiechec, E.; Hansson, K.T.; Alexandersson, L.; Jönsson, J.-I.; Roberg, K. Hypoxia Mediates Differential Response to Anti-EGFR Therapy in HNSCC Cells. Int. J. Mol. Sci. 2017, 18, 943. [Google Scholar] [CrossRef]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.-I.; Calderwood, S.K.; Okamoto, K.; Kozaki, K.-I. Carcinogenic Epithelial-Mesenchymal Transition Initiated by Oral Cancer Exosomes Is Inhibited by Anti-EGFR Antibody Cetuximab. Oral Oncol. 2018, 86, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Rebucci, M.; Peixoto, P.; Dewitte, A.; Wattez, N.; De Nuncques, M.-A.; Rezvoy, N.; Vautravers-Dewas, C.; Buisine, M.-P.; Guerin, E.; Peyrat, J.-P.; et al. Mechanisms Underlying Resistance to Cetuximab in the HNSCC Cell Line: Role of AKT Inhibition in Bypassing This Resistance. Int. J. Oncol. 2011, 38, 189–200. [Google Scholar] [PubMed]
- Swick, A.D.; Prabakaran, P.J.; Miller, M.C.; Javaid, A.M.; Fisher, M.M.; Sampene, E.; Ong, I.M.; Hu, R.; Iida, M.; Nickel, K.P.; et al. Cotargeting MTORC and EGFR Signaling as a Therapeutic Strategy in HNSCC. Mol. Cancer Ther. 2017, 16, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Eze, N.; Lee, J.-W.; Yang, D.-H.; Zhu, F.; Neumeister, V.; Sandoval-Schaefer, T.; Mehra, R.; Ridge, J.A.; Forastiere, A.; Chung, C.H.; et al. PTEN Loss Is Associated with Resistance to Cetuximab in Patients with Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2019, 91, 69–78. [Google Scholar] [CrossRef]
- Trivedi, S.; Concha-Benavente, F.; Srivastava, R.M.; Jie, H.B.; Gibson, S.P.; Schmitt, N.C.; Ferris, R.L. Immune Biomarkers of Anti-EGFR Monoclonal Antibody Therapy. Ann. Oncol. 2015, 26, 40–47. [Google Scholar] [CrossRef]
- Bagchi, A.; Haidar, J.N.; Eastman, S.W.; Vieth, M.; Topper, M.; Iacolina, M.D.; Walker, J.M.; Forest, A.; Shen, Y.; Novosiadly, R.D.; et al. Molecular Basis for Necitumumab Inhibition of EGFR Variants Associated with Acquired Cetuximab Resistance. Mol. Cancer Ther. 2018, 17, 521–531. [Google Scholar] [CrossRef]
- Sok, J.C.; Coppelli, F.M.; Thomas, S.M.; Lango, M.N.; Xi, S.; Hunt, J.L.; Freilino, M.L.; Graner, M.W.; Wikstrand, C.J.; Bigner, D.D.; et al. Mutant Epidermal Growth Factor Receptor (EGFRvIII) Contributes to Head and Neck Cancer Growth and Resistance to EGFR Targeting. Clin. Cancer Res. 2006, 12, 5064–5073. [Google Scholar] [CrossRef]
- Su, N.-W.; Leu, Y.-S.; Lee, J.-C.; Liu, C.-J.; Cheng, C.-Y.; Lin, J.-S.; Chen, Y.-J.; Chen, C.-K.; Fang, I.-C.; Hsieh, R.-K.; et al. EGF and EGFR Genetic Polymorphisms Predict Prognosis in Locally Advanced Pharyngolaryngeal Squamous Cell Carcinoma Patients Receiving Postoperative Concurrent Chemoradiotherapy. OncoTargets Ther. 2014, 7, 2197–2204. [Google Scholar] [CrossRef][Green Version]
- Braig, F.; Kriegs, M.; Voigtlaender, M.; Habel, B.; Grob, T.; Biskup, K.; Blanchard, V.; Sack, M.; Thalhammer, A.; Ben Batalla, I.; et al. Cetuximab Resistance in Head and Neck Cancer Is Mediated by EGFR-K521 Polymorphism. Cancer Res. 2017, 77, 1188–1199. [Google Scholar] [CrossRef]
- Stoehlmacher-Williams, J.; Obermann, L.; Ehninger, G.; Goekkurt, E. Polymorphisms of the Epidermal Growth Factor Receptor (EGFR) and Survival in Patients with Advanced Cancer of the Head and Neck (HNSCC). Anticancer Res. 2012, 32, 421–425. [Google Scholar]
- Klinghammer, K.; Knödler, M.; Schmittel, A.; Budach, V.; Keilholz, U.; Tinhofer, I. Association of Epidermal Growth Factor Receptor Polymorphism, Skin Toxicity, and Outcome in Patients with Squamous Cell Carcinoma of the Head and Neck Receiving Cetuximab-Docetaxel Treatment. Clin. Cancer Res. 2010, 16, 304–310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Szöőr, Á.; Tóth, G.; Zsebik, B.; Szabó, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab Derived HER2-Specific CARs for the Treatment of Trastuzumab-Resistant Breast Cancer: CAR T Cells Penetrate and Eradicate Tumors That Are Not Accessible to Antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Füredi, A.; Tóth, S.; Szebényi, K.; Pape, V.F.S.; Türk, D.; Kucsma, N.; Cervenak, L.; Tóvári, J.; Szakács, G. Identification and Validation of Compounds Selectively Killing Resistant Cancer: Delineating Cell Line-Specific Effects from P-Glycoprotein-Induced Toxicity. Mol. Cancer Ther. 2017, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Makabe, K.; Yokoyama, T.; Uehara, S.; Uchikubo-Kamo, T.; Shirouzu, M.; Kimura, K.; Tsumoto, K.; Asano, R.; Tanaka, Y.; Kumagai, I. Anti-EGFR Antibody 528 Binds to Domain III of EGFR at a Site Shifted from the Cetuximab Epitope. Sci. Rep. 2021, 11, 5790. [Google Scholar] [CrossRef]
- Tóth, G.; Szöőr, Á.; Simon, L.; Yarden, Y.; Szöllősi, J.; Vereb, G. The Combination of Trastuzumab and Pertuzumab Administered at Approved Doses May Delay Development of Trastuzumab Resistance by Additively Enhancing Antibody-Dependent Cell-Mediated Cytotoxicity. MAbs 2016, 8, 1361–1370. [Google Scholar] [CrossRef]
- Tóth, G.; Szöllősi, J.; Vereb, G. Quantitating ADCC against Adherent Cells: Impedance-Based Detection Is Superior to Release, Membrane Permeability, or Caspase Activation Assays in Resolving Antibody Dose Response. Cytom. A 2017, 91, 1021–1029. [Google Scholar] [CrossRef]
- Ladányi, A.; Kapuvári, B.; Papp, E.; Tóth, E.; Lövey, J.; Horváth, K.; Gődény, M.; Remenár, É. Local Immune Parameters as Potential Predictive Markers in Head and Neck Squamous Cell Carcinoma Patients Receiving Induction Chemotherapy and Cetuximab. Head Neck 2019, 41, 1237–1245. [Google Scholar] [CrossRef]
- Melchers, L.J.; Clausen, M.J.A.M.; Mastik, M.F.; Slagter-Menkema, L.; Langendijk, J.A.; van der Laan, B.F.A.M.; Van der Wal, J.E.; van der Vegt, B.; Roodenburg, J.L.N.; Schuuring, E. Head and Neck Squamous Cell Carcinomas Do Not Express EGFRvIII. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 454–462. [Google Scholar] [CrossRef]
- Khattri, A.; Zuo, Z.; Brägelmann, J.; Keck, M.K.; El Dinali, M.; Brown, C.D.; Stricker, T.; Munagala, A.; Cohen, E.E.W.; Lingen, M.W.; et al. Rare Occurrence of EGFRvIII Deletion in Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2015, 51, 53–58. [Google Scholar] [CrossRef]
- Gonzales, C.B.; De La Chapa, J.J.; Saikumar, P.; Singha, P.K.; Dybdal-Hargreaves, N.F.; Chavez, J.; Horning, A.M.; Parra, J.; Kirma, N.B. Co-Targeting ALK and EGFR Parallel Signaling in Oral Squamous Cell Carcinoma. Oral Oncol. 2016, 59, 12–19. [Google Scholar] [CrossRef]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and function-al characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Anikeeva, N.; Steblyanko, M.; Fayngerts, S.; Kopylova, N.; Marshall, D.J.; Powers, G.D.; Sato, T.; Campbell, K.S.; Sykulev, Y. Integrin receptors on tumor cells facilitate NK cell-mediated antibody-dependent cytotoxicity. Eur. J. Immunol. 2014, 44, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Binyamin, L.; Alpaugh, R.K.; Hughes, T.L.; Lutz, C.T.; Campbell, K.S.; Weiner, L.M. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J. Immunol. 2008, 180, 6392–6401. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Total 103 | EGFR wt 62 (60%) | EGFR R521K 41 (40%) | p |
|---|---|---|---|---|
| Men (n, %) Women (n, %) | 81 (79%) 22 (21%) | 54 (87%) 8 (13%) | 27 (66%) 14 (34%) | p < 0.05 |
| Age (median, years) | 59.5 | 60 | 58 | No correlation |
| Overall response rate (n, %) | 59 (57%) | 39 (63%) | 20 (49%) | No correlation |
| Disease control rate (n, %) | 89 (86%) | 52 (84%) | 37 (90%) | No correlation |
| Progression-free survival (median, weeks) | 29 | 29 | 28 | No correlation |
| Overall survival (median, weeks) | 50 | 48 | 49 | No correlation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cserepes, M.; Nelhűbel, G.A.; Meilinger-Dobra, M.; Herczeg, A.; Türk, D.; Hegedűs, Z.; Svajda, L.; Rásó, E.; Ladányi, A.; Csikó, K.G.; et al. EGFR R521K Polymorphism Is Not a Major Determinant of Clinical Cetuximab Resistance in Head and Neck Cancer. Cancers 2022, 14, 2407. https://doi.org/10.3390/cancers14102407
Cserepes M, Nelhűbel GA, Meilinger-Dobra M, Herczeg A, Türk D, Hegedűs Z, Svajda L, Rásó E, Ladányi A, Csikó KG, et al. EGFR R521K Polymorphism Is Not a Major Determinant of Clinical Cetuximab Resistance in Head and Neck Cancer. Cancers. 2022; 14(10):2407. https://doi.org/10.3390/cancers14102407
Chicago/Turabian StyleCserepes, Mihály, Györgyi A. Nelhűbel, Mónika Meilinger-Dobra, Adrienn Herczeg, Dóra Türk, Zita Hegedűs, Laura Svajda, Erzsébet Rásó, Andrea Ladányi, Kristóf György Csikó, and et al. 2022. "EGFR R521K Polymorphism Is Not a Major Determinant of Clinical Cetuximab Resistance in Head and Neck Cancer" Cancers 14, no. 10: 2407. https://doi.org/10.3390/cancers14102407
APA StyleCserepes, M., Nelhűbel, G. A., Meilinger-Dobra, M., Herczeg, A., Türk, D., Hegedűs, Z., Svajda, L., Rásó, E., Ladányi, A., Csikó, K. G., Kenessey, I., Szöőr, Á., Vereb, G., Remenár, É., & Tóvári, J. (2022). EGFR R521K Polymorphism Is Not a Major Determinant of Clinical Cetuximab Resistance in Head and Neck Cancer. Cancers, 14(10), 2407. https://doi.org/10.3390/cancers14102407