
Preventing and Overcoming Resistance to PARP Inhibitors: A Focus on the Clinical Landscape


Abstract
:Simple Summary
Abstract
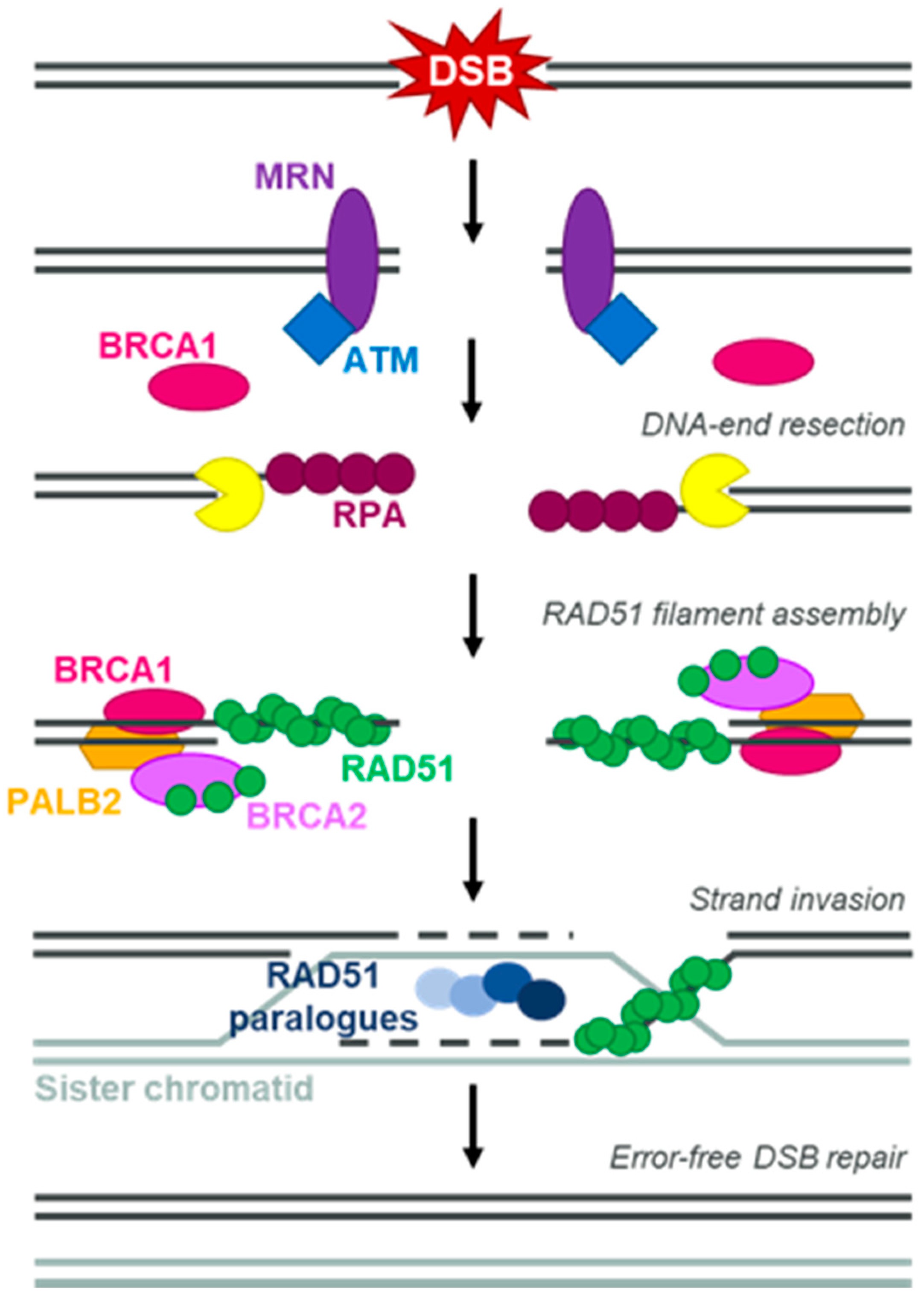
1. Introduction
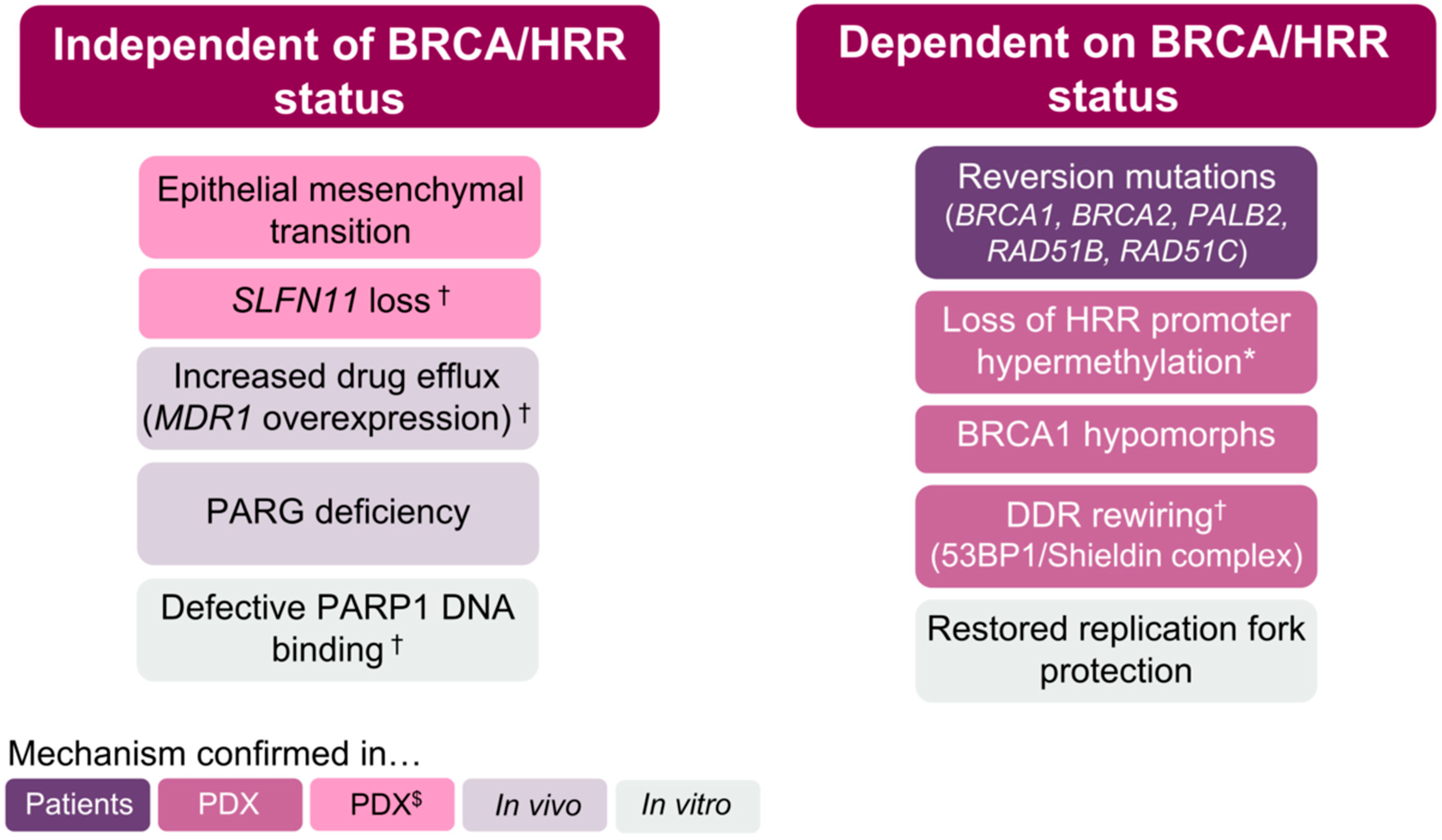
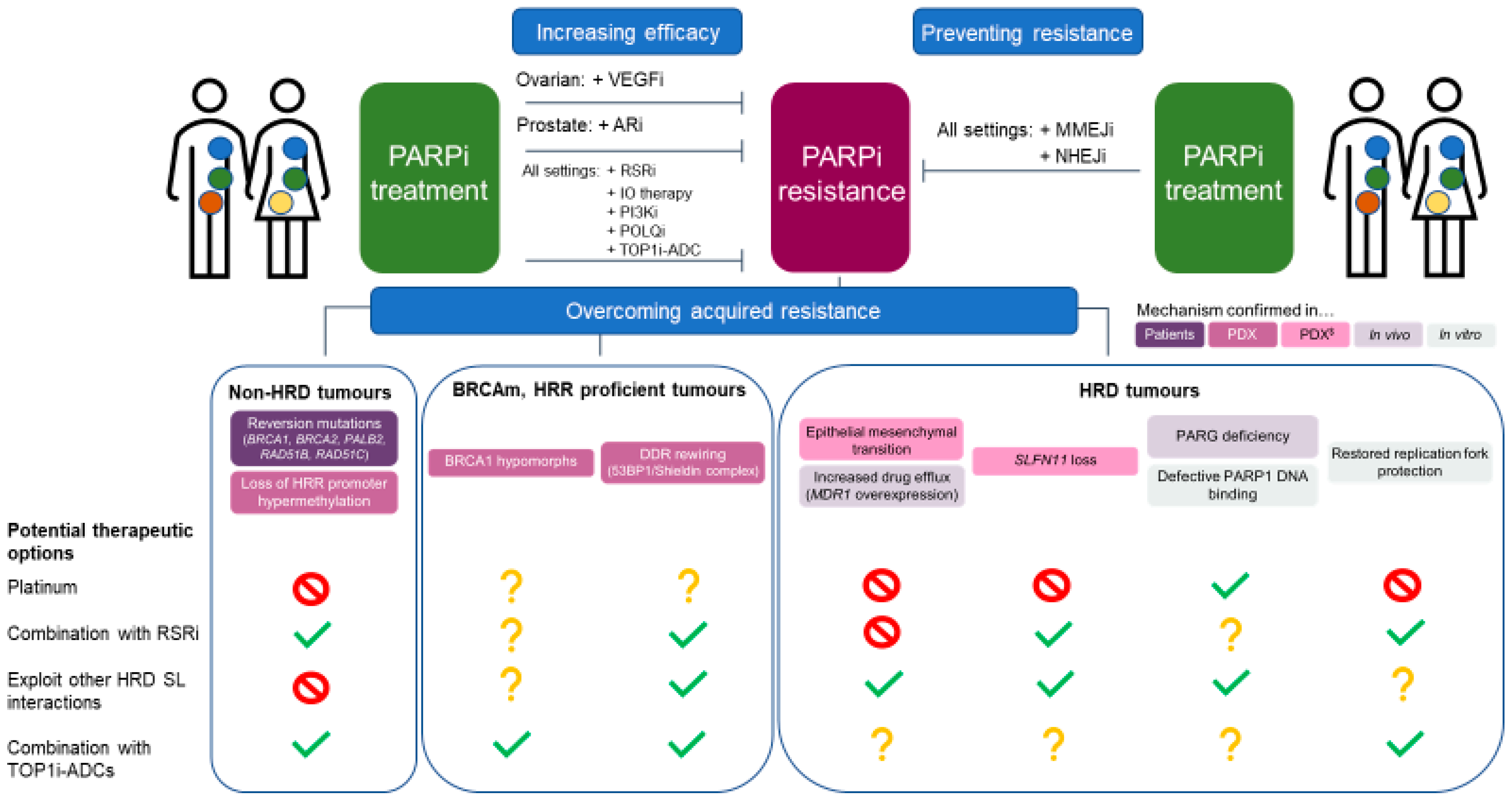
2. BRCA/HRR-Independent Mechanisms of PARPi Resistance
2.1. Epithelial–Mesenchymal Transition
2.2. SLFN11 Loss
2.3. P-Glycoprotein Overexpression
2.4. PARG Loss
2.5. PARP1 Mutations
3. BRCA/HRR-Dependent Mechanisms of PARPi Resistance
3.1. Dynamic Biomarkers of HRD
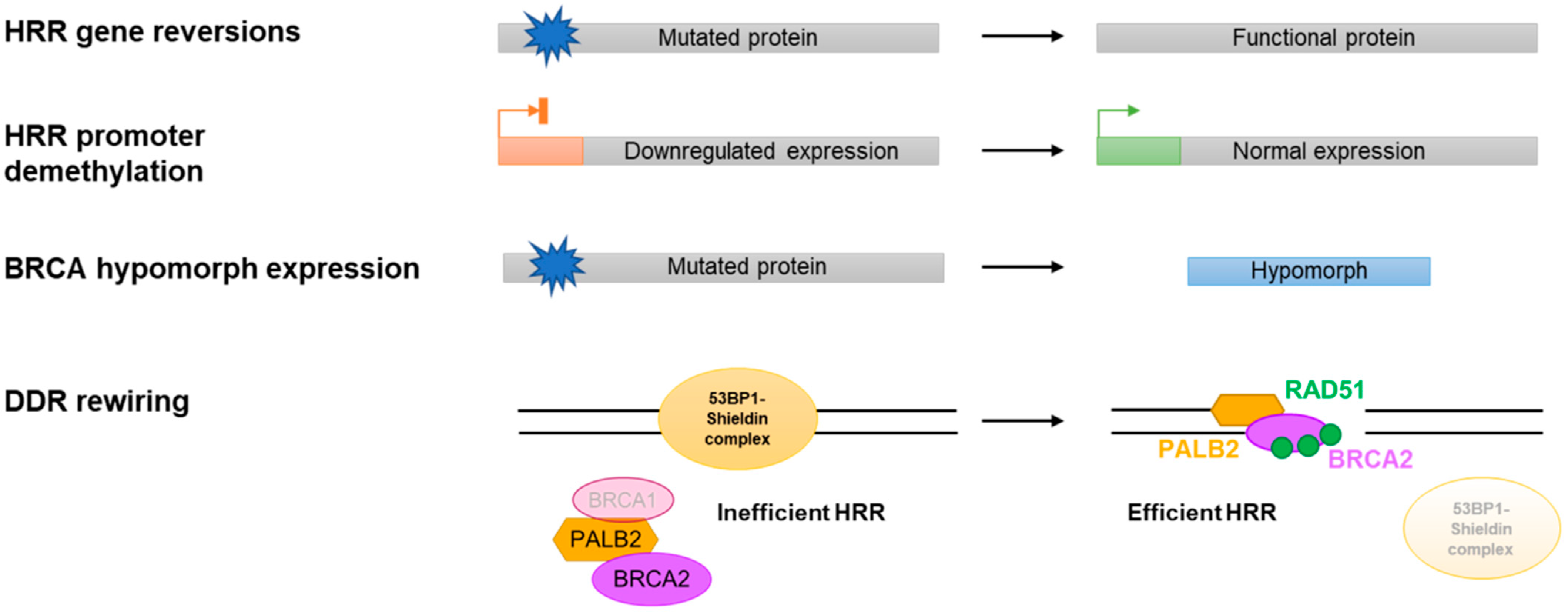
3.2. Reversion Mutations
3.3. Restored BRCA/HRR Gene Expression
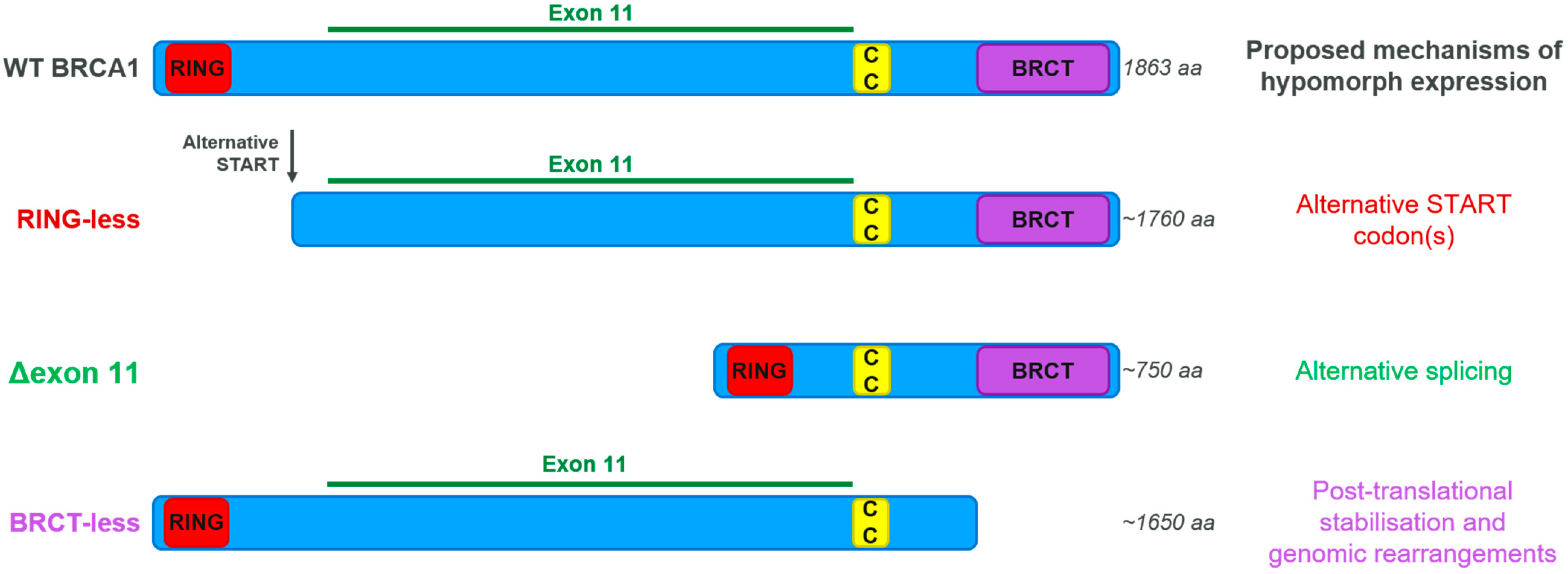
3.4. BRCA Hypomorphic Proteins
3.5. DDR Rewiring
3.6. Restoration of Replication Fork Protection
4. Preventing and Tackling PARPi Resistance
4.1. Non-HRD Tumours
4.2. BRCAm, HRR-Proficient Tumours
4.3. HRD Tumours
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, D.J.; Gaudet, M.M.; Pal, P.; Kirchhoff, T.; Balistreri, L.; Vora, K.; Bhatia, J.; Stadler, Z.; Fine, S.W.; Reuter, V.; et al. Germline BRCA Mutations Denote a Clinicopathologic Subset of Prostate Cancer. Clin. Cancer Res. 2010, 16, 2115–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, Z.K.; Salo-Mullen, E.; Patil, S.M.; Pietanza, M.C.; Vijai, J.; Saloustros, E.; Hansen, N.A.L.; Kauff, N.D.; Kurtz, R.C.; Kelsen, D.P.; et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer 2012, 118, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; van Overeem Hansen, T.; Sorensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Chen, C.C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef]
- Wang, X.; Liu, L.; Montagna, C.; Ried, T.; Deng, C.X. Haploinsufficiency of Parp1 accelerates Brca1-associated centrosome amplification, telomere shortening, genetic instability, apoptosis, and embryonic lethality. Cell Death Differ. 2007, 14, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.-y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Białkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordonez, L.D.; Hay, T.; McEwen, R.; Polanska, U.M.; Hughes, A.; Delpuech, O.; Cadogan, E.; Powell, S.; Dry, J.; Tornillo, G.; et al. Rapid activation of epithelial-mesenchymal transition drives PARP inhibitor resistance in Brca2-mutant mammary tumours. Oncotarget 2019, 10, 2586–2606. [Google Scholar] [CrossRef] [Green Version]
- Jaspers, J.E.; Sol, W.; Kersbergen, A.; Schlicker, A.; Guyader, C.; Xu, G.; Wessels, L.; Borst, P.; Jonkers, J.; Rottenberg, S. BRCA2-Deficient Sarcomatoid Mammary Tumors Exhibit Multidrug Resistance. Cancer Res. 2015, 75, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Allison Stewart, C.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoppoli, G.; Regairaz, M.; Leo, E.; Reinhold, W.C.; Varma, S.; Ballestrero, A.; Doroshow, J.H.; Pommier, Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. USA 2012, 109, 201205943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Tang, S.-W.; Leo, E.; Baechler, S.A.; Redon, C.E.; Zhang, H.; Al Abo, M.; Rajapakse, V.N.; Nakamura, E.; Jenkins, L.M.M.; et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 2018, 69, 371–384.e376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, C.; Armenia, J.; Jones, G.N.; Tobalina, L.; Sale, M.J.; Petreus, T.; Baird, T.; Serra, V.; Wang, A.T.; Lau, A.; et al. SLFN11 informs on standard of care and novel treatments in a wide range of cancer models. Br. J. Cancer 2021, 124, 951–962. [Google Scholar] [CrossRef]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.P.; Madariaga, A.; Dhani, N.C.; Bowering, V.; White, J.; Accardi, S.; Tan, Q.; et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin. Cancer Res. 2020, 26, 4206–4215. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M.; Ellenberger, T. The rise and fall of poly(ADP-ribose): An enzymatic perspective. DNA Repair 2015, 32, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e1012. [Google Scholar] [CrossRef] [Green Version]
- Pettitt, S.J.; Rehman, F.L.; Bajrami, I.; Brough, R.; Wallberg, F.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Chen, L.; Campbell, J.; et al. A Genetic Screen Using the PiggyBac Transposon in Haploid Cells Identifies Parp1 as a Mediator of Olaparib Toxicity. PLoS ONE 2013, 8, e61520. [Google Scholar] [CrossRef] [Green Version]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Herzog, M.; Puddu, F.; Coates, J.; Geisler, N.; Forment, J.V.; Jackson, S.P. Detection of functional protein domains by unbiased genome-wide forward genetic screening. Sci. Rep. 2018, 8, 6161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- West, S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003, 4, 435–445. [Google Scholar] [CrossRef]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef]
- Carreira, S.; Porta, N.; Arce-Gallego, S.; Seed, G.; Llop-Guevara, A.; Bianchini, D.; Rescigno, P.; Paschalis, A.; Bertan, C.; Baker, C.; et al. Biomarkers Associating with PARP Inhibitor Benefit in Prostate Cancer in the TOPARP-B Trial. Cancer Discov. 2021, 11, 2812–2827. [Google Scholar] [CrossRef] [PubMed]
- Guffanti, F.; Alvisi, M.F.; Anastasia, A.; Ricci, F.; Chiappa, M.; Llop-Guevara, A.; Serra, V.; Fruscio, R.; Degasperi, A.; Nik-Zainal, S.; et al. Basal expression of RAD51 foci predicts olaparib response in patient-derived ovarian cancer xenografts. Br. J. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.E.; Buendia Buendia, J.; Wander, S.; Helvie, K.; Lloyd, M.R.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 Mutations in BRCA1-Mutated Ovarian Carcinomas with Platinum Resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Frankum, J.R.; Punta, M.; Lise, S.; Alexander, J.; Chen, Y.; Yap, T.A.; Haider, S.; Tutt, A.N.J.; Lord, C.J. Clinical BRCA1/2 Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov. 2020, 10, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef]
- Swisher, E.M.; Kristeleit, R.S.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Burris, H.A.; Aghajanian, C.; O’Malley, D.M.; et al. Characterization of patients with long-term responses to rucaparib treatment in recurrent ovarian cancer. Gynecol. Oncol. 2021, 163, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.-Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, A.; Kim, K.; Jeong, K.; Choi, S.; Kim, S.; Suh, K.J.; Lee, K.-H.; Kim, S.; Im, S.-A. Homologous repair deficiency score for identifying breast cancers with defective DNA damage response. Sci. Rep. 2020, 10, 12506. [Google Scholar] [CrossRef] [PubMed]
- ter Brugge, P.; Kristel, P.; van der Burg, E.; Boon, U.; de Maaker, M.; Lips, E.; Mulder, L.; de Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. JNCI J. Natl. Cancer Inst. 2016, 108, djw148. [Google Scholar] [CrossRef]
- Hurley, R.M.; McGehee, C.D.; Nesic, K.; Correia, C.; Weiskittel, T.M.; Kelly, R.L.; Venkatachalam, A.; Hou, X.; Pathoulas, N.M.; Meng, X.W.; et al. Characterization of a RAD51C-silenced high-grade serous ovarian cancer model during development of PARP inhibitor resistance. NAR Cancer 2021, 3, zcab028. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bernhardy, A.J.; Nacson, J.; Krais, J.J.; Tan, Y.-F.; Nicolas, E.; Radke, M.R.; Handorf, E.; Llop-Guevara, A.; Balmaña, J.; et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun. 2019, 10, 5661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Krais, J.J.; Bernhardy, A.J.; Nicolas, E.; Cai, K.Q.; Harrell, M.I.; Kim, H.H.; George, E.; Swisher, E.M.; Simpkins, F.; et al. RING domain–deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Investig. 2016, 126, 3145–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drost, R.; Dhillon, K.K.; van der Gulden, H.; van der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918. [Google Scholar] [CrossRef]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.-C.; Choi, Y.-E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [Green Version]
- Park, P.H.; Yamamoto, T.M.; Li, H.; Alcivar, A.L.; Xia, B.; Wang, Y.; Bernhardy, A.J.; Turner, K.M.; Kossenkov, A.V.; Watson, Z.L.; et al. Amplification of the Mutation-Carrying BRCA2 Allele Promotes RAD51 Loading and PARP Inhibitor Resistance in the Absence of Reversion Mutations. Mol. Cancer Ther. 2020, 19, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [Green Version]
- Zong, D.; Adam, S.; Wang, Y.; Sasanuma, H.; Callén, E.; Murga, M.; Day, A.; Kruhlak, M.J.; Wong, N.; Munro, M.; et al. BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol. Cell 2019, 73, 1267–1281.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callen, E.; Zong, D.; Wu, W.; Wong, N.; Stanlie, A.; Ishikawa, M.; Pavani, R.; Dumitrache, L.C.; Byrum, A.K.; Mendez-Dorantes, C.; et al. 53BP1 Enforces Distinct Pre- and Post-resection Blocks on Homologous Recombination. Molecular Cell 2020, 77, 26–38.e27. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11, 819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.J.; et al. Isomerization of BRCA1–BARD1 promotes replication fork protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef]
- Clarke, N.W.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Ye, D.; Mateo, J.; Goessl, C.D.; Kang, J.; Liu, S.; Saad, F. PROPEL: A randomized, phase III trial evaluating the efficacy and safety of olaparib combined with abiraterone as first-line therapy in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2019, 37, TPS340. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.-R.; Hurst, K.E.; de Oliveira Taveira, M.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer 2020, 2, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmañà, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-Related Breast Cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [Green Version]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual Roles of PARP-1 Promote Cancer Growth and Progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced BRCAness and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal 2017, 10, eaam7479. [Google Scholar] [CrossRef] [Green Version]
- Saad, F.; Chi, K.N.; Shore, N.D.; Graff, J.N.; Posadas, E.M.; Lattouf, J.-B.; Espina, B.M.; Zhu, E.; Yu, A.; Hazra, A.; et al. Niraparib with androgen receptor-axis-targeted therapy in patients with metastatic castration-resistant prostate cancer: Safety and pharmacokinetic results from a phase 1b study (BEDIVERE). Cancer Chemother. Pharmacol. 2021, 88, 25–37. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I trial of the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2 and non-BRCA1/2 mutant cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Chowdhuri, S.P.; Das, B.B. Top1-PARP1 association and beyond: From DNA topology to break repair. NAR Cancer 2021, 3, zcab003. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Selle, F.; Scambia, G.; Asselain, B.; Marmé, F.; Lindemann, K.; Colombo, N.; Madry, R.; Glasspool, R.M.; Dubot, C.; et al. Maintenance olaparib rechallenge in patients (pts) with ovarian carcinoma (OC) previously treated with a PARP inhibitor (PARPi): Phase IIIb OReO/ENGOT Ov-38 trial. Ann. Oncol. 2021, 32, S1283–S1346. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e857. [Google Scholar] [CrossRef]
- Shah, P.D.; Zarrin, H.; Wethington, S.; Latif, N.; Martin, L.; Rodriguez, D.; Elkins, K.; Giuntoli, R.; Burger, R.; Tanyi, J.; et al. Abstract A72: Combination ATR and PARP inhibitor (CAPRI) for recurrent, platinum-resistant ovarian cancer. Clin. Cancer Res. 2020, 26, A72. [Google Scholar] [CrossRef]
- Westin, S.N.; Coleman, R.L.; Fellman, B.M.; Yuan, Y.; Sood, A.K.; Soliman, P.T.; Wright, A.A.; Horowitz, N.S.; Campos, S.M.; Konstantinopoulos, P.A.; et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: A randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J. Clin. Oncol. 2021, 39, 5505. [Google Scholar] [CrossRef]
- Wethington, S.L.; Shah, P.D.; Martin, L.P.; Tanyi, J.L.; Latif, N.A.; Morgan, M.A.; Torigian, D.A.; Pagan, C.; Rodriguez, D.; Domchek, S.M.; et al. Combination of PARP and ATR inhibitors (olaparib and ceralasertib) shows clinical activity in acquired PARP inhibitor-resistant recurrent ovarian cancer. J. Clin. Oncol. 2021, 39, 5516. [Google Scholar] [CrossRef]
- Harnor, S.J.; Brennan, A.; Cano, C. Targeting DNA-Dependent Protein Kinase for Cancer Therapy. Chem. Med. Chem. 2017, 12, 895–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, H.; Chiang, T.-W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Fugger, K.; Hewitt, G.; West, S.C.; Boulton, S.J. Tackling PARP inhibitor resistance. Trends Cancer 2021, 7, 1102–1118. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PARP Inhibitor | Olaparib (Lynparza)–AstraZeneca | Rucaparib (Rubraca)—Clovis Oncology | Niraparib (Zejula)—GSK | Talazoparib (Talzenna)—Pfizer | |
|---|---|---|---|---|---|
| Cancer type | Monotherapy | Combination | Monotherapy | Monotherapy | Monotherapy |
| Ovarian | Treatment setting—patients with recurrent gBRCAm advanced cancer who have been treated with 3L+ of chemotherapy Maintenance setting—patients in CR or PR to platinum-based chemotherapy (recurrent disease) and germline or somatic BRCAm advanced cancer (1L) | Maintenance setting—with bevacizumab (VEGFi) in patients in CR or PR to platinum-based chemotherapy and HRD-positive status | Treatment setting—patients with BRCAm (germline and/or somatic) cancer who have been treated with 2L+ of chemotherapies Maintenance setting—patients with recurrent cancer who are in a CR or PR to platinum-based chemotherapy | Treatment setting—patients with advanced cancer who have been treated with 3L+ of chemotherapy and whose cancer is associated with HRD Maintenance setting—patients with advanced cancer who are in a CR or PR to 1L+ platinum-based chemotherapy | N/A |
| Breast | Treatment setting—patients with gBRCAm, HER2-negative mBC who have been treated with chemotherapy | None | N/A | N/A | Treatment setting—patients with gBRCAm, HER2-negative locally advanced or mBC |
| Pancreatic | Maintenance setting—patients with gBRCAm mPA whose disease has not progressed on at least 16 weeks of 1L platinum-based chemotherapy | None | N/A | N/A | N/A |
| Prostate | Treatment setting—patients with germline or somatic HRR gene-mutated mCRPC who have progressed following prior treatment with enzalutamide or abiraterone | None | Treatment setting—patients with BRCAm (germline and/or somatic)-associated mCRPC who have been treated with androgen receptor therapy and a taxane-based chemotherapy | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prados-Carvajal, R.; Irving, E.; Lukashchuk, N.; Forment, J.V. Preventing and Overcoming Resistance to PARP Inhibitors: A Focus on the Clinical Landscape. Cancers 2022, 14, 44. https://doi.org/10.3390/cancers14010044
Prados-Carvajal R, Irving E, Lukashchuk N, Forment JV. Preventing and Overcoming Resistance to PARP Inhibitors: A Focus on the Clinical Landscape. Cancers. 2022; 14(1):44. https://doi.org/10.3390/cancers14010044
Chicago/Turabian StylePrados-Carvajal, Rosario, Elsa Irving, Natalia Lukashchuk, and Josep V. Forment. 2022. "Preventing and Overcoming Resistance to PARP Inhibitors: A Focus on the Clinical Landscape" Cancers 14, no. 1: 44. https://doi.org/10.3390/cancers14010044
APA StylePrados-Carvajal, R., Irving, E., Lukashchuk, N., & Forment, J. V. (2022). Preventing and Overcoming Resistance to PARP Inhibitors: A Focus on the Clinical Landscape. Cancers, 14(1), 44. https://doi.org/10.3390/cancers14010044