
Secretory Autophagy Forges a Therapy Resistant Microenvironment in Melanoma


 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
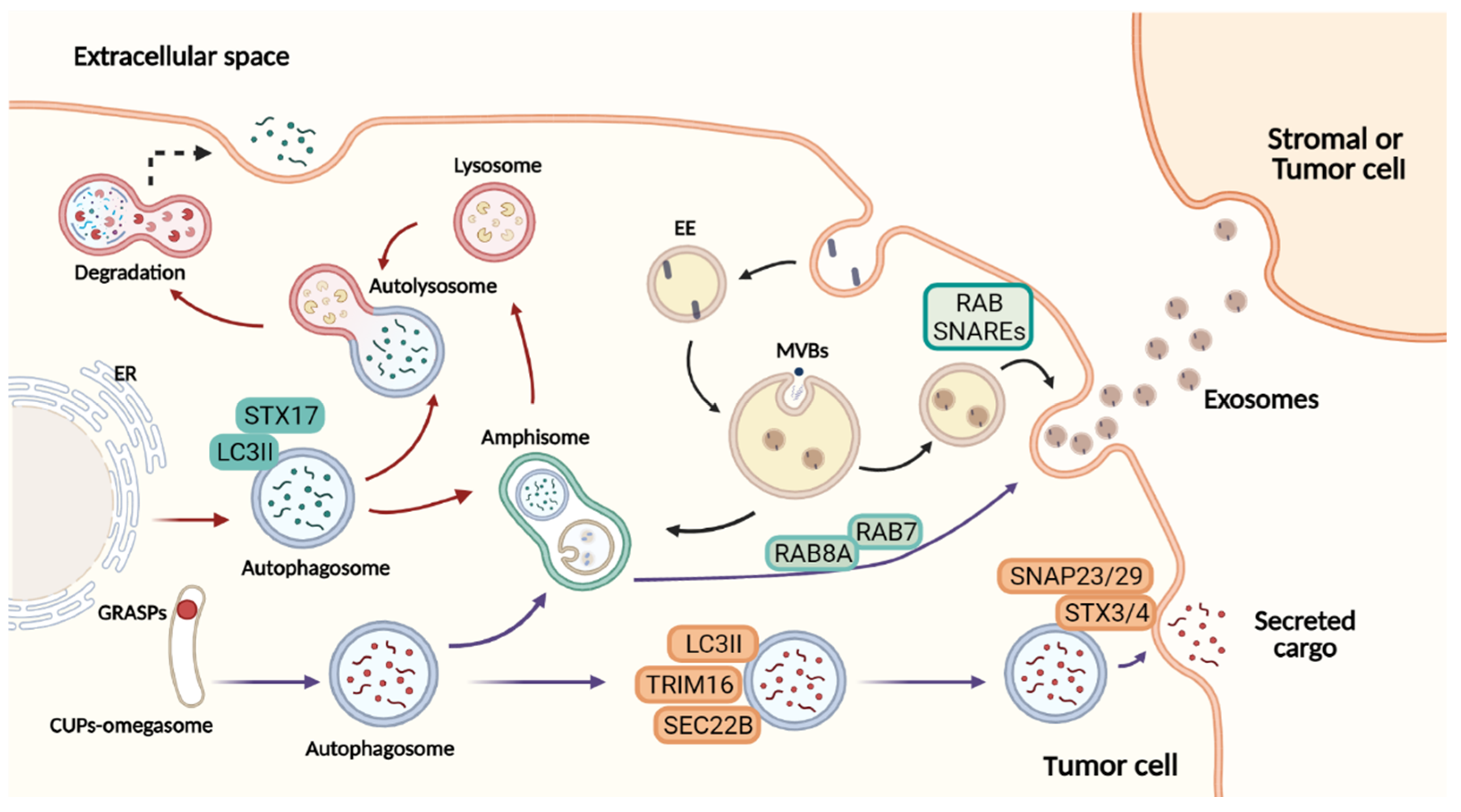
2. Secretory Mechanisms: A Complex Network for Cargo Release
2.1. Secretory Autophagy
2.2. Branches of the Secretory Autophagy System
2.3. Crosstalk between Extracellular Vesicles and the Autophagy Pathway
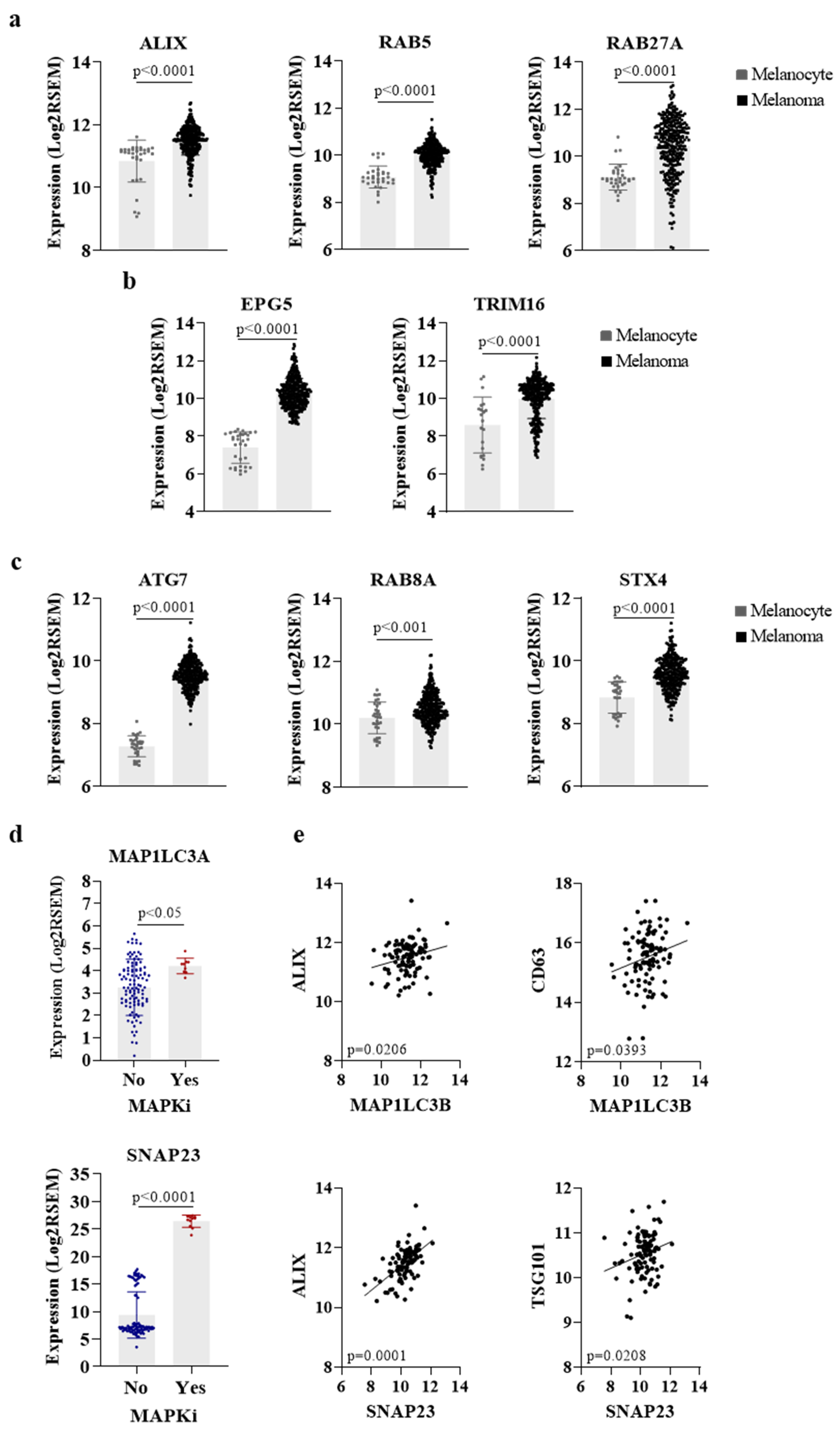
3. Role of Secretory Autophagy and Exosomes in Melanocytes Biology
4. Melanoma Progression, Tumor Microenvironment, and Autophagy
5. Autophagy and Exosome Signaling Modulates Melanoma Behavior and Therapy Outcome
6. Secretory Autophagy and Exosome Biogenesis in Melanoma: Two Sides of the Same Coin?
7. How to Deal with Secretory Autophagy and Exosome Release in Cancer? A Targetable or Not Targetable Pathway: This Is the Question!
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Haas, L.; Elewaut, A.; Gerard, C.L.; Umkehrer, C.; Leiendecker, L.; Pedersen, M.; Krecioch, I.; Hoffmann, D.; Novatchkova, M.; Kuttke, M.; et al. Acquired resistance to anti-MAPK targeted therapy confers an immune-evasive tumor microenvironment and cross-resistance to immunotherapy in melanoma. Nat. Cancer 2021, 2, 693–708. [Google Scholar] [CrossRef]
- Patel, M.; Eckburg, A.; Gantiwala, S.; Hart, Z.; Dein, J.; Lam, K.; Puri, N. Resistance to Molecularly Targeted Therapies in Melanoma. Cancers 2021, 13, 1115. [Google Scholar] [CrossRef]
- Verykiou, S.; Alexander, M.; Edwards, N.; Plummer, R.; Chaudhry, B.; Lovat, P.; Hill, D.S. Harnessing autophagy to overcome mitogen-activated protein kinase kinase inhibitor-induced resistance in metastatic melanoma. Br. J. Dermatol. 2019, 180, 346–356. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.-H.; Piao, S.-F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress–induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Jang, G.-B.; Yang, X.; Wang, Q.; He, S.; Li, S.; Quach, C.; Zhao, S.; Li, F.; Yuan, Z.; et al. Central role of autophagic UVRAG in melanogenesis and the suntan response. Proc. Natl. Acad. Sci. USA 2018, 115, E7728–E7737. [Google Scholar] [CrossRef] [Green Version]
- Möller, K.; Sigurbjornsdottir, S.; Arnthorsson, A.O.; Pogenberg, V.; Dilshat, R.; Fock, V.; Brynjolfsdottir, S.H.; Bindesboll, C.; Bessadottir, M.; Ogmundsdottir, H.M.; et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 2019, 9, 1055. [Google Scholar] [CrossRef] [Green Version]
- Katheder, N.; Khezri, R.; Ofarrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef]
- Kraya, A.A.; Piao, S.; Xu, X.; Zhang, G.; Herlyn, M.; Gimotty, P.; Levine, B.; Amaravadi, R.K.; Speicher, D.W. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy 2015, 11, 60–74. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Moreiras, H.; Seabra, M.; Barral, D. Melanin Transfer in the Epidermis: The Pursuit of Skin Pigmentation Control Mechanisms. Int. J. Mol. Sci. 2021, 22, 4466. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Somasundaram, R.; Herlyn, M. Pre-clinical modeling of cutaneous melanoma. Nat. Commun. 2020, 11, 2858. [Google Scholar] [CrossRef]
- Chen, Y.-D.; Fang, Y.-T.; Cheng, Y.-L.; Lin, C.-F.; Hsu, L.-J.; Wang, S.-Y.; Anderson, R.; Chang, C.-P.; Lin, Y.-S. Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-γ-stimulated lung epithelial cells. Sci. Rep. 2017, 7, 5676. [Google Scholar] [CrossRef]
- Leidal, A.M.; Debnath, J. Emerging roles for the autophagy machinery in extracellular vesicle biogenesis and secretion. FASEB BioAdv. 2021, 3, 377–386. [Google Scholar] [CrossRef]
- Gee, H.Y.; Noh, S.H.; Tang, B.L.; Kim, K.H.; Lee, M.G. Rescue of ΔF508-CFTR Trafficking via a GRASP-Dependent Unconventional Secretion Pathway. Cell 2011, 146, 746–760. [Google Scholar] [CrossRef] [Green Version]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Popa, S.; Stewart, S.E.; Moreau, K. Unconventional secretion of annexins and galectins. Semin. Cell Dev. Biol. 2018, 83, 42–50. [Google Scholar] [CrossRef]
- Takenouchi, T.; Nakai, M.; Iwamaru, Y.; Sugama, S.; Tsukimoto, M.; Fujita, M.; Wei, J.; Sekigawa, A.; Sato, M.; Kojima, S.; et al. The Activation of P2X7 Receptor Impairs Lysosomal Functions and Stimulates the Release of Autophagolysosomes in Microglial Cells. J. Immunol. 2009, 182, 2051–2062. [Google Scholar] [CrossRef] [Green Version]
- Ponpuak, M.; Mandell, M.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Bruns, C.; McCaffery, J.M.; Curwin, A.; Duran, J.M.; Malhotra, V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J. Cell Biol. 2011, 195, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahat, E.; Li, J.; Wang, Y. New Insights into the Golgi Stacking Proteins. Front. Cell Dev. Biol. 2019, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. eLife 2015, 4, e11205. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrow, L.; Malhotra, R.; Debnath, J. ATG12–ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat. Cell Biol. 2015, 17, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNARE s and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Leidal, A.M.; Huang, H.H.; Marsh, T.; Solvik, T.; Zhang, D.; Ye, J.; Kai, F.; Goldsmith, J.; Liu, J.Y.; Huang, Y.-H.; et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat. Cell Biol. 2020, 22, 187–199. [Google Scholar] [CrossRef]
- Solvik, T.A.; Nguyen, T.A.; Lin, Y.-H.T.; Marsh, T.; Huang, E.J.; Wiita, A.P.; Debnath, J.; Leidal, A.M. Autophagy cargo receptors are secreted via extracellular vesicles and particles in response to endolysosomal inhibition or impaired autophagosome maturation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Singh, R.K.; Varney, M.L. Regulation of interleukin 8 expression in human malignant melanoma cells. Cancer Res. 1998, 58, 1532–1537. [Google Scholar] [PubMed]
- Luca, M.; Huang, S.; Gershenwald, J.E.; Singh, R.K.; Reich, R.; Bar-Eli, M. Expression of interleukin-8 by human melanoma cells up-regulates MMP-2 activity and increases tumor growth and metastasis. Am. J. Pathol. 1997, 151, 1105–1113. [Google Scholar]
- Singh, S.; Singh, A.P.; Sharma, B.; Owen, L.B.; Singh, R.K. CXCL8 and its cognate receptors in melanoma progression and metastasis. Futur. Oncol. 2010, 6, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Scheibenbogen, C.; Möhler, T.; Haefele, J.; Hunstein, W.; Keilholz, U. Serum interleukin-8 (IL-8) is elevated in patients with metastatic melanoma and correlates with tumour load. Melanoma Res. 1995, 5, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Tengesdal, I.W.; Dinarello, A.; Powers, N.E.; Burchill, M.A.; Joosten, L.A.B.; Marchetti, C.; Dinarello, C.A. Tumor NLRP3-Derived IL-1β Drives the IL-6/STAT3 Axis Resulting in Sustained MDSC-Mediated Immunosuppression. Front. Immunol. 2021, 12, 3439. [Google Scholar] [CrossRef]
- Möller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef]
- Teng, F.; Fussenegger, M. Shedding Light on Extracellular Vesicle Biogenesis and Bioengineering. Adv. Sci. 2021, 8, 2003505. [Google Scholar] [CrossRef] [PubMed]
- Gudbergsson, J.M.; Johnsen, K.B. Exosomes and autophagy: Rekindling the vesicular waste hypothesis. J. Cell Commun. Signal. 2019, 13, 443–450. [Google Scholar] [CrossRef]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borràs, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A Compendium for Extracellular Vesicles with Continuous Community Annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.K.; Miyoshi, H.; Beatty, W.L.; Head, R.D.; Malvin, N.P.; Cadwell, K.; Guan, J.-L.; Saitoh, T.; Akira, S.; O Seglen, P.; et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013, 32, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Jimbow, K.; Hua, C.; Gomez, P.F.; Hirosaki, K.; Shinoda, K.; Salopek, T.G.; Matsusaka, H.; Jin, H.-Y.; Yamashita, T. Intracellular Vesicular Trafficking of Tyrosinase Gene Family Protein in Eu- and Pheomelanosome Biogenesis. Pigment. Cell Res. 2000, 13, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Sitaram, A.; Marks, M.S. Mechanisms of Protein Delivery to Melanosomes in Pigment Cells. Physiology 2012, 27, 85–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramkumar, A.; Murthy, D.; Raja, D.A.; Singh, A.; Krishnan, A.; Khanna, S.; Vats, A.; Thukral, L.; Sharma, P.; Sivasubbu, S.; et al. Classical autophagy proteins LC3B and ATG4B facilitate melanosome movement on cytoskeletal tracks. Autophagy 2017, 13, 1331–1347. [Google Scholar] [CrossRef] [Green Version]
- Ohbayashi, N.; Fukuda, M. SNARE dynamics during melanosome maturation. Biochem. Soc. Trans. 2018, 46, 911–917. [Google Scholar] [CrossRef]
- Shen, Z.; Sun, J.; Shao, J.; Xu, J. Ultraviolet B irradiation enhances the secretion of exosomes by human primary melanocytes and changes their exosomal miRNA profile. PLoS ONE 2020, 15, e0237023. [Google Scholar] [CrossRef] [PubMed]
- Sha, J.; Arbesman, J.; Harter, M.L. Premature senescence in human melanocytes after exposure to solar UVR: An exosome and UV-miRNA connection. Pigment. Cell Melanoma Res. 2020, 33, 671–684. [Google Scholar] [CrossRef]
- Wäster, P.; Eriksson, I.; Vainikka, L.; Rosdahl, I.; Öllinger, K. Extracellular vesicles are transferred from melanocytes to keratinocytes after UVA irradiation. Sci. Rep. 2016, 6, 27890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-F.; Gruber, F.; Ni, C.; Mildner, M.; Koenig, U.; Karner, S.; Barresi, C.; Rossiter, H.; Narzt, M.-S.; Nagelreiter, I.M.; et al. Suppression of Autophagy Dysregulates the Antioxidant Response and Causes Premature Senescence of Melanocytes. J. Investig. Dermatol. 2015, 135, 1348–1357. [Google Scholar] [CrossRef] [Green Version]
- Ni, C.; Narzt, M.-S.; Nagelreiter, I.M.; Zhang, C.F.; Larue, L.; Rossiter, H.; Grillari, J.; Tschachler, E.; Gruber, F. Autophagy deficient melanocytes display a senescence associated secretory phenotype that includes oxidized lipid mediators. Int. J. Biochem. Cell Biol. 2016, 81, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; He, Z.; Von Rütte, T.; Yousefi, S.; Hunger, R.E.; Simon, H.-U. Down-Regulation of Autophagy-Related Protein 5 (ATG5) Contributes to the Pathogenesis of Early-Stage Cutaneous Melanoma. Sci. Transl. Med. 2013, 5, 202ra123. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Horsman, M. Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacol. Ther. 2015, 153, 107–124. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Olmeda, D.; Soengas, M.S.; Agostinis, P. Vesicular trafficking mechanisms in endothelial cells as modulators of the tumor vasculature and targets of antiangiogenic therapies. FEBS J. 2015, 283, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, J.; Horita, H.; Redzic, J.; Hansen, K.; E Frankel, A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2008, 16, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Damsky, J.W.E.; Rosenbaum, L.E.; Bosenberg, M. Decoding Melanoma Metastasis. Cancers 2010, 3, 126–163. [Google Scholar] [CrossRef]
- Pandita, A.; Ekstrand, M.; Bjursten, S.; Zhao, Z.; Fogelstrand, P.; Le Gal, K.; Ny, L.; Bergo, M.O.; Karlsson, J.; Nilsson, J.A.; et al. Intussusceptive Angiogenesis in Human Metastatic Malignant Melanoma. Am. J. Pathol. 2021, 191, 2023–2038. [Google Scholar] [CrossRef] [PubMed]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Di Bella, V.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef]
- Liu, D.; Yang, X.; Wu, X. Tumor Immune Microenvironment Characterization Identifies Prognosis and Immunotherapy-Related Gene Signatures in Melanoma. Front. Immunol. 2021, 12, 663495. [Google Scholar] [CrossRef]
- Di Leo, L.; Bodemeyer, V.; De Zio, D. The Complex Role of Autophagy in Melanoma Evolution: New Perspectives from Mouse Models. Front. Oncol. 2020, 9, 1506. [Google Scholar] [CrossRef] [Green Version]
- Leonce, C.; Saintigny, P.; Ortiz-Cuaran, S. Cell-Intrinsic Mechanisms of Drug Tolerance to Systemic Therapies in Cancer. Mol. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Zhu, X.; Li, S.; Xu, B.; Luo, H. Cancer evolution: A means by which tumors evade treatment. Biomed. Pharmacother. 2021, 133, 111016. [Google Scholar] [CrossRef]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.-F.; et al. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef]
- Cesi, G.; Philippidou, D.; Kozar, I.; Kim, Y.J.; Bernardin, F.; Van Niel, G.; Wienecke-Baldacchino, A.; Felten, P.; Letellier, E.; Dengler, S.; et al. A new ALK isoform transported by extracellular vesicles confers drug resistance to melanoma cells. Mol. Cancer 2018, 17, 145. [Google Scholar] [CrossRef]
- Ma, Y.; Yuwen, D.; Chen, J.; Zheng, B.; Gao, J.; Fan, M.; Xue, W.; Wang, Y.; Li, W.; Shu, Y.; et al. Exosomal Transfer of Cisplatin-Induced miR-425-3p Confers Cisplatin Resistance in NSCLC Through Activating Autophagy. Int. J. Nanomed. 2019, 14, 8121–8132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago-O’Farrill, J.M.; Weroha, S.J.; Hou, X.; Oberg, A.L.; Bs, E.P.H.; Ms, M.J.M.; Pang, L.; Rask, P.; Amaravadi, R.K.; Becker, S.E.; et al. Poly (adenosine diphosphate ribose) polymerase inhibitors induce autophagy-mediated drug resistance in ovarian cancer cells, xenografts, and patient-derived xenograft models. Cancer 2020, 126, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Cho, H.; Hong, S.O.; Oh, S.J.; Lee, H.J.; Cho, E.; Woo, S.R.; Song, J.S.; Chung, J.Y.; Son, S.W.; et al. LC3B upregulation by NANOG promotes immune resistance and stem-like property through hyperactivation of EGFR signaling in immune-refractory tumor cells. Autophagy 2021, 17, 1978–1997. [Google Scholar] [CrossRef]
- Wu, S.; Luo, M.; To, K.K.W.; Zhang, J.; Su, C.; Zhang, H.; An, S.; Wang, F.; Chen, D.; Fu, L. Intercellular transfer of exosomal wild type EGFR triggers osimertinib resistance in non-small cell lung cancer. Mol. Cancer 2021, 20, 17. [Google Scholar] [CrossRef]
- Demirsoy, S.; Martin, S.; Maes, H.; Agostinis, P. Adapt, Recycle, and Move on: Proteostasis and Trafficking Mechanisms in Melanoma. Front. Oncol. 2016, 6, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curbelo, D.A.; Riveiro-Falkenbach, E.; Pérez-Guijarro, E.; Cifdaloz, M.; Karras, P.; Osterloh, L.; Megias, D.; Cañón, E.; Calvo, T.G.; Olmeda, D.; et al. RAB7 Controls Melanoma Progression by Exploiting a Lineage-Specific Wiring of the Endolysosomal Pathway. Cancer Cell 2014, 26, 61–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Ma, X.-H.; Piao, S.; Wang, D.; McAfee, Q.W.; Nathanson, K.; Lum, J.J.; Li, L.; Amaravadi, R.K. Measurements of Tumor Cell Autophagy Predict Invasiveness, Resistance to Chemotherapy, and Survival in Melanoma. Clin. Cancer Res. 2011, 17, 3478–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Zahedi, S.; Griesinger, A.M. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef] [PubMed]
- Goulielmaki, M.; Koustas, E.; Moysidou, E.; Vlassi, M.; Sasazuki, T.; Shirasawa, S.; Zografos, G.; Oikonomou, E.; Pintzas, A. BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget 2016, 7, 9188–9221. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.-B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An autophagy-driven pathway of ATP secretion supports the aggressive phenotype of BRAF. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.-F.; Liu, H.; Gao, R.; Zhou, M.; Ma, J.; Zhang, Y.; Zhao, J.; Chen, Y.; Zhang, T.; Huang, F.; et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J. Immunother. Cancer 2018, 6, 151. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, H.-T.; Yang, J.-L.; Tseng, Y.-J.; Lee, C.-H.; Chen, W.-J.; Chyuan, I.-T. Plasminogen Activator Inhibitor-1 Secretion by Autophagy Contributes to Melanoma Resistance to Chemotherapy through Tumor Microenvironment Modulation. Cancers 2021, 13, 1253. [Google Scholar] [CrossRef]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Lunavat, T.A.-O.; Cheng, L.; Einarsdottir, B.A.-O.; Bagge, R.O.; Muralidharan, S.V.; Sharples, R.A.; Lässer, C.; Gho, Y.S.; Hill, A.F.; Nilsson, J.A.; et al. BRAF(V600) inhibition alters the microRNA cargo in the vesicular secretome of malignant melanoma cells (1091-6490 (Electronic)). Proc. Natl. Acad. Sci. USA 2017, 114, E5930–E5939. [Google Scholar] [CrossRef] [Green Version]
- Gad, S.A.; Ali, H.E.A.; Gaballa, R.; Abdelsalam, R.M.; Zerfaoui, M.; Ali, H.I.; Salama, S.H.; Kenawy, S.A.; Kandil, E.; Elmageed, Z.Y.A. Targeting CDC7 sensitizes resistance melanoma cells to BRAF. Sci. Rep. 2019, 9, 14197. [Google Scholar] [CrossRef]
- Vella, L.J.; Behren, A.; Coleman, B.; Greening, D.W.; Hill, A.F.; Cebon, J. Intercellular Resistance to BRAF Inhibition Can Be Mediated by Extracellular Vesicle-Associated PDGFRβ. Neoplasia 2017, 19, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, J.; Yin, M.; Liu, H.; Zhang, X.; Li, J.; Yan, B.; Guo, Y.; Zhou, J.; Tao, J.; et al. Inhibition of xCT suppresses the efficacy of anti-PD-1/L1 melanoma treatment through exosomal PD-L1-induced macrophage M2 polarization. Mol. Ther. 2021, 29, 2321–2334. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.N.D.S.; Otake, A.H.; Cardim, S.G.B.; Da Silva, F.I.; Sakamoto, M.M.I.; Furuya, T.K.; Uno, M.; Pasini, F.S.; Chammas, R. Extracellular Vesicles Shedding Promotes Melanoma Growth in Response to Chemotherapy. Sci. Rep. 2019, 9, 14482. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xie, L.; Li, B.; Sang, W.; Yan, J.; Li, J.; Tian, H.; Li, W.; Zhang, Z.; Tian, Y.; et al. A nanounit strategy reverses immune suppression of exosomal PD-L1 and is associated with enhanced ferroptosis. Nat. Commun. 2021, 12, 5733. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Noh, S.H.; Gee, H.Y.; Kim, Y.; Piao, H.; Kim, J.; Kang, C.M.; Lee, G.; Mook-Jung, I.; Lee, Y.; Cho, J.W.; et al. Specific autophagy and ESCRT components participate in the unconventional secretion of CFTR. Autophagy 2018, 14, 1761–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atienzar-Aroca, S.; Serrano-Heras, G.; Valls, A.F.; De Almodovar, C.R.; Muriach, M.; Barcia, J.M.; Garcia-Verdugo, J.M.; Romero, F.J.; Sancho-Pelluz, J. Role of retinal pigment epithelium-derived exosomes and autophagy in new blood vessel formation. J. Cell. Mol. Med. 2018, 22, 5244–5256. [Google Scholar] [CrossRef]
- Nam, S.-E.; Cheung, Y.W.S.; Nguyen, T.N.; Gong, M.; Chan, S.; Lazarou, M.; Yip, C.K. Insights on autophagosome–lysosome tethering from structural and biochemical characterization of human autophagy factor EPG5. Commun. Biol. 2021, 4, 291. [Google Scholar] [CrossRef]
- Qian, L.; Yang, X.; Li, S.; Zhao, H.; Gao, Y.; Zhao, S.; Lv, X.; Zhang, X.; Li, L.; Zhai, L.; et al. Reduced O-GlcNAcylation of SNAP-23 promotes cisplatin resistance by inducing exosome secretion in ovarian cancer. Cell Death Discov. 2021, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Verweij, F.J.; Bebelman, M.P.; Jimenez, C.R.; Garcia-Vallejo, J.J.; Janssen, H.; Neefjes, J.; Knol, J.C.; De Haas, R.G.; Piersma, S.R.; Baglio, S.R.; et al. Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J. Cell Biol. 2018, 217, 1129–1142. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Peng, X.; Li, Y.; Zhang, X.; Ma, Y.; Wu, C.; Fan, Q.; Wei, S.; Li, H.; Liu, J. Long non-coding RNA HOTAIR promotes exosome secretion by regulating RAB35 and SNAP23 in hepatocellular carcinoma. Mol. Cancer 2019, 18, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.-Q.; Wang, S.-B.; Shao, Y.-F.; Shi, J.-N.; Wang, W.; Chen, W.-Y.; Ye, Z.-Q.; Jiang, J.-Y.; Fang, Q.-X.; Zhang, G.-B.; et al. Hydroxychloroquine potentiates the anti-cancer effect of bevacizumab on glioblastoma via the inhibition of autophagy. Biomed. Pharmacother. 2019, 118, 109339. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.-S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Sheen, J.-H.; Zoncu, R.; Kim, D.; Sabatini, D.M. Defective Regulation of Autophagy upon Leucine Deprivation Reveals a Targetable Liability of Human Melanoma Cells In Vitro and In Vivo. Cancer Cell 2011, 19, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor Vessel Normalization by Chloroquine Independent of Autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [Green Version]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, J.; Liu, J.; Zhang, G.; Lu, A. Advances in the discovery of exosome inhibitors in cancer. J. Enzym. Inhib. Med. Chem. 2020, 35, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Isola, A.L.; Eddy, K.; Chen, S. Biology, Therapy and Implications of Tumor Exosomes in the Progression of Melanoma. Cancers 2016, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH Is a Key Factor for Exosome Traffic in Tumor Cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target. Ther. 2020, 5, 242. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Protein | Times Identified in Melanoma EVs | Vesiclepedia Experiment IDs |
|---|---|---|
| LAMP2 | 11 | (71, 72, 274, 275, 276, 617, 618, 621, 622, 623, 624) |
| HSPA8 | 7 | (12, 24, 453, 620, 621, 623, 986) |
| MTOR | 6 | (617, 618, 619, 620, 624, 625) |
| PSMD4 | 6 | (617, 620, 621, 622, 624, 625) |
| ATG7 | 5 | (617, 618, 619, 622, 624) |
| HSP90AA1 | 5 | (453, 621, 622, 623, 986) |
| LAMP1 | 5 | (74, 621, 622, 623, 624) |
| GABARAP | 5 | (617, 618, 619, 620, 621) |
| RAB7A | 5 | (453, 621, 622, 625, 986) |
| SQSTM1 | 5 | (453, 617, 618, 619, 625) |
| SEC22A | 5 | (617, 619, 620, 621, 622) |
| SNAP23 | 5 | (617, 619, 620, 622, 624) |
| STX4 | 5 | (617, 618,620, 624, 625) |
| GARASP | 4 | (619, 620, 621, 622) |
| ACBD3 | 4 | (617, 618, 620, 622) |
| ACBD5 | 4 | (618, 619, 621, 623) |
| BCL2 | 4 | (619, 621, 622, 624) |
| PHB2 | 4 | (621, 623, 624, 986) |
| SNX18 | 4 | (620, 621, 623, 624) |
| TAX1BP1 | 4 | (453, 622, 623, 625) |
| TGM2 | 4 | (617, 619, 623, 624) |
| TRIM16 | 4 | (617, 619, 621, 624) |
| VCP | 4 | (453, 622, 625, 986) |
| SNAP29 | 4 | (617, 619, 620, 625) |
| RAB8A | 4 | (617, 619, 621, 623) |
| EI24 | 3 | (617, 620,624) |
| EPG5 | 3 | (617, 618, 624) |
| LGALS3 | 3 | (620, 625, 986) |
| OPTN | 3 | (621, 623, 624) |
| PIK3R4 | 3 | (617, 624, 625) |
| RAB7B | 3 | (621, 622, 624) |
| SNX4 | 3 | (627, 621, 625) |
| TBK1 | 3 | (617, 619, 622) |
| TOLLIP | 3 | (617, 625, 986) |
| UVRAG | 3 | (623, 624, 625) |
| SEC22B | 3 | (618, 623, 624). |
| ATG3 | 2 | (618, 620) |
| BNIP3 | 2 | (619, 621) |
| RAB11A | 2 | (621, 622) |
| SNX3 | 2 | (617, 623) |
| TFEB | 2 | (621, 623) |
| WDFY3 | 2 | (621, 623) |
| ATG9A | 1 | (617) |
| BCL2L13 | 1 | (621) |
| FUNDC1 | 1 | (625) |
| GFAP | 1 | (617) |
| LGALS8 | 1 | (618) |
| PEX14 | 1 | (621) |
| STX3 | 1 | (624) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustos, S.O.; Leal Santos, N.; Chammas, R.; Andrade, L.N.d.S. Secretory Autophagy Forges a Therapy Resistant Microenvironment in Melanoma. Cancers 2022, 14, 234. https://doi.org/10.3390/cancers14010234
Bustos SO, Leal Santos N, Chammas R, Andrade LNdS. Secretory Autophagy Forges a Therapy Resistant Microenvironment in Melanoma. Cancers. 2022; 14(1):234. https://doi.org/10.3390/cancers14010234
Chicago/Turabian StyleBustos, Silvina Odete, Nathalia Leal Santos, Roger Chammas, and Luciana Nogueira de Sousa Andrade. 2022. "Secretory Autophagy Forges a Therapy Resistant Microenvironment in Melanoma" Cancers 14, no. 1: 234. https://doi.org/10.3390/cancers14010234
APA StyleBustos, S. O., Leal Santos, N., Chammas, R., & Andrade, L. N. d. S. (2022). Secretory Autophagy Forges a Therapy Resistant Microenvironment in Melanoma. Cancers, 14(1), 234. https://doi.org/10.3390/cancers14010234