
Biomarkers of Response to Neoadjuvant Androgen Deprivation in Localised Prostate Cancer

Abstract
Simple Summary
Abstract
1. Introduction
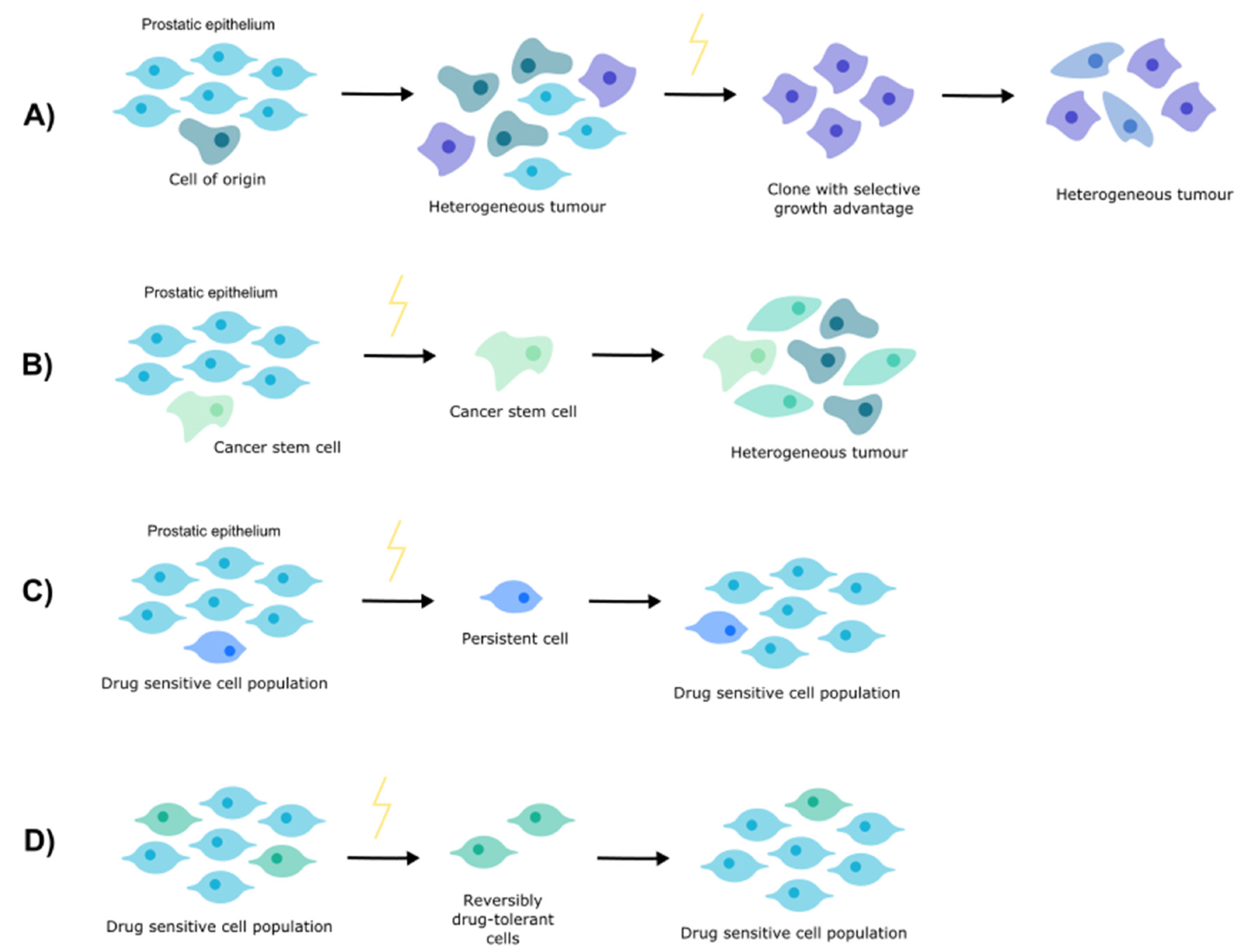
2. Clonal Evolution
3. Cancer Stem Cells
4. Cell Persistence
5. Drug Tolerance
6. Castration Resistance
7. Neoadjuvant Androgen Deprivation Therapy Resistance
8. Biomarkers of Resistance
9. Combination Therapy to Overcome Resistance
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Lyou, Y.; Dorff, T.B. Hormonal manipulation in androgen signaling: A narrative review on using novel androgen therapy agents to optimize clinical outcomes and minimize side effects for prostate cancer patients. Transl. Androl. Urol. 2021, 10, 3199–3207. [Google Scholar] [CrossRef] [PubMed]
- Prostate Cancer Foundation. Risk Groups. 2020. Available online: https://www.pcf.org/about-prostate-cancer/diagnosis-staging-prostate-cancer/risk-groups/ (accessed on 12 September 2020).
- McKay, R.R.; Choueiri, T.K.; Taplin, M.E. Rationale for and review of neoadjuvant therapy prior to radical prostatectomy for patients with high-risk prostate cancer. Drugs 2013, 73, 1417–1430. [Google Scholar] [CrossRef]
- Ashrafi, A.N.; Yip, W.; Aron, M. Neoadjuvant Therapy in High-Risk Prostate Cancer. Indian J. Urol. 2020, 36, 251–261. [Google Scholar] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Hu, Z.; Curtis, C. Big Bang Tumor Growth and Clonal Evolution. Cold Spring Harb. Perspect. Med. 2018, 8, a028381. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Schwartz, M.; Zlotorynski, E.; Kerem, B. The molecular basis of common and rare fragile sites. Cancer Lett. 2006, 232, 13–26. [Google Scholar] [CrossRef]
- Varley, K.E.; Mutch, D.G.; Edmonston, T.B.; Goodfellow, P.J.; Mitra, R.D. Intra-tumor heterogeneity of MLH1 promoter methylation revealed by deep single molecule bisulfite sequencing. Nucleic Acids Res. 2009, 37, 4603–4612. [Google Scholar] [CrossRef]
- Van Etten, J.L.; Dehm, S.M. Clonal origin and spread of metastatic prostate cancer. Endocr. Relat. Cancer 2016, 23, R207–R217. [Google Scholar] [CrossRef]
- Campbell, L.L.; Polyak, K. Breast tumor heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle 2007, 6, 2332–2338. [Google Scholar] [CrossRef]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1107. [Google Scholar]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Dudás, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.I. Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol. 2018, 53, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea–A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Kleffel, S.; Schatton, T. Tumor dormancy and cancer stem cells: Two sides of the same coin? In Systems Biology of Tumor Dormancy; Springer: Berlin/Heidelberg, Germany, 2013; pp. 145–179. [Google Scholar]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, I.; Debbage, P.; Kumar, V.; Skvortsov, S. (Eds.) Radiation resistance: Cancer stem cells (CSCs) and their enigmatic pro-survival signaling. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Goff, D.J.; Recart, A.C.; Sadarangani, A.; Chun, H.-J.; Barrett, C.L.; Krajewska, M.; Leu, H.; Low-Marchelli, J.; Ma, W.; Shih, A.Y.; et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 2013, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; Iglesia-Vicente, J.d.l.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch-and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7. [Google Scholar]
- Borovski, T.; De Sousa, E.M.F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef]
- Glickman, M.S.; Sawyers, C.L. Converting cancer therapies into cures: Lessons from infectious diseases. Cell 2012, 148, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.; Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 230, 13–18. [Google Scholar] [CrossRef]
- Maisonneuve, E.; Gerdes, K. Molecular mechanisms underlying bacterial persisters. Cell 2014, 157, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Kurata, T.; Tamura, K.; Kaneda, H.; Nogami, T.; Uejima, H.; Asai Go, G.; Nakagawa, K.; Fukuoka, M. Effect of re-treatment with gefitinib (‘Iressa’, ZD1839) after acquisition of resistance. Ann. Oncol. 2004, 15, 173–174. [Google Scholar] [CrossRef]
- Yano, S.; Nakataki, E.; Ohtsuka, S.; Inayama, M.; Tomimoto, H.; Edakuni, N.; Kakiuchi, S.; Nishikubo, N.; Muguruma, H.; Sone, S. Retreatment of lung adenocarcinoma patients with gefitinib who had experienced favorable results from their initial treatment with this selective epidermal growth factor receptor inhibitor: A report of three cases. Oncol. Res. 2005, 15, 107–111. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R.; Gangurde, P.; et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-K.; Kang, S.-K. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007, 16, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, R.H.; Lorenzi, T.; Lorz, A.; Larsen, A.K.; de Almeida, L.N.; Escargueil, A.; Clairambault, E. Emergence of drug tolerance in cancer cell populations: An evolutionary outcome of selection, nongenetic instability, and stress-induced adaptation. Cancer Res. 2015, 75, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef]
- Lou, D.Y.; Fong, L. Neoadjuvant therapy for localized prostate cancer: Examining mechanism of action and efficacy within the tumor. Urol. Oncol. 2016, 34, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, N.; Granata, I.; Capaia, M.; Piccirillo, M.; Guarracino, M.R.; Venè, R.; Brizzolara, A.; Petretto, A.; Inglese, E.; Morini, M.; et al. Adaptive phenotype drives resistance to androgen deprivation therapy in prostate cancer. Cell Commun. Signal. 2017, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, P.A. Neuroendocrine cells in tumour growth of the prostate. Endocr. Relat. Cancer 1999, 6, 503–519. [Google Scholar] [CrossRef]
- Miao, L.; Yang, L.; Li, R.; Rodrigues, D.N.; Crespo, M.; Hsieh, J.T.; Tilley, W.D.; Bono, J.d.; Selth, L.A.; Raj, G.A. Disrupting Androgen Receptor Signaling Induces Snail-Mediated Epithelial-Mesenchymal Plasticity in Prostate Cancer. Cancer Res. 2017, 77, 3101–3112. [Google Scholar] [CrossRef] [PubMed]
- Isaacsson Velho, P.; Fu, W.; Wang, H.; Mirkheshti, N.; Qazi, F.; Lima, F.A.S.; Shaukat, F.; Carducci, M.A.; Denmeade, S.R.; Paller, C.J.; et al. Wnt-pathway Activating Mutations Are Associated with Resistance to First-line Abiraterone and Enzalutamide in Castration-resistant Prostate Cancer. Eur. Urol. 2020, 77, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lucas, J.M.; Lam, H.-M.; Dumpit, R.; Corey, E.; Chéry, L.; et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 4698–4708. [Google Scholar] [CrossRef]
- Aprikian, A.G.; Cordon-Cardo, C.; Fair, W.R.; Reuter, V.E. Characterization of neuroendocrine differentiation in human benign prostate and prostatic adenocarcinoma. Cancer 1993, 71, 3952–3965. [Google Scholar] [CrossRef]
- Vashchenko, N.; Abrahamsson, P.A. Neuroendocrine differentiation in prostate cancer: Implications for new treatment modalities. Eur. Urol. 2005, 47, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Nelson, E.C.; Cambio, A.J.; Yang, J.C.; Ok, J.H.; Lara, P.N., Jr.; Evans, C.P. Clinical implications of neuroendocrine differentiation in prostate cancer. Prostate Cancer Prostatic Dis. 2007, 10, 6–14. [Google Scholar] [CrossRef][Green Version]
- Klotz, L.H.; Goldenberg, S.L.; Jewett, M.A.; Fradet, Y.; Nam, R.; Barkin, J.; Chin, J.; Chatterjee, S.; Canadian Uro-Oncology Group. Long-term followup of a randomized trial of 0 versus 3 months of neoadjuvant androgen ablation before radical prostatectomy. J. Urol. 2003, 170, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Dalkin, B.L.; Ahmann, F.R.; Nagle, R.; Johnson, C.S. Randomized study of neoadjuvant testicular androgen ablation therapy before radical prostatectomy in men with clinically localized prostate cancer. J. Urol. 1996, 155, 1357–1360. [Google Scholar] [CrossRef]
- Schulman, C.C.; Debruyne, F.M.; Forster, G.; Selvaggi, F.P.; Zlotta, A.R.; Witjes, W.P. 4-Year follow-up results of a European prospective randomized study on neoadjuvant hormonal therapy prior to radical prostatectomy in T2-3N0M0 prostate cancer. European Study Group on Neoadjuvant Treatment of Prostate Cancer. Eur. Urol. 2000, 38, 706–713. [Google Scholar] [CrossRef]
- Labrie, F.; Cusan, L.; Gomez, J.L.; Diamond, P.; Suburu, R.; Lemay, M.; Tetu, B.; Fradet, Y.; Bélanger, A.; Candas, B. Neoadjuvant hormonal therapy: The Canadian experience. Urology 1997, 49, 56–64. [Google Scholar] [CrossRef]
- Bandini, M.; Fossati, N.; Gandaglia, G.; Preisser, F.; Dell’Oglio, P.; Zaffuto, E.; Stabile, A.; Gallina, A.; Suardi, N.; Shariat, S.F.; et al. Neoadjuvant and adjuvant treatment in high-risk prostate cancer. Expert Rev. Clin. Pharmacol. 2018, 11, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Aus, G.; Abrahamsson, P.A.; Ahlgren, G.; Hugosson, J.; Lundberg, S.; Schain, M.; Schelin, S.; Pedersen, K. Three-month neoadjuvant hormonal therapy before radical prostatectomy: A 7-year follow-up of a randomized controlled trial. BJU Int. 2002, 90, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Gleave, M.E.; Goldenberg, S.L.; Chin, J.L.; Warner, J.; Saad, F.; Klotz, L.H.; Jewett, M.; Kassabian, V.; Chetner, M.; Dupont, C.; et al. Randomized comparative study of 3 versus 8-month neoadjuvant hormonal therapy before radical prostatectomy: Biochemical and pathological effects. J. Urol. 2001, 166, 500–506. [Google Scholar] [CrossRef]
- Prezioso, D.; Lotti, T.; Polito, M.; Montironi, R. Neoadjuvant hormone treatment with leuprolide acetate depot 3.75 mg and cyproterone acetate, before radical prostatectomy: A randomized study. Urol. Int. 2004, 72, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Selli, C.; Montironi, R.; Bono, A.; Pagano, F.; Zattoni, F.; Manganelli, A.; Selvaggi, F.P.; Comeri, G.; Fiaccavento, G.; Guazzieri, S.; et al. Effects of complete androgen blockade for 12 and 24 weeks on the pathological stage and resection margin status of prostate cancer. J. Clin. Pathol. 2002, 55, 508–513. [Google Scholar] [CrossRef]
- Soloway, M.S.; Pareek, K.; Sharifi, R.; Wajsman, Z.; McLeod, D.; Wood, D.P., Jr.; Puras-Baez, A.; Lupron Depot Neoadjuvant Prostate Cancer Study Group. Neoadjuvant androgen ablation before radical prostatectomy in cT2bNxMo prostate cancer: 5-year results. J. Urol. 2002, 167, 112–116. [Google Scholar] [CrossRef]
- Van der Kwast, T.H.; Têtu, B.; Candas, B.; Gomez, J.L.; Cusan, L.; Labrie, F. Prolonged neoadjuvant combined androgen blockade leads to a further reduction of prostatic tumor volume: Three versus six months of endocrine therapy. Urology 1999, 53, 523–529. [Google Scholar] [CrossRef]
- Yee, D.S.; Lowrance, W.T.; Eastham, J.A.; Maschino, A.C.; Cronin, A.M.; Rabbani, F. Long-term follow-up of 3-month neoadjuvant hormone therapy before radical prostatectomy in a randomized trial. BJU Int. 2010, 105, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qi, M.; Zhang, J.; Sun, X.; Guo, H.; Pang, Y.; Zhang, Q.; Chen, X.; Zhang, R.; Liu, Z.; et al. Differential response to neoadjuvant hormonal therapy in prostate cancer: Predictive morphological parameters and molecular markers. Prostate 2019, 79, 709–719. [Google Scholar] [CrossRef]
- McKay, R.R.; Ye, H.; Xie, W.; Lis, R.; Calagua, C.; Zhang, Z.; Trinh, Q.-D.; Chang, S.L.; Harshman, L.C.; Ross, A.E.; et al. Evaluation of Intense Androgen Deprivation Before Prostatectomy: A Randomized Phase II Trial of Enzalutamide and Leuprolide with or Without Abiraterone. J. Clin. Oncol. 2019, 37, 923–931. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.R.; Werner, L.; Mostaghel, E.A.; Lis, R.; Voznesensky, O.; Zhang, Z.; Marck, B.T.; Matsumoto, A.M.; Domachevsky, L.; Zukotynski, K.A.; et al. A Phase II Trial of Abiraterone Combined with Dutasteride for Men with Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2017, 23, 935–945. [Google Scholar] [CrossRef]
- Sowalsky, A.G.; Ye, H.; Bhasin, M.; Van Allen, E.M.; Loda, M.; Lis, R.T.; Montaser-Kouhsari, L.; Calagua, C.; Ma, F.; Russo, J.W.; et al. Neoadjuvant-Intensive Androgen Deprivation Therapy Selects for Prostate Tumor Foci with Diverse Subclonal Oncogenic Alterations. Cancer Res. 2018, 78, 4716–4730. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Cmero, M.; Kurganovs, N.J.; Stuchbery, R.; McCoy, P.; Grima, C.; Ngyuen, A.; Chow, K.; Mangiola, S.; Macintyre, G.; Howard, N.; et al. Loss of SNAI2 in Prostate Cancer Correlates with Clinical Response to Androgen Deprivation Therapy. JCO Precis. Oncol. 2021, 5, 1048–1059. [Google Scholar] [CrossRef]
- Deep, G.; Jain, A.K.; Ramteke, A.; Ting, H.; Vijendra, K.C.; Gangar, S.C.; Agarwal, C.; Agarwal, R. SNAI1 is critical for the aggressiveness of prostate cancer cells with low E-cadherin. Mol. Cancer 2014, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Neal, C.L.; Henderson, V.; Smith, B.N.; McKeithen, D.; Graham, T.; Vo, B.T.; Odero-Marah, V.A. Snail transcription factor negatively regulates maspin tumor suppressor in human prostate cancer cells. BMC Cancer 2012, 12, 336. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Smith, B.N.; Odero-Marah, V.A. The role of Snail in prostate cancer. Cell Adh. Migr. 2012, 6, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Takkunen, M.; Grenman, R.; Hukkanen, M.; Korhonen, M.; García de Herreros, A.; Virtanen, I. Snail-dependent and -independent epithelial-mesenchymal transition in oral squamous carcinoma cells. J. Histochem. Cytochem. 2006, 54, 1263–1275. [Google Scholar] [CrossRef]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef]
- Kajita, M.; McClinic, K.N.; Wade, P.A. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol. Cell. Biol. 2004, 24, 7559–7566. [Google Scholar] [CrossRef]
- Esposito, S.; Russo, M.V.; Airoldi, I.; Tupone, M.G.; Sorrentino, C.; Barbarito, G.; Meo, S.D.; Carlo, E.D. SNAI2/Slug gene is silenced in prostate cancer and regulates neuroendocrine differentiation, metastasis-suppressor and pluripotency gene expression. Oncotarget 2015, 6, 17121–17134. [Google Scholar] [CrossRef] [PubMed]
- Emadi Baygi, M.; Soheili, Z.S.; Essmann, F.; Deezagi, A.; Engers, R.; Goering, W.; Schulz, W.A. Slug/SNAI2 regulates cell proliferation and invasiveness of metastatic prostate cancer cell lines. Tumour Biol. 2010, 31, 297–307. [Google Scholar] [CrossRef]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef]
- Wu, W.S.; Heinrichs, S.; Xu, D.; Garrison, S.P.; Zambetti, G.P.; Adams, J.M.; Look, A.T. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 2005, 123, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhou, L.; Wei, C.; Zhao, W.; Yu, D. Slug inhibition increases radiosensitivity of oral squamous cell carcinoma cells by upregulating PUMA. Int. J. Oncol. 2016, 49, 709–719. [Google Scholar] [CrossRef]
- Vitali, R.; Mancini, C.; Cesi, V.; Tanno, B.; Mancuso, M.; Bossi, G.; Zhang, Y.; Martinez, R.V.; Calabretta, B.; Dominici, C.; et al. Slug (SNAI2) down-regulation by RNA interference facilitates apoptosis and inhibits invasive growth in neuroblastoma preclinical models. Clin. Cancer Res. 2008, 14, 4622–4630. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.H.; Tsai, M.F.; Su, K.Y.; Wu, S.G.; Huang, C.P.; Yu, S.L.; Yu, Y.-L.; Lan, C.C.; Yang, C.-H.; Lin, S.-B.; et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am. J. Respir. Crit. Care Med. 2011, 183, 1071–1079. [Google Scholar] [CrossRef]
- Yang, H.W.; Menon, L.G.; Black, P.M.; Carroll, R.S.; Johnson, M.D. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer 2010, 10, 301. [Google Scholar] [CrossRef]
- Bhat-Nakshatri, P.; Appaiah, H.; Ballas, C.; Pick-Franke, P.; Goulet, R., Jr.; Badve, S.; Srour, E.F.; Nakshatri, H. SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24- phenotype. BMC Cancer 2010, 10, 411. [Google Scholar] [CrossRef]
- Shih, J.-Y.; Tsai, M.-F.; Chang, T.-H.; Chang, Y.-L.; Yuan, A.; Yu, C.-J.; Lin, S.-B.; Liou, G.-Y.; Lee, M.-L.; Chen, J.J.W.; et al. Transcription repressor slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin. Cancer Res. 2005, 11, 8070–8078. [Google Scholar] [CrossRef]
- Shioiri, M.; Shida, T.; Koda, K.; Oda, K.; Seike, K.; Nishimura, M.; Takano, S.; Miyazaki, M. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br. J. Cancer 2006, 94, 1816–1822. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.-H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.-N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Eastham, J.A.; Heller, G.; Halabi, S.; Monk, J.P., 3rd; Beltran, H.; Gleave, M.; Evans, C.P.; Clinton, S.K.; Szmulewitz, R.Z.; Coleman, J.; et al. Cancer and Leukemia Group B 90203 (Alliance): Radical Prostatectomy with or Without Neoadjuvant Chemohormonal Therapy in Localized, High-Risk Prostate Cancer. J. Clin. Oncol. 2020, 38, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Taplin, M.E.; Montgomery, B.; Logothetis, C.J.; Bubley, G.J.; Richie, J.P.; Dalkin, B.L.; Sanda, M.G.; Davis, J.W.; Loda, M.; True, L.D.; et al. Intense androgen-deprivation therapy with abiraterone acetate plus leuprolide acetate in patients with localized high-risk prostate cancer: Results of a randomized phase II neoadjuvant study. J. Clin. Oncol. 2014, 32, 3705–3715. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Castration Resistant Prostate Cancer | Neoadjuvant ADT Resistant Prostate Cancer |
|---|---|
| Amplification and/or mutation of the AR | Cribriform growth pattern |
| Constitutively active AR splice variants | Macro-nucleoli |
| Increased intracrine androgen synthesis | Ductal adenocarcinoma differentiation |
| Altered expression or activity of AR coactivators or corepressors | PTEN loss |
| Increased androgen biosynthesis | ERG-positive and PTEN loss |
| Neuroendocrine differentiation | RB1 loss |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pechlivanis, M.; Campbell, B.K.; Hovens, C.M.; Corcoran, N.M. Biomarkers of Response to Neoadjuvant Androgen Deprivation in Localised Prostate Cancer. Cancers 2022, 14, 166. https://doi.org/10.3390/cancers14010166
Pechlivanis M, Campbell BK, Hovens CM, Corcoran NM. Biomarkers of Response to Neoadjuvant Androgen Deprivation in Localised Prostate Cancer. Cancers. 2022; 14(1):166. https://doi.org/10.3390/cancers14010166
Chicago/Turabian StylePechlivanis, Maree, Bethany K. Campbell, Christopher M. Hovens, and Niall M. Corcoran. 2022. "Biomarkers of Response to Neoadjuvant Androgen Deprivation in Localised Prostate Cancer" Cancers 14, no. 1: 166. https://doi.org/10.3390/cancers14010166
APA StylePechlivanis, M., Campbell, B. K., Hovens, C. M., & Corcoran, N. M. (2022). Biomarkers of Response to Neoadjuvant Androgen Deprivation in Localised Prostate Cancer. Cancers, 14(1), 166. https://doi.org/10.3390/cancers14010166