
Neutrophil Extracellular Traps Are Increased in Chronic Myeloid Leukemia and Are Differentially Affected by Tyrosine Kinase Inhibitors

 ,
,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Blood Samples and Isolation of Neutrophils
2.2. BCR-ABL1 Transduced ER-HOXB8 Cell Line
2.3. Treatment with TKIs
2.4. NET Stimulation and Formation Assay
2.5. Immunostaining, Fluorescence Microscopy and NET Quantification
2.6. ROS Production Assays
2.7. Statistical Analysis
3. Results
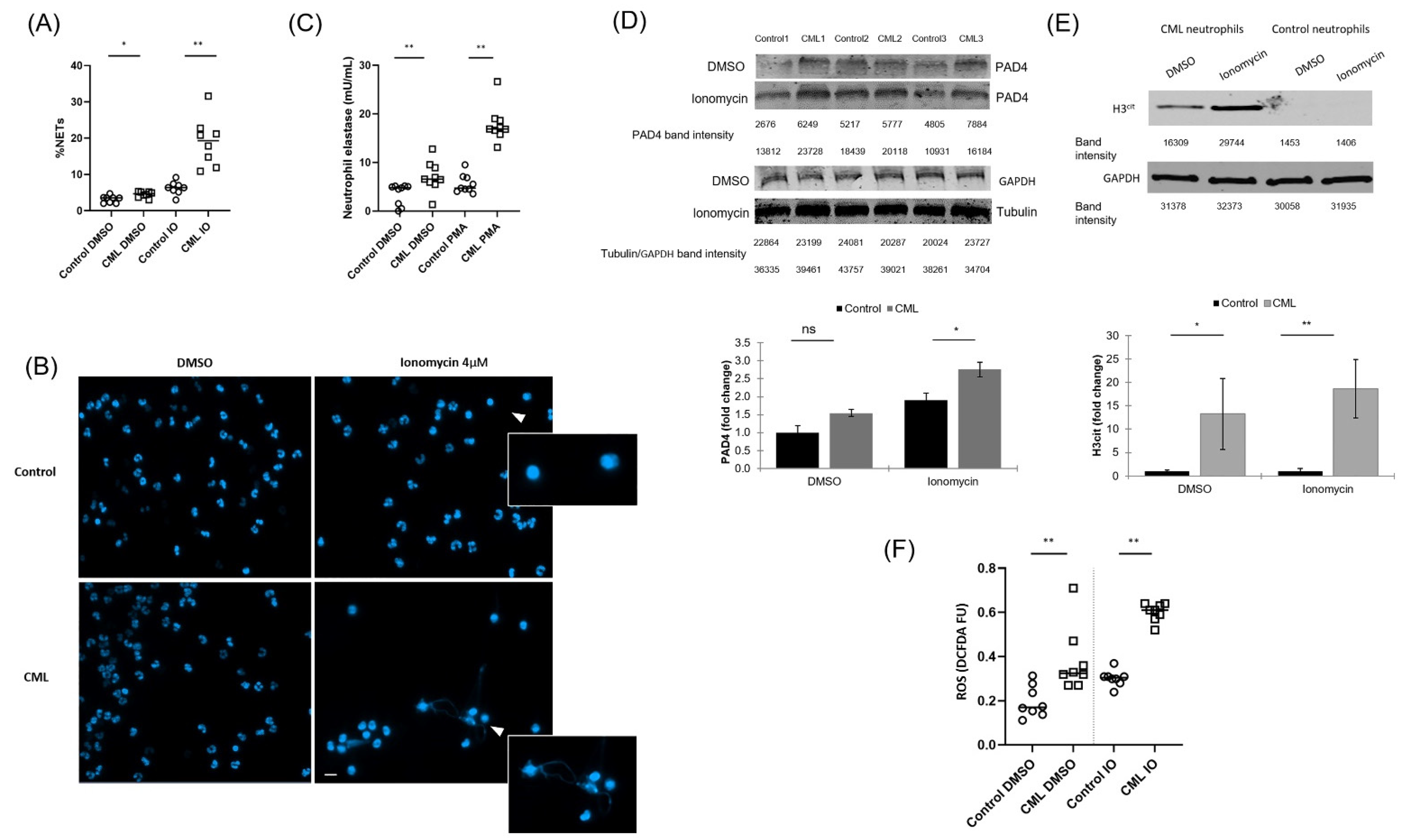
3.1. NETs Are Increased in Neutrophils from Patients with CML
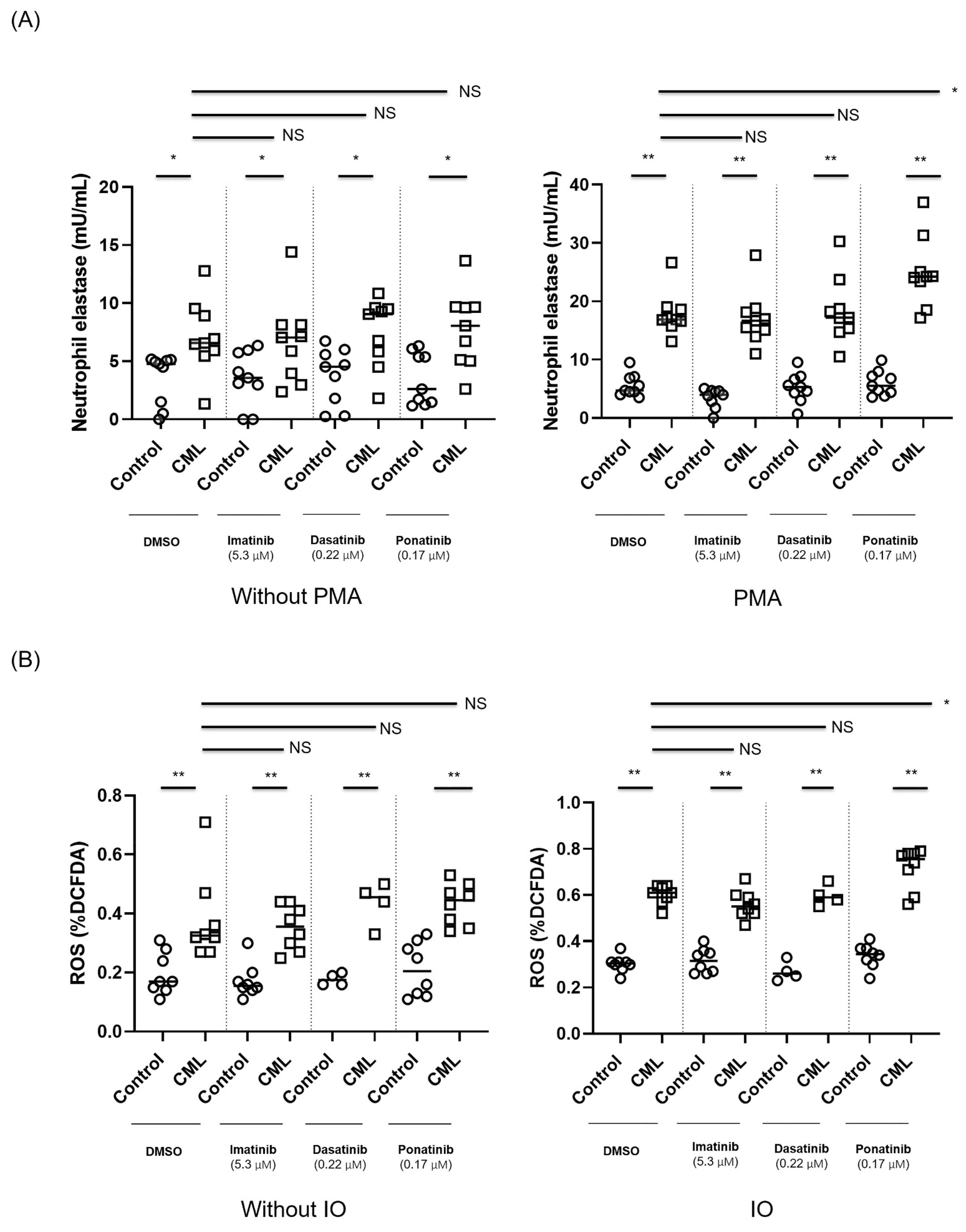
3.2. Ponatinib Augments NET Formation in Neutrophils Derived from Patients with CML
3.3. BCR-ABL1 Transduced ER-HoxB8 Cell Line System Recapitulates NET Phenotype in CML, TKI Effects and Reveals PAD4 Dependency
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raanani, P.; Granot, G.; Ben-Bassat, I. Is cure of chronic myeloid leukemia in the third millennium a down to earth target (ed) or a castle in the air? Cancer Lett. 2014, 352, 21–27. [Google Scholar] [CrossRef]
- Zeng, P.; Schmaier, A. Ponatinib and other CML Tyrosine Kinase Inhibitors in Thrombosis. Int. J. Mol. Sci. 2020, 21, 6556. [Google Scholar] [CrossRef]
- Steegmann, J.L.; Baccarani, M.; Breccia, M.; Casado, L.F.; García-Gutiérrez, V.; Hochhaus, A.; Kim, D.W.; Kim, T.D.; Khoury, H.J.; Le Coutre, P.; et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia 2016, 30, 1648–1671. [Google Scholar] [CrossRef] [Green Version]
- Damrongwatanasuk, R.; Fradley, M.G. Cardiovascular Complications of Targeted Therapies for Chronic Myeloid Leukemia. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 24. [Google Scholar] [CrossRef] [PubMed]
- Gover-Proaktor, A.; Granot, G.; Shapira, S.; Raz, O.; Pasvolsky, O.; Nagler, A.; Lev, D.L.; Inbal, A.; Lubin, I.; Raanani, P.; et al. Ponatinib reduces viability, migration, and functionality of human endothelial cells. Leuk. Lymphoma 2017, 58, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Gover-Proaktor, A.; Granot, G.; Pasmanik-Chor, M.; Pasvolsky, O.; Shapira, S.; Raz, O.; Raanani, P.; Leader, A. Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib differentially affect the vascular molecular pathways and functionality of human endothelial cells. Leuk. Lymphoma 2019, 60, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Granger, V.; Peyneau, M.; Chollet-Martin, S.; de Chaisemartin, L. Neutrophil Extracellular Traps in Autoimmunity and Allergy: Immune Complexes at Work. Front. Immunol. 2019, 10, 2824. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, A.S.; Bardoel, B.W.; Harbort, C.J.; Zychlinsky, A. Induction and quantification of neutrophil extracellular traps. Methods Mol. Biol. 2014, 1124, 307–318. [Google Scholar] [CrossRef]
- Wang, G.G.; Calvo, K.R.; Pasillas, M.P.; Sykes, D.B.; Häcker, H.; Kamps, M.P. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods 2006, 3, 287–293. [Google Scholar] [CrossRef]
- Granot, G.; (Rabin Medical Center, Petah-Tikva, Israel); Partouche, S.; (Rabin Medical Center, Petah-Tikva, Israel); Telerman, A.; (Rabin Medical Center, Petah-Tikva, Israel); Wolach, O.; (Tel Aviv University, Ramat-Aviv, Israel). Personal communication, 2021.
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Kanthi, Y.; Knight, J.S.; Kim, A.H.J. The interplay between neutrophils, complement, and microthrombi in COVID-19. Best Pract. Res. Clin. Rheumatol. 2021, 35, 101661. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, H.; Qu, M.; Nan, K.; Cao, H.; Cata, J.P.; Chen, W.; Miao, C. Review: The Emerging Role of Neutrophil Extracellular Traps in Sepsis and Sepsis-Associated Thrombosis. Front. Cell Infect. Microbiol. 2021, 11, 653228. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [Green Version]
- Antoszewska-Smith, J.; Pawlowska, E.; Blasiak, J. Reactive oxygen species in BCR-ABL1-expressing cells—Relevance to chronic myeloid leukemia. Acta Biochim. Pol. 2017, 64, 1–10. [Google Scholar] [CrossRef]
- Valent, P.; Hadzijusufovic, E.; Hoermann, G.; Füreder, W.; Schernthaner, G.H.; Sperr, W.R.; Kirchmair, R.; Wolf, D. Risk factors and mechanisms contributing to TKI-induced vascular events in patients with CML. Leuk. Res. 2017, 59, 47–54. [Google Scholar] [CrossRef]
- Hamadi, A.; Grigg, A.P.; Dobie, G.; Burbury, K.L.; Schwarer, A.P.; Kwa, F.A.; Jackson, D.E. Ponatinib Tyrosine Kinase Inhibitor Induces a Thromboinflammatory Response. Thromb. Haemost. 2019, 119, 1112–1123. [Google Scholar] [CrossRef]
- Latifi, Y.; Moccetti, F.; Wu, M.; Xie, A.; Packwood, W.; Qi, Y.; Ozawa, K.; Shentu, W.; Brown, E.; Shirai, T.; et al. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood 2019, 133, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkulova, A.; Mitchell, S.C.; Stavrou, E.X.; Forbes, G.L.; Schmaier, A.H. Ponatinib treatment promotes arterial thrombosis and hyperactive platelets. Blood Adv. 2019, 3, 2312–2316. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462.e445. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Hurtado-Nedelec, M.; Csillag-Grange, M.J.; Boussetta, T.; Belambri, S.A.; Fay, M.; Cassinat, B.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. Increased reactive oxygen species production and p47phox phosphorylation in neutrophils from myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica 2013, 98, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Paech, F.; Mingard, C.; Grünig, D.; Abegg, V.F.; Bouitbir, J.; Krähenbühl, S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology 2018, 395, 34–44. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.L.; Wagner, D.D. Peptidylarginine deiminase 4: A nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. Faseb J. 2018, 32, fj201800691R. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telerman, A.; Granot, G.; Leibovitch, C.; Yarchovsky-Dolberg, O.; Shacham-Abulafia, A.; Partouche, S.; Yeshurun, M.; Ellis, M.H.; Raanani, P.; Wolach, O. Neutrophil Extracellular Traps Are Increased in Chronic Myeloid Leukemia and Are Differentially Affected by Tyrosine Kinase Inhibitors. Cancers 2022, 14, 119. https://doi.org/10.3390/cancers14010119
Telerman A, Granot G, Leibovitch C, Yarchovsky-Dolberg O, Shacham-Abulafia A, Partouche S, Yeshurun M, Ellis MH, Raanani P, Wolach O. Neutrophil Extracellular Traps Are Increased in Chronic Myeloid Leukemia and Are Differentially Affected by Tyrosine Kinase Inhibitors. Cancers. 2022; 14(1):119. https://doi.org/10.3390/cancers14010119
Chicago/Turabian StyleTelerman, Alona, Galit Granot, Chiya Leibovitch, Osnat Yarchovsky-Dolberg, Adi Shacham-Abulafia, Shirly Partouche, Moshe Yeshurun, Martin H. Ellis, Pia Raanani, and Ofir Wolach. 2022. "Neutrophil Extracellular Traps Are Increased in Chronic Myeloid Leukemia and Are Differentially Affected by Tyrosine Kinase Inhibitors" Cancers 14, no. 1: 119. https://doi.org/10.3390/cancers14010119
APA StyleTelerman, A., Granot, G., Leibovitch, C., Yarchovsky-Dolberg, O., Shacham-Abulafia, A., Partouche, S., Yeshurun, M., Ellis, M. H., Raanani, P., & Wolach, O. (2022). Neutrophil Extracellular Traps Are Increased in Chronic Myeloid Leukemia and Are Differentially Affected by Tyrosine Kinase Inhibitors. Cancers, 14(1), 119. https://doi.org/10.3390/cancers14010119