
Genetic Analysis Reveals Rare Variants in T-Cell Response Gene MR1 Associated with Poor Overall Survival after Urothelial Cancer Diagnosis


Abstract
Simple Summary
Abstract
1. Introduction
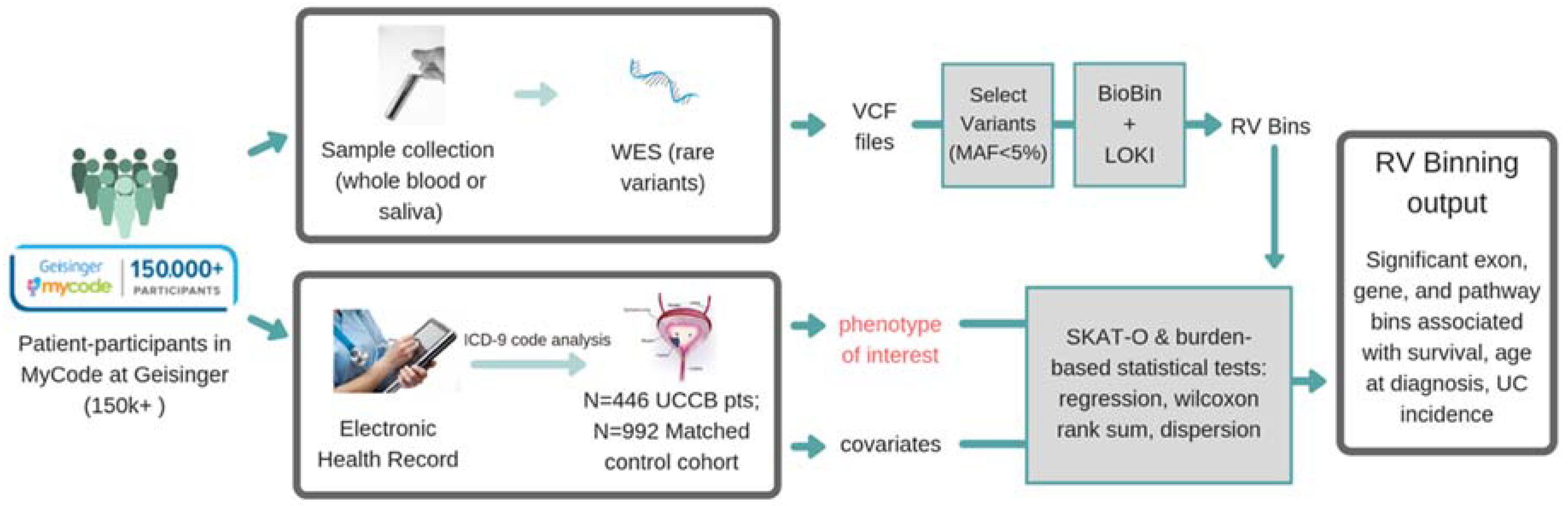
2. Materials and Methods
2.1. Geisinger MyCode/DiscovEHR Study Participants
2.2. DiscovEHR Study Variant Data (SNP Array and Exome Sequencing)
2.3. Knowledge-Driven Binning Approach for Rare Variant Association Analysis
2.4. eQTL Analysis
3. Results
3.1. Phenotypic Data
3.2. Gene-Based Rare Variant Binning Associations with UC Diagnosis Using a Control Group
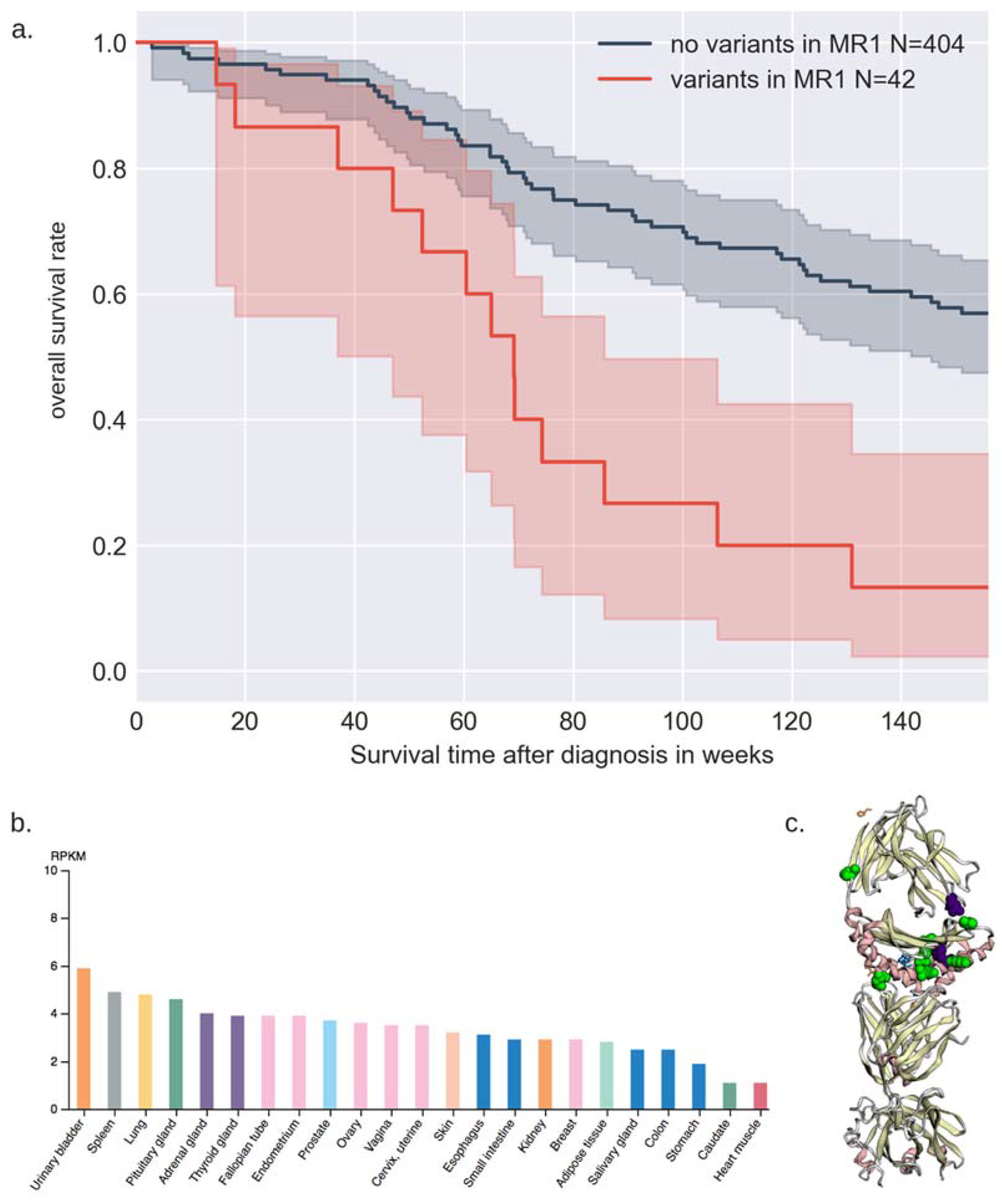
3.3. Gene-Based Binned Rare Variant Associations with Survival and Age of Diagnosis
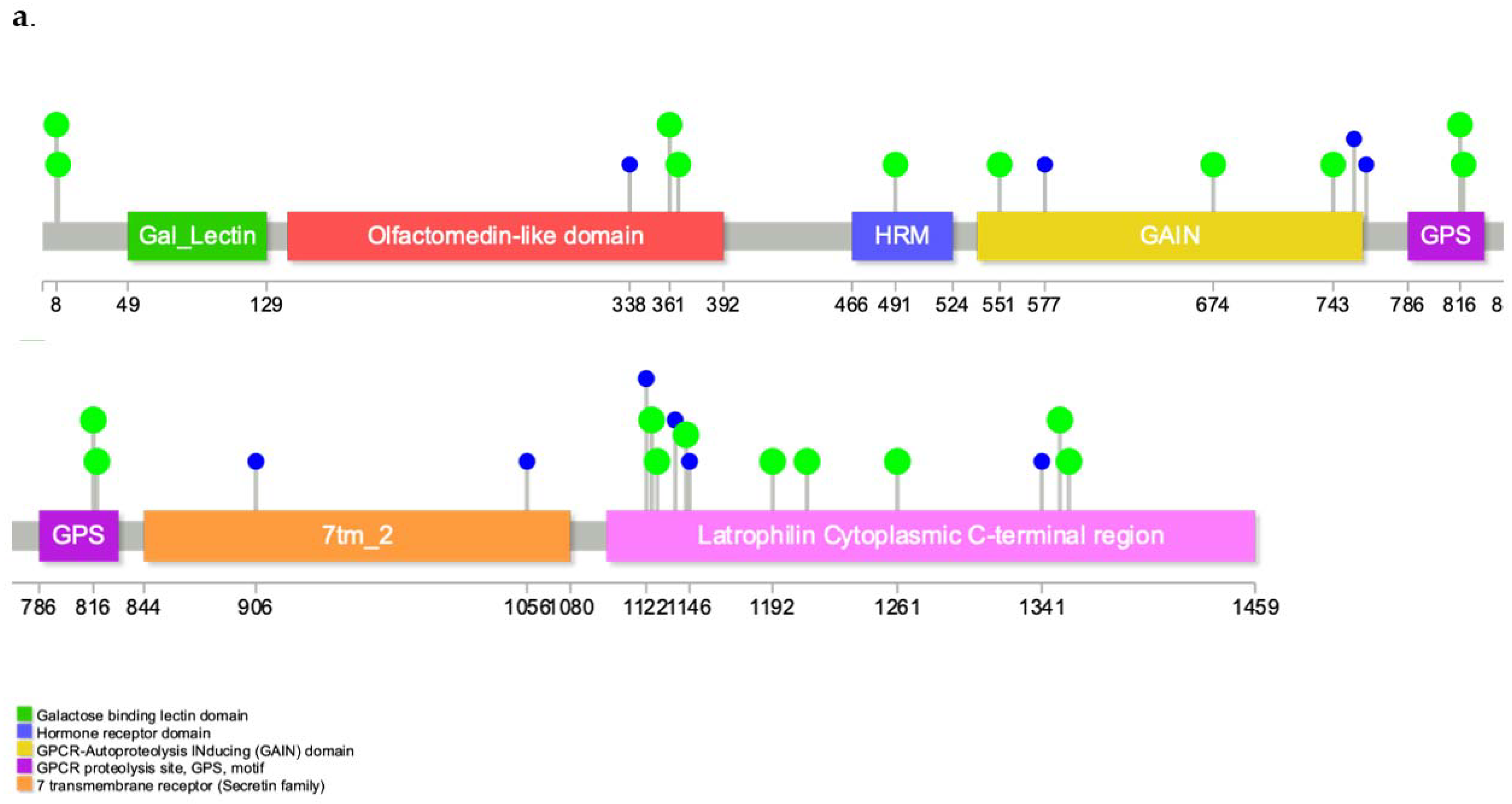
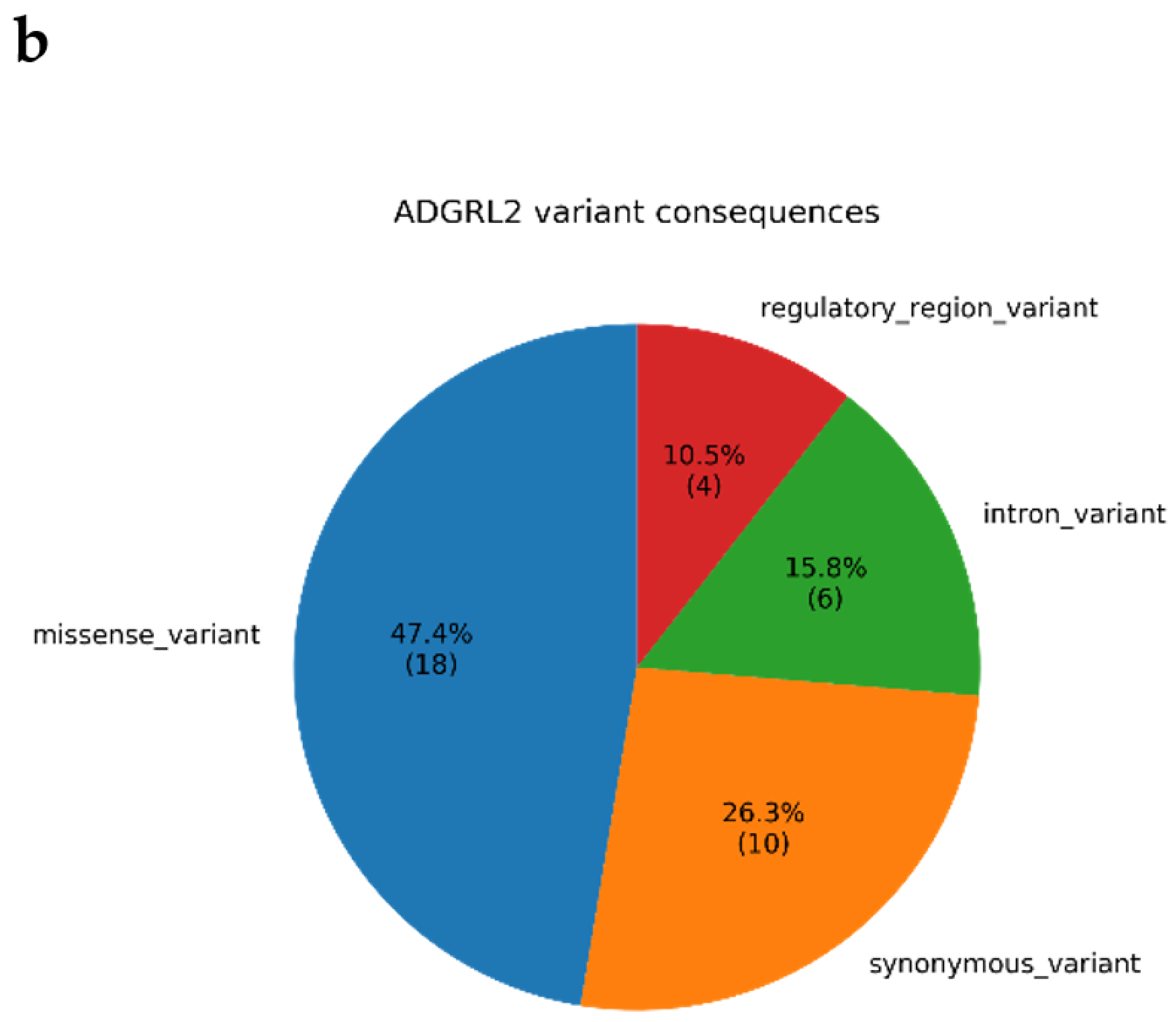
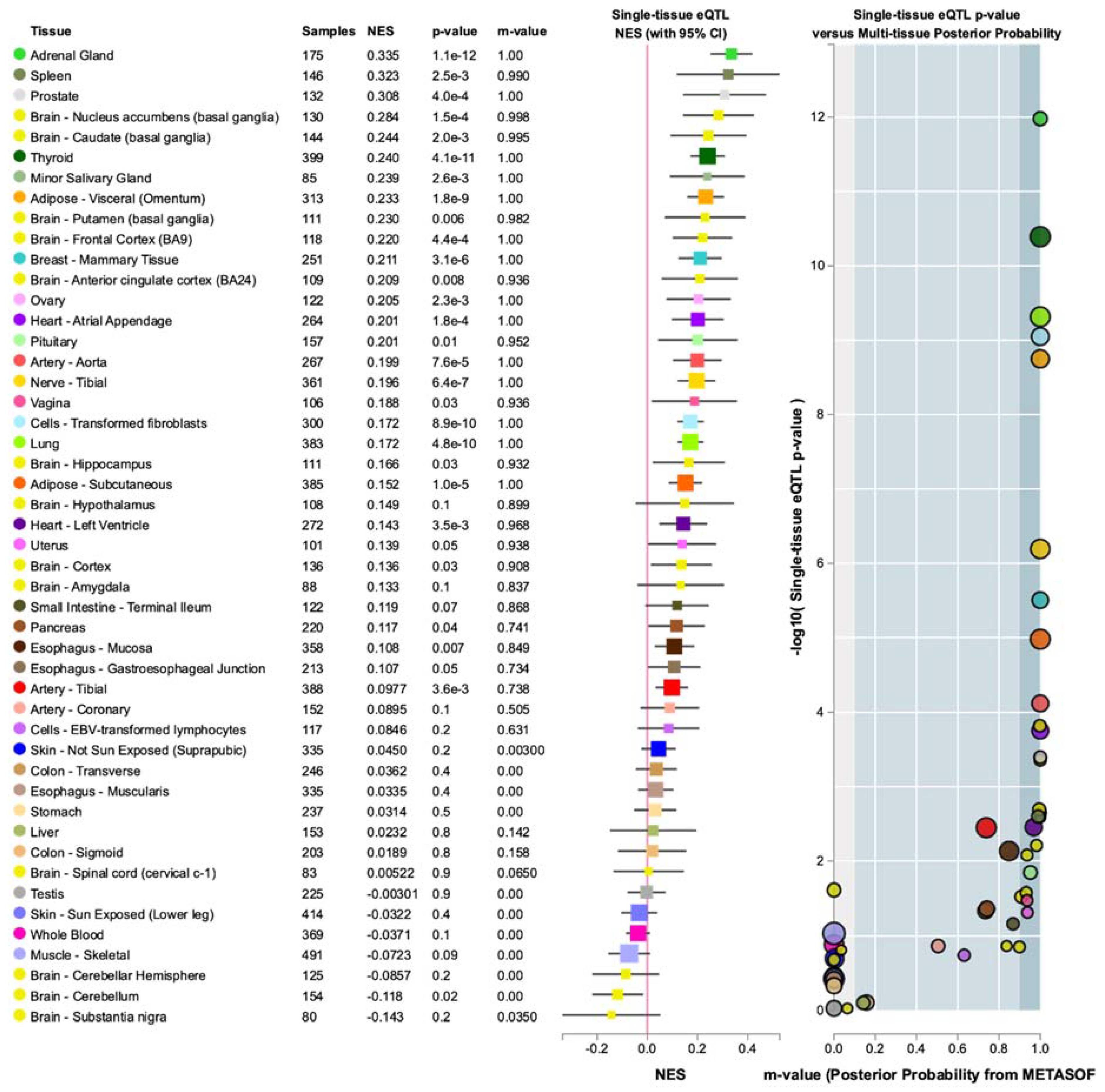
3.4. Annotations of Variants in MR1 and GTEx eQTL Analysis
4. Discussion
4.1. Biological Relevance of ADGRL2
4.2. Biological Relevance of MR1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volanis, D.; Kadiyska, T.; Galanis, A.; Delakas, D.; Logotheti, S.; Zoumpourlis, V. Environmental factors and genetic susceptibility promote urinary bladder cancer. Toxicol. Lett. 2010, 193, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Catto, J.W.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA A Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, Y.; Tsang, E.K.; Davis, J.R.; Damani, F.N.; Chiang, C.; Hess, G.T.; Zappala, Z.; Strober, B.J.; Scott, A.J.; et al. The impact of rare variation on gene expression across tissues. Nature 2017, 550, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Schork, N.J.; Murray, S.S.; Frazer, K.A.; Topol, E.J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 2009, 19, 212–219. [Google Scholar] [CrossRef]
- Lu, C.; Xie, M.; Wendl, M.C.; Wang, J.; McLellan, M.D.; Leiserson, M.D.M.; Huang, K.-l.; Wyczalkowski, M.A.; Jayasinghe, R.; Banerjee, T.; et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun. 2015, 6, 10086. [Google Scholar] [CrossRef]
- Verma, S.S.; Josyula, N.; Verma, A.; Zhang, X.; Veturi, Y.; Dewey, F.E.; Hartzel, D.N.; Lavage, D.R.; Leader, J.; Ritchie, M.D.; et al. Rare variants in drug target genes contributing to complex diseases, phenome-wide. Sci. Rep. 2018, 8, 4624. [Google Scholar] [CrossRef]
- Hart, S.N.; Ellingson, M.S.; Schahl, K.; Vedell, P.T.; Carlson, R.E.; Sinnwell, J.P.; Barman, P.; Sicotte, H.; Eckel-Passow, J.E.; Wang, L.; et al. Determining the frequency of pathogenic germline variants from exome sequencing in patients with castrate-resistant prostate cancer. BMJ Open 2016, 6, e010332. [Google Scholar] [CrossRef]
- Tiao, G.; Improgo, M.R.; Kasar, S.; Poh, W.; Kamburov, A.; Landau, D.A.; Tausch, E.; Taylor-Weiner, A.; Cibulskis, C.; Bahl, S.; et al. Rare germline variants in ATM are associated with chronic lymphocytic leukemia. Leukemia 2017, 31, 2244–2247. [Google Scholar] [CrossRef]
- Teo, M.T.W.; Dyrskjøt, L.; Nsengimana, J.; Buchwald, C.; Snowden, H.; Morgan, J.; Jensen, J.B.; Knowles, M.A.; Taylor, G.; Barrett, J.H.; et al. Next-generation sequencing identifies germline MRE11A variants as markers of radiotherapy outcomes in muscle-invasive bladder cancer. Ann. Oncol. 2014, 25, 877–883. [Google Scholar] [CrossRef]
- Huang, S.; Martin, E.; Kim, S.; Yu, L.; Soudais, C.; Fremont, D.H.; Lantz, O.; Hansen, T.H. MR1 antigen presentation to mucosal-associated invariant T cells was highly conserved in evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 8290–8295. [Google Scholar] [CrossRef] [PubMed]
- Lepore, M.; Kalinichenko, A.; Calogero, S.; Kumar, P.; Paleja, B.; Schmaler, M.; Narang, V.; Zolezzi, F.; Poidinger, M.; Mori, L.; et al. Functionally diverse human T cells recognize non-microbial antigens presented by MR1. eLife 2017, 6, e24476. [Google Scholar] [CrossRef]
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.A.E.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR–Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Im Munology 2020, 21, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; Van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016, 354, aaf6814. [Google Scholar] [CrossRef]
- Basile, A.O.; Wallace, J.R.; Peissig, P.; McCarty, C.A.; Brilliant, M.; Ritchie, M.D. Knowledge Driven Binning and Phewas Analysis in Marshfield Personalized Medicine Research Project Using Biobin. Pac. Symp. Biocomput. 2016, 21, 249–260. [Google Scholar] [PubMed]
- Madsen, B.E.; Browning, S.R. A Groupwise Association Test for Rare Mutations Using a Weighted Sum Statistic. PLoS Genet. 2009, 5, e1000384. [Google Scholar] [CrossRef]
- Lee, S.; Wu, M.C.; Lin, X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics 2012, 13, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Niknafs, N.; Kim, D.; Kim, R.; Diekhans, M.; Ryan, M.; Stenson, P.D.; Cooper, D.N.; Karchin, R. MuPIT interactive: Webserver for mapping variant positions to annotated, interactive 3D structures. Hum. Genet. 2013, 132, 1235–1243. [Google Scholar] [CrossRef]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef] [PubMed]
- Garg, T.; Pinheiro, L.C.; Atoria, C.L.; Donat, S.M.; Weissman, J.S.; Herr, H.W.; Elkin, E.B. Gender disparities in hematuria evaluation and bladder cancer diagnosis: A population based analysis. J. Urol. 2014, 192, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Kubica, P.; Lara-Velazquez, M.; Bam, M.; Sira, S.; Ong, I.; Liu, P.; Priya, R.; Salamat, S.; Brutkiewicz, R.R.; Dey, M. MR1 overexpression correlates with poor clinical prognosis in glioma patients. Neuro Oncol. Adv. 2021. [CrossRef]
- White, G.R.M.; Varley, J.M.; Heighway, J. Genomic structure and expression profile of LPHH1, a 7TM gene variably expressed in breast cancer cell lines11Accession numbers: AJ242517–8, AJ44492–AJ44516. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2000, 1491, 75–92. [Google Scholar] [CrossRef]
- Jeon, M.S.; Song, S.H.; Yun, J.; Kang, J.Y.; Kim, H.P.; Han, S.W.; Kim, T.Y. Aberrant Epigenetic Modifications of LPHN2 Function as a Potential Cisplatin-Specific Biomarker for Human Gastrointestinal Cancer. Cancer Res. Treat. 2016, 48, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Gorski, M.; van der Most, P.J.; Teumer, A.; Chu, A.Y.; Li, M.; Mijatovic, V.; Nolte, I.M.; Cocca, M.; Taliun, D.; Gomez, F.; et al. 1000 Genomes-based meta-analysis identifies 10 novel loci for kidney function. Sci. Rep. 2017, 7, 45040. [Google Scholar] [CrossRef] [PubMed]
- Lowrance, W.T.; Ordoñez, J.; Udaltsova, N.; Russo, P.; Go, A.S. CKD and the risk of incident cancer. J. Am. Soc. Nephrol. 2014, 25, 2327–2334. [Google Scholar] [CrossRef]
- Vezain, M.; Lecuyer, M.; Rubio, M.; Dupé, V.; Ratié, L.; David, V.; Pasquier, L.; Odent, S.; Coutant, S.; Tournier, I.; et al. A de novo variant in ADGRL2 suggests a novel mechanism underlying the previously undescribed association of extreme microcephaly with severely reduced sulcation and rhombencephalosynapsis. Acta Neuropathol. Commun 2018, 6, 109. [Google Scholar] [CrossRef]
- Huang, S.; Gilfillan, S.; Cella, M.; Miley, M.J.; Lantz, O.; Lybarger, L.; Fremont, D.H.; Hansen, T.H. Evidence for MR1 Antigen Presentation to Mucosal-associated Invariant T Cells. J. Biol. Chem. 2005, 280, 21183–21193. [Google Scholar] [CrossRef]
- Yao, T.; Shooshtari, P.; Haeryfar, S.M.M. Leveraging Public Single-Cell and Bulk Transcriptomic Datasets to Delineate MAIT Cell Roles and Phenotypic Characteristics in Human Malignancies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Kawachi, I.; Maldonado, J.; Strader, C.; Gilfillan, S. MR1-restricted V alpha 19i mucosal-associated invariant T cells are innate T cells in the gut lamina propria that provide a rapid and diverse cytokine response. J. Immunol. 2006, 176, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All in Cohort | Males (%) | Females (%) | ||
|---|---|---|---|---|
| Age at Diagnosis | <50 | 21 (4.7%) | 14 (66.7%) | 7 (33.3%) |
| 50–59.9 | 66 (14.8%) | 57 (86.4%) | 9 (13.6%) | |
| 60–69.9 | 135 (30.2%) | 98 (72.5%) | 37 (27.4%) | |
| 70+ | 224 (50.2%) | 184 (82.1%) | 40 (17.9%) | |
| Sex | 446 | 353 (79.1%) | 93 (20.9%) | |
| Race | White | 446 (100%) | ||
| Smoking Status | Smoking | 302 (67.7%) | 257 (85.1%) | 48 (14.9%) |
| Nonsmoking | 144 (32.3%) | 97 (67.4%) | 45 (32.6%) | |
| Grade | Grade I | 42 (9.4%) | 32 (76.2%) | 10 (23.8%) |
| Grade II | 117 (26.2%) | 88 (75.2%) | 29 (24.8%) | |
| Grade III | 29 (6.5%) | 24 (82.8%) | 5 (18.2%) | |
| Grade IV | 233 (52.2%) | 190 (81.5%) | 43 (18.5%) | |
| Other | 25 (5.6%) | 19 (76.0%) | 6 (24.0%) | |
| Stage | Ta | 213 (47.8%) | 163 (76.5%) | 50 (23.5%) |
| Tis | 9 (2%) | 6 (66.7%) | 3 (33.3%) | |
| T1 | 99 (22.2%) | 81 (81.8%) | 18 (18.2%) | |
| T2 | 42 (9.4%) | 34 (81.0%) | 6 (19.0%) | |
| T3 | 17 (3.8%) | 13 (76.4%) | 4 (23.6%) | |
| T4 | 7 (1.6%) | 6 (85.7%) | 1 (14.3%) | |
| Unknown | 59 (13.2%) | 50 (84.7%) | 9 (15.3%) | |
| Vital Status | Alive | 315 (70.6%) | 246 (78.1%) | 69 (21.9%) |
| Deceased | 131 (29.4%) | 107 (81.7%) | 24 (18.3%) | |
| Recurrence | None or Unknown | 425 (95.3%) | 334 (73.1%) | 90 (26.9%) |
| 1+ | 21 (4.7%) | 18 (85.7%) | 3 (14.3%) |
| Bin | Num_Loci | SKAT_Logistic | SKAT-O | FDR |
|---|---|---|---|---|
| ADGRL2 | 59 | 0.000385 | 7.32 × 10−6 | 0.11797 |
| ARHGAP4 | 67 | 0.000129 | 6.53 × 10−5 | 0.460917 |
| INSRR | 70 | 4.01 × 10−5 | 8.58 × 10−5 | 0.460917 |
| EMX2 | 9 | 0.000203 | 0.000203 | 0.59549 |
| PAICS | 19 | 0.000125 | 0.000262 | 0.59549 |
| CHPF | 50 | 0.000118 | 0.00027 | 0.59549 |
| CCDC155 | 35 | 0.000138 | 0.000282 | 0.59549 |
| NTRK1 | 133 | 0.000129 | 0.000307 | 0.59549 |
| GNL2 | 28 | 0.000332 | 0.000332 | 0.59549 |
| Coefficient | eCoefficient | Coefficient SE | z | p | Lower 0.95 | Upper 0.95 | |
|---|---|---|---|---|---|---|---|
| Sex | −0.05 | 0.95 | 0.24 | −0.20 | 0.84 | −0.51 | 0.41 |
| Smoking | −0.17 | 0.85 | 0.22 | −0.77 | 0.44 | −0.60 | 0.26 |
| MR1 | −1.44 | 0.24 | 0.31 | −4.72 | <0.005 | −2.04 | −0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bang, L.; Shivakumar, M.; Garg, T.; Kim, D. Genetic Analysis Reveals Rare Variants in T-Cell Response Gene MR1 Associated with Poor Overall Survival after Urothelial Cancer Diagnosis. Cancers 2021, 13, 1864. https://doi.org/10.3390/cancers13081864
Bang L, Shivakumar M, Garg T, Kim D. Genetic Analysis Reveals Rare Variants in T-Cell Response Gene MR1 Associated with Poor Overall Survival after Urothelial Cancer Diagnosis. Cancers. 2021; 13(8):1864. https://doi.org/10.3390/cancers13081864
Chicago/Turabian StyleBang, Lisa, Manu Shivakumar, Tullika Garg, and Dokyoon Kim. 2021. "Genetic Analysis Reveals Rare Variants in T-Cell Response Gene MR1 Associated with Poor Overall Survival after Urothelial Cancer Diagnosis" Cancers 13, no. 8: 1864. https://doi.org/10.3390/cancers13081864
APA StyleBang, L., Shivakumar, M., Garg, T., & Kim, D. (2021). Genetic Analysis Reveals Rare Variants in T-Cell Response Gene MR1 Associated with Poor Overall Survival after Urothelial Cancer Diagnosis. Cancers, 13(8), 1864. https://doi.org/10.3390/cancers13081864