
Roles of Inflammasomes in Epstein–Barr Virus-Associated Nasopharyngeal Cancer

 , , ,
, , ,
Abstract
Simple Summary
Abstract
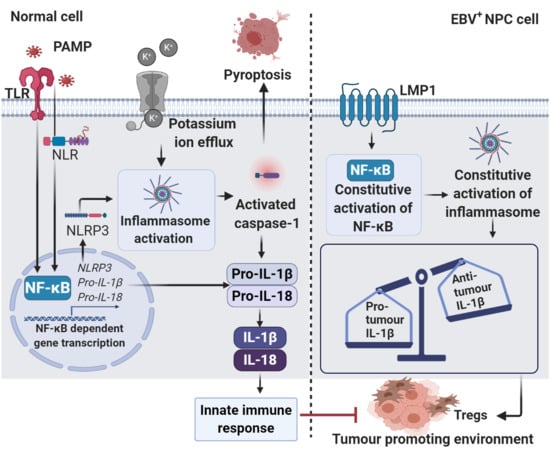
1. Introduction
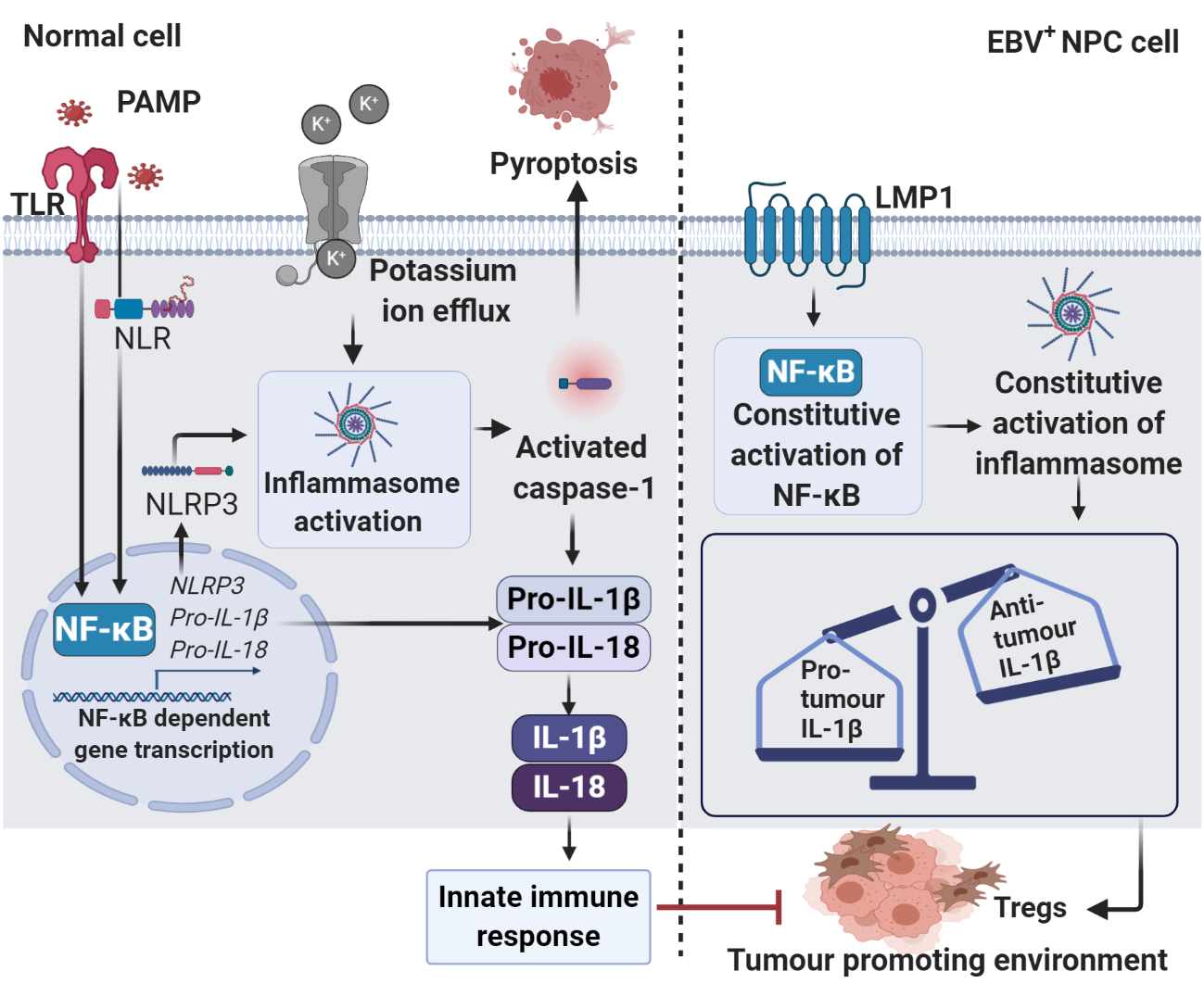
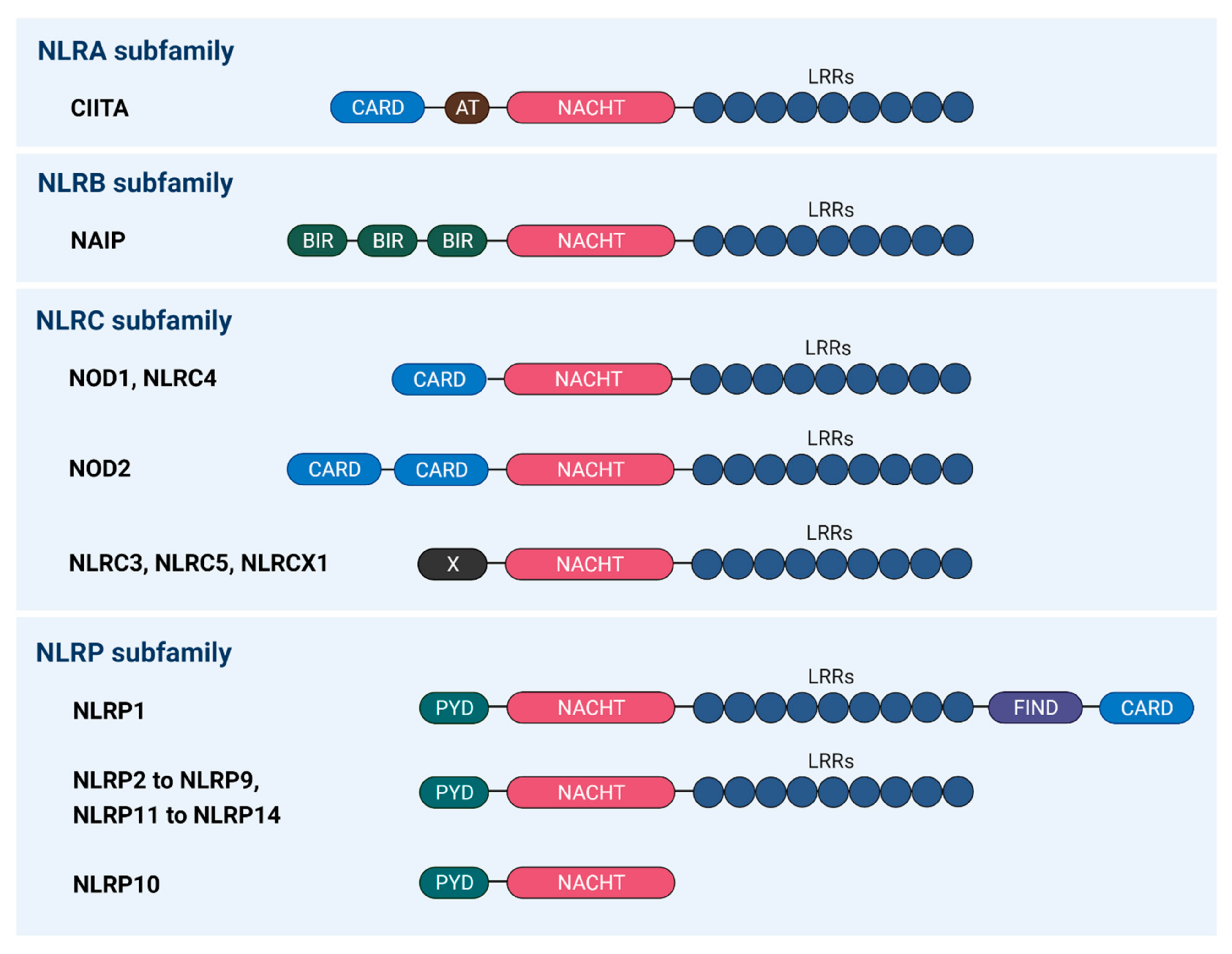
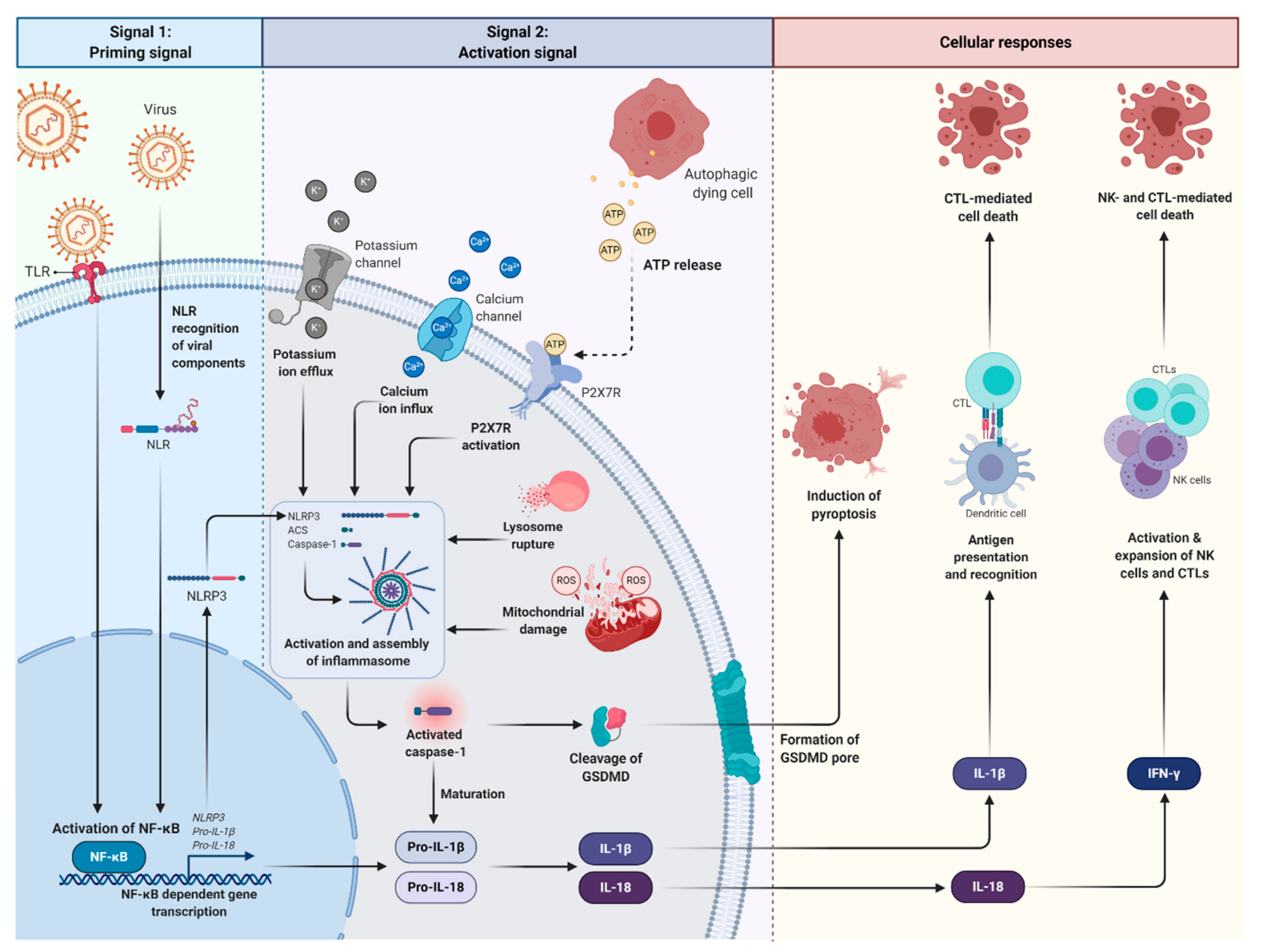
2. Roles of Inflammasomes during Viral Infection
3. Virus Evasion of Inflammasome-Mediated Immune Response
3.1. Inhibition of Inflammasome Assembly and Activation
3.2. Degradation of Inflammasome and Its Components
3.3. Dysregulation of NF-κB Signalling
3.4. Aberrant Production of Pro-Inflammatory Cytokines
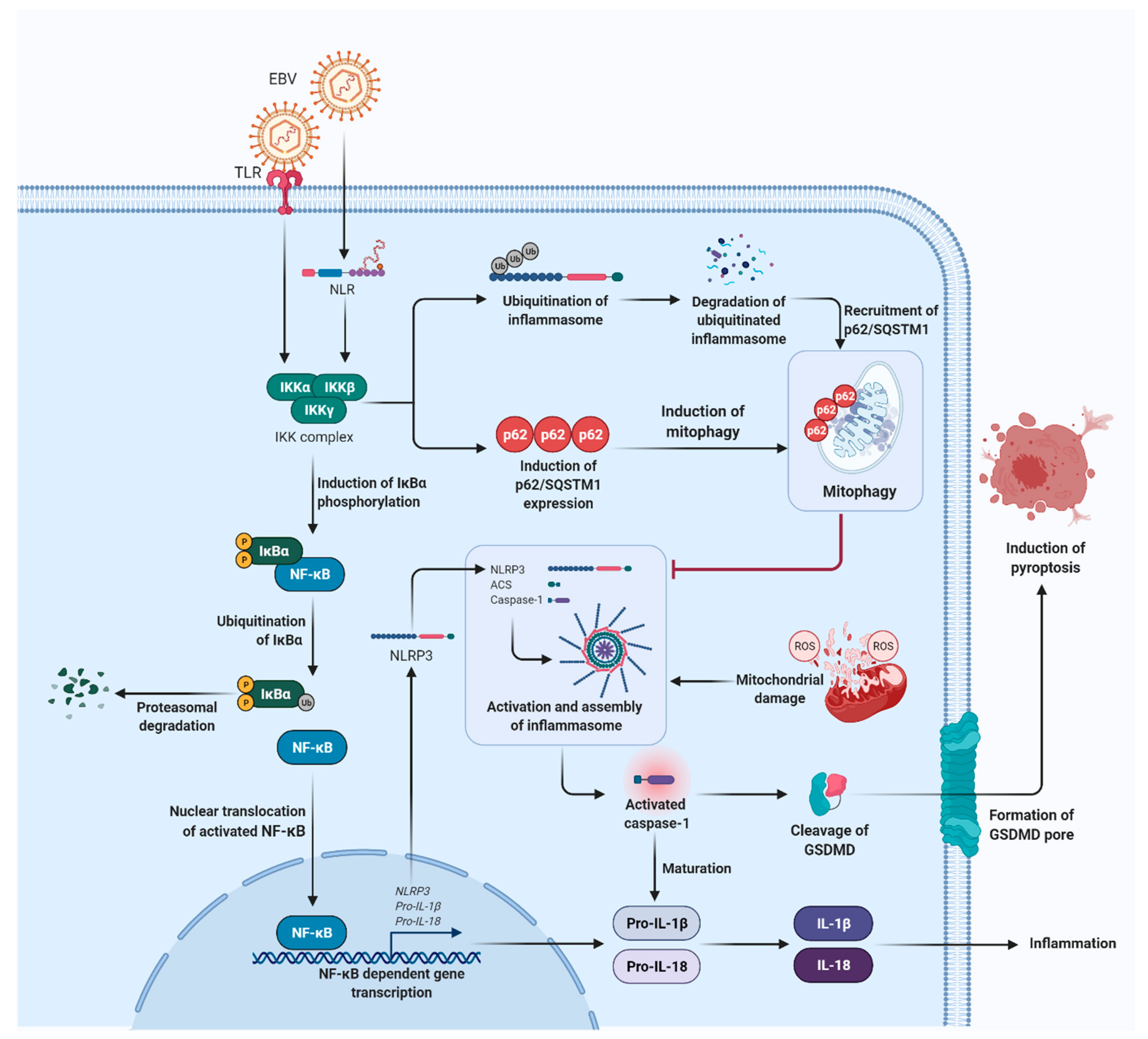
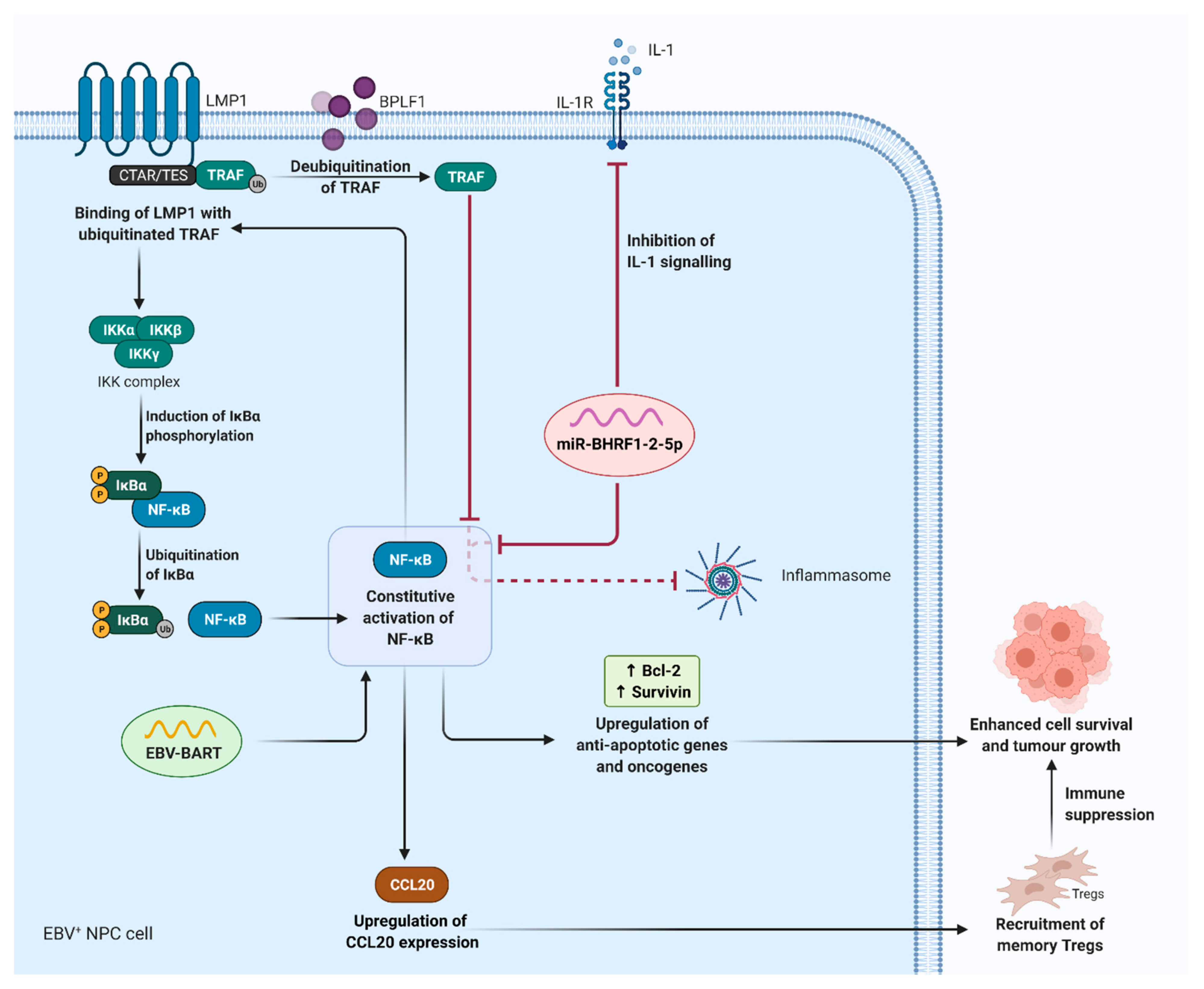
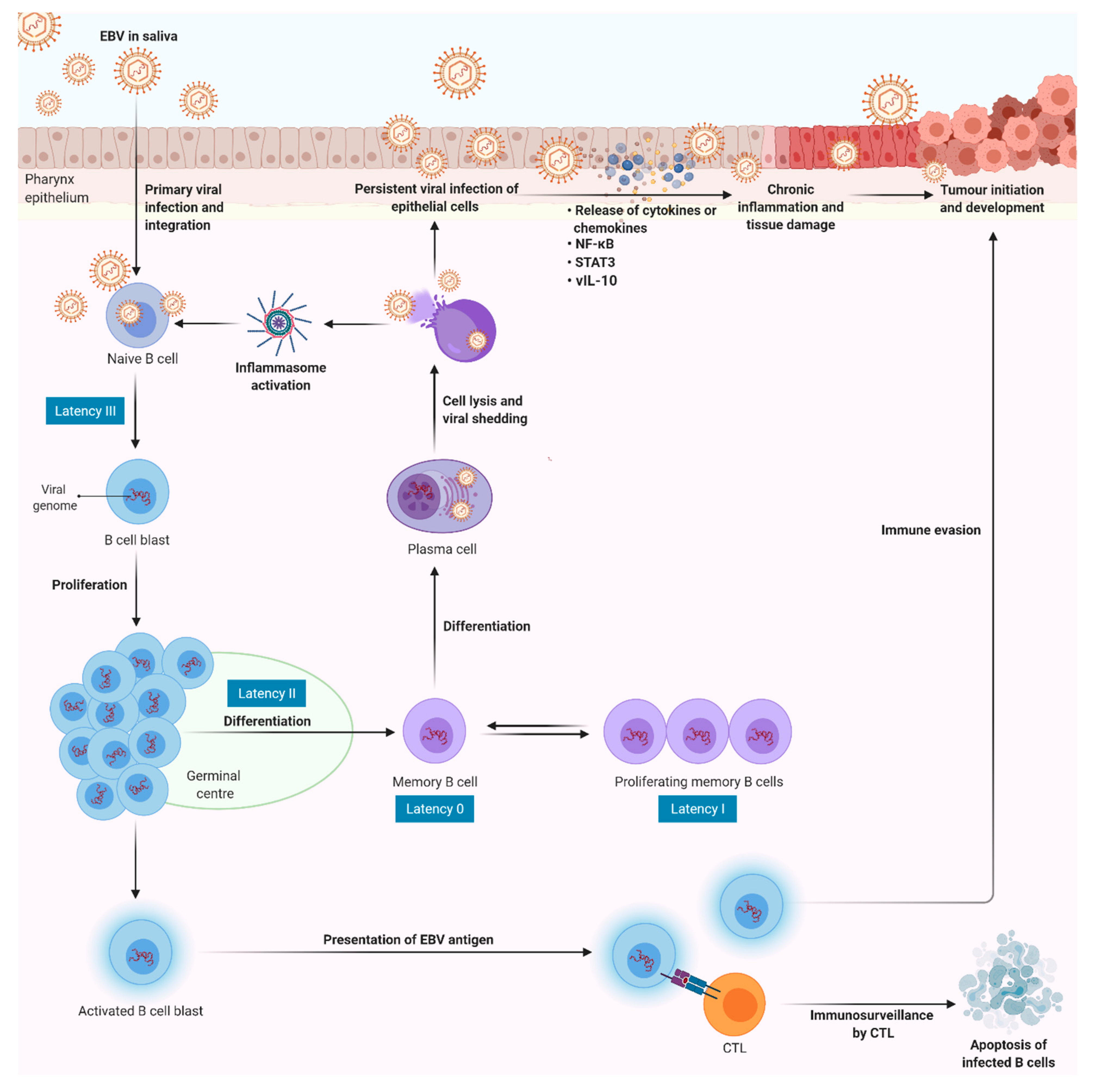
4. Epstein–Barr Virus (EBV)-Induced Sustained Inflammation in NPC Carcinogenesis
5. Roles of Inflammasome in NPC and Other Malignancies
6. Therapeutic Potential of the Inflammasomes in Cancer Immunotherapy
7. Roles of Other Inflammasome Inhibitors in Cancer Therapy
8. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–441. [Google Scholar]
- Massini, G.; Siemer, D.; Hohaus, S. EBV in Hodgkin Lymphoma. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009013. [Google Scholar] [CrossRef]
- Brady, G.; Macarthur, G.J.; Farrell, P.J. Epstein-Barr virus and Burkitt lymphoma. Postgrad. Med J. 2008, 84, 372–377. [Google Scholar] [CrossRef]
- Xiong, F.; Deng, S.; Huang, H.B.; Li, X.Y.; Zhang, W.L.; Liao, Q.J.; Ma, J.; Li, X.L.; Xiong, W.; Li, G.Y.; et al. Effects and mechanisms of innate immune molecules on inhibiting nasopharyngeal carcinoma. Chin. Med J. 2019, 132, 749–752. [Google Scholar] [CrossRef]
- Thompson, L.D. Update on nasopharyngeal carcinoma. Head Neck Pathol. 2007, 1, 81–86. [Google Scholar] [CrossRef]
- Peterson, B.R.; Nelson, B.L. Nonkeratinizing undifferentiated nasopharyngeal carcinoma. Head Neck Pathol. 2013, 7, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.M.; Lui, V.W.Y.; Bruce, J.P.; Pugh, T.J.; Lo, K.W. Translational genomics of nasopharyngeal cancer. Semin. Cancer Biol. 2020, 61, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.F.; Sheng, W.; Cheng, W.M.; Ng, M.H.; Wu, B.H.; Yu, X.; Wei, K.R.; Li, F.G.; Lian, S.F.; Wang, P.P. Incidence and mortality of nasopharyngeal carcinoma: Interim analysis of a cluster randomized controlled screening trial (PRO-NPC-001) in southern China. Ann. Oncol. 2019, 30, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, C.; Pan, L. Nasopharyngeal carcinoma: A review of current updates. Exp. Ther. Med. 2018, 15, 3687–3692. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Q.-Y.; Liu, H.; Tang, L.-Q.; Mai, H.-Q. Emerging treatment options for nasopharyngeal carcinoma. Drug Des. Dev. Ther. 2013, 7, 37. [Google Scholar]
- Bossi, P.; Chan, A.T.; Licitra, L.; Trama, A.; Orlandi, E.; Hui, E.P.; Halámková, J.; Mattheis, S.; Baujat, B.; Hardillo, J. Nasopharyngeal carcinoma: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Pfister, D.G.; Spencer, S.; Adelstein, D.; Adkins, D.; Anzai, Y.; Brizel, D.M.; Bruce, J.Y.; Busse, P.M.; Caudell, J.J.; Cmelak, A.J. Head and neck cancers, version 2.2020, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 873–898. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Y.; Wen, Y.-F.; Guo, L.; Liu, H.; Huang, P.-Y.; Mo, H.-Y.; Li, N.-W.; Xiang, Y.-Q.; Luo, D.-H.; Qiu, F. Concurrent chemoradiotherapy vs radiotherapy alone in stage II nasopharyngeal carcinoma: Phase III randomized trial. J. Natl. Cancer Inst. 2011, 103, 1761–1770. [Google Scholar] [CrossRef]
- Ahn, Y.C. Less is more: Role of additional chemotherapy to concurrent chemoradiotherapy in locoregionally advanced nasopharyngeal cancer management. Radiat. Oncol. J. 2019, 37, 67–72. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, C.; Wang, L.; Yan, F.; Sun, Q.; Ye, Z.; Liu, T.; Fu, Z.; Jiang, Y. Influence of concurrent chemotherapy on locoregionally advanced nasopharyngeal carcinoma treated with neoadjuvant chemotherapy plus intensity-modulated radiotherapy: A retrospective matched analysis. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Thi, H.T.H.; Hong, S. Inflammasome as a therapeutic target for cancer prevention and treatment. J. Cancer Prev. 2017, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kassem, S.; Cleynen, A.; Chrétien, M.-L.; Guillerey, C.; Putz, E.M.; Bald, T.; Förster, I.; Vuckovic, S.; Hill, G.R. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell 2018, 33, 634–648.e635. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; de Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Kantono, M.; Guo, B. Inflammasomes and cancer: The dynamic role of the inflammasome in tumor development. Front. Immunol. 2017, 8, 1132. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Fu, Y.; Tian, D.; Yan, W. The contrasting roles of inflammasomes in cancer. Am. J. Cancer Res. 2018, 8, 566–583. [Google Scholar]
- Mai, C.W.; Kang, Y.B.; Pichika, M.R. Should a Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: Its expression and effects in the ten most common cancers. OncoTargets Ther. 2013, 6, 1573–1587. [Google Scholar] [CrossRef]
- Shaima′a Hamarsheh, R.Z. NLRP3 inflammasome activation in cancer: A double-edged sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome—A key player in antiviral responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef]
- Dagenais, M.; Skeldon, A.; Saleh, M. The inflammasome: In memory of Dr. Jurg Tschopp. Cell Death Differ. 2012, 19, 5–12. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Yi, Y.S. Caspase-11 non-canonical inflammasome: A critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 2017, 152, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Ciążyńska, M.; Bednarski, I.A.; Wódz, K.; Narbutt, J.; Lesiak, A. NLRP1 and NLRP3 inflammasomes as a new approach to skin carcinogenesis. Oncol. Lett. 2020, 19, 1649–1656. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Liu, X.-L.; Zhao, R. Induction of pyroptosis and its implications in cancer management. Front. Oncol. 2019, 9, 971. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, L.; Lu, L.; Liu, J.; Li, X.; Zhao, H.; Hou, Z.; Zheng, B. The Role of Caspase-4 and NLRP1 in MCF7 Cell Pyroptosis Induced by hUCMSC-Secreted Factors. Stem Cells Int. 2020, 2020, 8867115. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.-S. Caspase-11 non-canonical inflammasome: Emerging activator and regulator of infection-mediated inflammatory responses. Int. J. Mol. Sci. 2020, 21, 2736. [Google Scholar] [CrossRef]
- Lupfer, C.; Malik, A.; Kanneganti, T.-D. Inflammasome control of viral infection. Curr. Opin. Virol. 2015, 12, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Gram, A.M.; Frenkel, J.; Ressing, M.E. Inflammasomes and viruses: Cellular defence versus viral offence. J. Gen. Virol. 2012, 93, 2063–2075. [Google Scholar] [CrossRef]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.Y. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Jangra, S.; Yuen, K.-S.; Botelho, M.G.; Jin, D.-Y. Epstein–Barr Virus and Innate Immunity: Friends or Foes? Microorganisms 2019, 7, 183. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Karan, D. Inflammasomes: Emerging central players in cancer immunology and immunotherapy. Front. Immunol. 2018, 9, 3028. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and Mitophagy Connection in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef]
- Chen, L.C.; Wang, L.J.; Tsang, N.M.; Ojcius, D.M.; Chen, C.C.; OuYang, C.N.; Hsueh, C.; Liang, Y.; Chang, K.P.; Chen, C.C. Tumour inflammasome-derived IL-1β recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma. EMBO Mol. Med. 2012, 4, 1276–1293. [Google Scholar] [CrossRef]
- Lee, C.; Do, H.T.T.; Her, J.; Kim, Y.; Seo, D.; Rhee, I. Inflammasome as a promising therapeutic target for cancer. Life Sci. 2019, 231, 116593. [Google Scholar] [CrossRef]
- Couillin, I.; Gombault, A.; Baron, L. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 2013, 3, 414. [Google Scholar]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Di Virgilio, F. The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol. Rev. 2013, 65, 872–905. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.B.; Netea, M.G.; Dinarello, C.A. Interleukin-1β in innate inflammation, autophagy and immunity. Semin. Immunol. 2013, 25, 416–424. [Google Scholar] [CrossRef]
- Nambu, A.; Nakae, S.; Iwakura, Y. IL-1β, but not IL-1α, is required for antigen-specific T cell activation and the induction of local inflammation in the delayed-type hypersensitivity responses. Int. Immunol. 2006, 18, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sasson, S.Z.; Wang, K.; Cohen, J.; Paul, W.E. IL-1beta strikingly enhances antigen-driven CD4 and CD8 T-cell responses. Cold Spring Harb. Symp. Quant. Biol. 2013, 78, 117–124. [Google Scholar] [CrossRef]
- Ramos-Junior, E.S.; Morandini, A.C. Gasdermin: A new player to the inflammasome game. Biomed. J. 2017, 40, 313–316. [Google Scholar] [CrossRef]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Müller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Strittmatter, G.E.; Garstkiewicz, M.; Sand, J.; Beer, H.-D. Caspase-1: The inflammasome and beyond. Innate immunity 2014, 20, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Condotta, S.A.; Richer, M.J. The immune battlefield: The impact of inflammatory cytokines on CD8 T-cell immunity. PLoS Pathog. 2017, 13, e1006618. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, S.; Cao, M.; Fan, X.; Yang, T.; Huang, Y.; Song, X.; Li, Y.; Ye, L.; Shen, N. Antigen-specific CD8 T cell feedback activates NLRP3 inflammasome in antigen-presenting cells through perforin. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y. A critical role of NLRP3 inflammasome in boosting antigen-specific immunity. J. Immunol. 2018, 200 (Suppl. 1), 111.3. [Google Scholar]
- Yao, Y.; Wang, Y.; Chen, F.; Huang, Y.; Zhu, S.; Leng, Q.; Wang, H.; Shi, Y.; Qian, Y. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 2012, 22, 836–847. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Modulation of inflammasome pathways by bacterial and viral pathogens. J. Immunol. 2011, 187, 597–602. [Google Scholar] [CrossRef]
- Komatsu, T.; Tanaka, Y.; Kitagawa, Y.; Koide, N.; Naiki, Y.; Morita, N.; Gotoh, B.; Yokochi, T. Sendai Virus V Protein Inhibits the Secretion of Interleukin-1beta by Preventing NLRP3 Inflammasome Assembly. J. Virol. 2018, 92, e00842-18. [Google Scholar] [CrossRef] [PubMed]
- Le, H.T.; Harton, J.A. Pyrin-and CARD-only proteins as regulators of NLR functions. Front. Immunol. 2013, 4, 275. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, G.; León-Juárez, M.; García-Cordero, J.; Meza-Sánchez, D.E.; Cedillo-Barrón, L. Inflammasomes and its importance in viral infections. Immunol. Res. 2016, 64, 1101–1117. [Google Scholar] [CrossRef]
- de Almeida, L.; Khare, S.; Misharin, A.V.; Patel, R.; Ratsimandresy, R.A.; Wallin, M.C.; Perlman, H.; Greaves, D.R.; Hoffman, H.M.; Dorfleutner, A. The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 2015, 43, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Ratsimandresy, R.A.; Chu, L.H.; Khare, S.; de Almeida, L.; Gangopadhyay, A.; Indramohan, M.; Misharin, A.V.; Greaves, D.R.; Perlman, H.; Dorfleutner, A. The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Johnston, J.B.; Barrett, J.W.; Nazarian, S.H.; Goodwin, M.; Ricuttio, D.; Wang, G.; McFadden, G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 2005, 23, 587–598. [Google Scholar] [CrossRef] [PubMed]
- de Castro-Jorge, L.A.; de Carvalho, R.V.H.; Klein, T.M.; Hiroki, C.H.; Lopes, A.H.; Guimarães, R.M.; Fumagalli, M.J.; Floriano, V.G.; Agostinho, M.R.; Slhessarenko, R.D. The NLRP3 inflammasome is involved with the pathogenesis of Mayaro virus. PLoS Pathog. 2019, 15, e1007934. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.-A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O′Neill, L.A.J.; Masters, S.L. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 2020, 287, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.; Zhang, Q.; Liu, W.; Jia, Y.; Ai, S.; Wang, T.; Wang, W.; Pan, P.; Yang, G.; Xiang, Q.; et al. Cullin1 binds and promotes NLRP3 ubiquitination to repress systematic inflammasome activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 5793–5807. [Google Scholar] [CrossRef]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lear, T.B.; Jerome, J.A.; Rajbhandari, S.; Snavely, C.A.; Gulick, D.L.; Gibson, K.F.; Zou, C.; Chen, B.B.; Mallampalli, R.K. Lipopolysaccharide Primes the NALP3 Inflammasome by Inhibiting Its Ubiquitination and Degradation Mediated by the SCFFBXL2 E3 Ligase. J. Biol. Chem. 2015, 290, 18124–18133. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Karasawa, T.; Tago, K.; Kimura, H.; Kamata, R.; Usui-Kawanishi, F.; Watanabe, S.; Ohta, S.; Funakoshi-Tago, M.; Yanagisawa, K.; et al. ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J. Immunol. 2017, 199, 3614–3622. [Google Scholar] [CrossRef]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Soby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef]
- Wang, H.; Lei, X.; Xiao, X.; Yang, C.; Lu, W.; Huang, Z.; Leng, Q.; Jin, Q.; He, B.; Meng, G. Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep. 2015, 12, 42–48. [Google Scholar] [CrossRef]
- Shil, N.K.; Pokharel, S.M.; Banerjee, A.K.; Hoffman, M.; Bose, S. Inflammasome Antagonism by Human Parainfluenza Virus Type 3 C Protein. J. Virol. 2018, 92, e01776-17. [Google Scholar] [CrossRef]
- Niebler, M.; Qian, X.; Höfler, D.; Kogosov, V.; Kaewprag, J.; Kaufmann, A.M.; Ly, R.; Böhmer, G.; Zawatzky, R.; Rösl, F. Post-translational control of IL-1β via the human papillomavirus type 16 E6 oncoprotein: A novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog. 2013, 9, e1003536. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, Z.; Xiao, X.; Qi, J.; He, B.; Wang, J. Enterovirus 71 Inhibits Pyroptosis through Cleavage of Gasdermin, D. J. Virol. 2017, 91, e01069-17. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Zeligs, K.P.; Neuman, M.K.; Annunziata, C.M. Molecular Pathways: The Balance between Cancer and the Immune System Challenges the Therapeutic Specificity of Targeting Nuclear Factor-kappaB Signaling for Cancer Treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4302–4308. [Google Scholar] [CrossRef]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory kappa B Kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-κB and IκB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Poudel, B.; Gurung, P. An update on cell intrinsic negative regulators of the NLRP3 inflammasome. J. Leukoc. Biol. 2018, 103, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Chung, G.T.Y.; Lou, W.P.K.; Chow, C.; To, K.F.; Choy, K.W.; Leung, A.W.C.; Tong, C.Y.K.; Yuen, J.W.F.; Ko, C.W.; Yip, T.T.C. Constitutive activation of distinct NF-κB signals in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 311–322. [Google Scholar] [CrossRef]
- Ersing, I.; Bernhardt, K.; Gewurz, B.E. NF-κB and IRF7 pathway activation by Epstein-Barr virus latent membrane protein 1. Viruses 2013, 5, 1587–1606. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.; Keck, F.; Bailey, C.; Narayanan, A. The role of the IKK complex in viral infections. Pathog. Dis. 2014, 72, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Lin, Y.C.; Kim, J.; You, L.; Xu, Z.; He, B.; Jablons, D.M. Nasopharyngeal carcinoma—review of the molecular mechanisms of tumorigenesis. Head Neck J. Sci. Spec. Head Neck 2008, 30, 946–963. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C.; Cesarman, E. NF-κB as a target for oncogenic viruses. In NF-kB in Health and Disease; Springer: New York, NY, USA, 2010; pp. 197–244. [Google Scholar]
- van Gent, M.; Braem, S.G.E.; de Jong, A.; Delagic, N.; Peeters, J.G.C.; Boer, I.G.J.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef]
- Saito, S.; Murata, T.; Kanda, T.; Isomura, H.; Narita, Y.; Sugimoto, A.; Kawashima, D.; Tsurumi, T. Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-kappaB signaling during productive replication. J. Virol. 2013, 87, 4060–4070. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, R.J.A.; Tong, S.; Zong, J.; Chen, Y.; Tsao, S.-W.; Pan, J.; Chen, H. NF-κB Signaling Regulates Epstein–Barr Virus BamHI-Q-Driven EBNA1 Expression. Cancers 2018, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, R.J.; Tong, S.; Zhang, G.; Zong, J.; Chen, Y.; Jin, D.Y.; Chen, M.R.; Pan, J.; Chen, H. NF-kappaB Signaling Regulates Expression of Epstein-Barr Virus BART MicroRNAs and Long Noncoding RNAs in Nasopharyngeal Carcinoma. J. Virol. 2016, 90, 6475–6488. [Google Scholar] [CrossRef] [PubMed]
- Skinner, C.M.; Ivanov, N.S.; Barr, S.A.; Chen, Y.; Skalsky, R.L. An Epstein-Barr Virus MicroRNA Blocks Interleukin-1 (IL-1) Signaling by Targeting IL-1 Receptor 1. J. Virol. 2017, 91, e00530-17. [Google Scholar] [CrossRef]
- Yue, W.; Zhu, M.; Zuo, L.; Xin, S.; Zhang, J.; Liu, L.; Li, S.; Dang, W.; Zhang, S.; Xie, Y. Early pattern of Epstein-Barr virus infection in gastric epithelial cells by “cell-in-cell”. Virol. Sin. 2019, 34, 253–261. [Google Scholar] [CrossRef]
- Poeck, H.; Ruland, J. From virus to inflammation: Mechanisms of RIG-I-induced IL-1β production. Eur. J. Cell Biol. 2012, 91, 59–64. [Google Scholar] [CrossRef]
- Dinarello, C.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Inoue, K.; Adachi-Takasawa, K.; Takada, K. Epstein—Barr virus RNA confers resistance to interferon-α-induced apoptosis in Burkitt’s lymphoma. EMBO J. 2002, 21, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Singh, V.V.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chikoti, L.; Lu, J.; Everly, D.; Chandran, B. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J. Virol. 2013, 87, 8606–8623. [Google Scholar] [CrossRef]
- Bodaghi, B.; Jones, T.R.; Zipeto, D.; Vita, C.; Sun, L.; Laurent, L.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Michelson, S. Chemokine sequestration by viral chemoreceptors as a novel viral escape strategy: Withdrawal of chemokines from the environment of cytomegalovirus-infected cells. J. Exp. Med. 1998, 188, 855–866. [Google Scholar] [CrossRef]
- Chen, L.; Li, C.; Peng, Z.; Zhao, J.; Gong, G.; Tan, D. miR-197 Expression in Peripheral Blood Mononuclear Cells from Hepatitis B Virus-Infected Patients. Gut Liver 2013, 7, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Bae, Y.S.; Kim, H.R.; Kim, Y.S.; Kim, D.J.; Cho, B.J.; Yang, H.-K.; Hwang, Y.-I.; Kim, K.J.; Park, H.S. Interleukin-18 increases metastasis and immune escape of stomach cancer via the downregulation of CD70 and maintenance of CD44. Carcinogenesis 2009, 30, 1987–1996. [Google Scholar] [CrossRef]
- Terme, M.; Ullrich, E.; Aymeric, L.; Meinhardt, K.; Desbois, M.; Delahaye, N.; Viaud, S.; Ryffel, B.; Yagita, H.; Kaplanski, G.; et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 2011, 71, 5393–5399. [Google Scholar] [CrossRef] [PubMed]
- Liou, A.K.-F.; Soon, G.; Tan, L.; Peng, Y.; Cher, B.M.; Goh, B.C.; Wang, S.; Lim, C.M. Elevated IL18 levels in Nasopharyngeal carcinoma induced PD-1 expression on NK cells in TILS leading to poor prognosis. Oral Oncol. 2020, 104, 104616. [Google Scholar] [CrossRef]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007, 67, 10019–10026. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.P.; Alcami, A. Expression of secreted cytokine and chemokine inhibitors by ectromelia virus. J. Virol. 2000, 74, 8460–8471. [Google Scholar] [CrossRef][Green Version]
- Balahura, L.R.; Selaru, A.; Dinescu, S.; Costache, M. Inflammation and Inflammasomes: Pros and Cons in Tumorigenesis. J. Immunol. Res. 2020, 2020, 1–15. [Google Scholar] [CrossRef]
- Abdulamir, A.S.; Hafidh, R.R.; Abdulmuhaimen, N.; Abubakar, F.; Abbas, K.A. The distinctive profile of risk factors of nasopharyngeal carcinoma in comparison with other head and neck cancer types. BMC Public Health 2008, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.B. Viruses and human cancer. Yale J. Biol. Med. 2006, 79, 115–122. [Google Scholar]
- Thorley-Lawson, D.A. EBV persistence—Introducing the virus. Epstein Barr Virus 2015, 1, 151–209. [Google Scholar]
- Shannon-Lowe, C.; Rowe, M. Epstein-Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 2011, 7, e1001338. [Google Scholar] [CrossRef] [PubMed]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The interplay between Epstein–Barr virus and B lymphocytes: Implications for infection, immunity, and disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef]
- Hochberg, D.; Souza, T.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Acute infection with Epstein-Barr virus targets and overwhelms the peripheral memory B-cell compartment with resting, latently infected cells. J. Virol. 2004, 78, 5194–5204. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.D.; Neuhierl, B.; Baldwin, G.; Rickinson, A.B.; Delecluse, H.J. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Albanese, M.; Tagawa, T.; Bouvet, M.; Maliqi, L.; Lutter, D.; Hoser, J.; Hastreiter, M.; Hayes, M.; Sugden, B.; Martin, L. Epstein–Barr virus microRNAs reduce immune surveillance by virus-specific CD8 T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6467–E6475. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Read, S.A.; Douglas, M.W. Virus induced inflammation and cancer development. Cancer Lett. 2014, 345, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.M.; Goldbach-Mansky, R.; Bhaduri-McIntosh, S. A promiscuous inflammasome sparks replication of a common tumor virus. Proc. Natl. Acad. Sci. USA 2020, 117, 1722–1730. [Google Scholar] [CrossRef]
- Li, Z.; Duan, Y.; Cheng, S.; Chen, Y.; Hu, Y.; Zhang, L.; He, J.; Liao, Q.; Yang, L.; Sun, L.Q. EBV-encoded RNA via TLR3 induces inflammation in nasopharyngeal carcinoma. Oncotarget 2015, 6, 24291–24303. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yeh, T.H.; Lai, H.C.; Wu, S.Y.; Su, I.J.; Takada, K.; Chang, Y. Epstein-Barr virus Zta-induced immunomodulators from nasopharyngeal carcinoma cells upregulate interleukin-10 production from monocytes. J. Virol. 2011, 85, 7333–7342. [Google Scholar] [CrossRef]
- Hsu, M.; Wu, S.Y.; Chang, S.S.; Su, I.J.; Tsai, C.H.; Lai, S.J.; Shiau, A.L.; Takada, K.; Chang, Y. Epstein-Barr virus lytic transactivator Zta enhances chemotactic activity through induction of interleukin-8 in nasopharyngeal carcinoma cells. J. Virol. 2008, 82, 3679–3688. [Google Scholar] [CrossRef]
- Slobedman, B.; Barry, P.A.; Spencer, J.V.; Avdic, S.; Abendroth, A. Virus-encoded homologs of cellular interleukin-10 and their control of host immune function. J. Virol. 2009, 83, 9618–9629. [Google Scholar] [CrossRef]
- Salek-Ardakani, S.; Arrand, J.R.; Mackett, M. Epstein–Barr virus encoded interleukin-10 inhibits HLA-class I, ICAM-1, and B7 expression on human monocytes: Implications for immune evasion by EBV. Virology 2002, 304, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.M.; Avia, M.; Martín, V.; Sevilla, N. IL-10: A multifunctional cytokine in viral infections. J. Immunol. Res. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Hsu, D.H.; de Waal-Malefyt, R.; Fiorentino, D.F.; Dang, M.N.; Vieira, P.; de Vries, J.; Spits, H.; Mosmann, T.R.; Moore, K.W. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science 1990, 250, 830–832. [Google Scholar] [CrossRef] [PubMed]
- Schaeffner, M.; Mrozek-Gorska, P.; Buschle, A.; Woellmer, A.; Tagawa, T.; Cernilogar, F.M.; Schotta, G.; Krietenstein, N.; Lieleg, C.; Korber, P.; et al. BZLF1 interacts with chromatin remodelers promoting escape from latent infections with EBV. Life Sci. Alliance 2019, 2, e201800108. [Google Scholar] [CrossRef]
- Lu, J.; Chua, H.H.; Chen, S.Y.; Chen, J.Y.; Tsai, C.H. Regulation of matrix metalloproteinase-1 by Epstein-Barr virus proteins. Cancer Res. 2003, 63, 256–262. [Google Scholar] [PubMed]
- Yoshizaki, T.; Sato, H.; Murono, S.; Pagano, J.S.; Furukawa, M. Matrix metalloproteinase 9 is induced by the Epstein–Barr virus BZLF1 transactivator. Clin. Exp. Metastasis 1999, 17, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Jochum, S.; Moosmann, A.; Lang, S.; Hammerschmidt, W.; Zeidler, R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012, 8, e1002704. [Google Scholar] [CrossRef]
- Croft, N.P.; Shannon-Lowe, C.; Bell, A.I.; Horst, D.; Kremmer, E.; Ressing, M.E.; Wiertz, E.J.H.J.; Middeldorp, J.M.; Rowe, M.; Rickinson, A.B. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog. 2009, 5, e1000490. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 beta—a friend or foe in malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, Q.; Feng, X.; Zhang, R.; Li, J.; Chen, F. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer 2018, 18, 500. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Huang, H.; Qiu, Z.; Li, H.; Tan, J.; Ren, G.; Wang, X. NLRP1 overexpression is correlated with the tumorigenesis and proliferation of human breast tumor. BioMed Res. Int. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, Y. Expression of Caspase-1 in breast cancer tissues and its effects on cell proliferation, apoptosis and invasion. Oncol. Lett. 2018, 15, 6431–6435. [Google Scholar] [CrossRef]
- Wang, Y.; Kong, H.; Zeng, X.; Liu, W.; Wang, Z.; Yan, X.; Wang, H.; Xie, W. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol. Rep. 2016, 35, 2053–2064. [Google Scholar] [CrossRef]
- Lasithiotaki, I.; Tsitoura, E.; Samara, K.D.; Trachalaki, A.; Charalambous, I.; Tzanakis, N.; Antoniou, K.M. NLRP3/Caspase-1 inflammasome activation is decreased in alveolar macrophages in patients with lung cancer. PLoS ONE 2018, 13, e0205242. [Google Scholar]
- Wang, H.; Wang, Y.; Du, Q.; Lu, P.; Fan, H.; Lu, J.; Hu, R. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp. Cell Res. 2016, 342, 184–192. [Google Scholar] [CrossRef]
- Dagenais, M.; Saleh, M. Linking cancer-induced Nlrp3 inflammasome activation to efficient NK cell-mediated immunosurveillance. Oncoimmunology 2016, 5, e1129484. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 2010, 185, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Chen, L.; Li, Y.-C.; Wu, L.; Yu, G.-T.; Zhang, W.-F.; Sun, Z.-J. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 116. [Google Scholar] [CrossRef]
- Bae, J.Y.; Lee, S.W.; Shin, Y.H.; Lee, J.H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972–48982. [Google Scholar] [CrossRef]
- Yaw, A.C.K.; Chan, E.W.L.; Yap, J.K.Y.; Mai, C.W. The effects of NLRP3 inflammasome inhibition by MCC950 on LPS-induced pancreatic adenocarcinoma inflammation. J. Cancer Res. Clin. Oncol. 2020, 146, 2219–2229. [Google Scholar] [CrossRef]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.D.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.-M.T.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P.Y. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Leeth, R.A.; Rothschild, D.E.; Coutermarsh-Ott, S.L.; McDaniel, D.K.; Simmons, A.E.; Heid, B.; Cecere, T.E.; Allen, I.C. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J. Immunol. 2015, 194, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.-D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Struzik, J.; Szulc-Dąbrowska, L. NF-κB Signaling in Targeting Tumor Cells by Oncolytic Viruses—Therapeutic Perspectives. Cancers 2018, 10, 426. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, H.; Brenner, M.K. Immunotherapy against cancer-related viruses. Cell Res. 2017, 27, 59–73. [Google Scholar] [CrossRef]
- Ma, B.B.Y.; Lim, W.T.; Goh, B.C.; Hui, E.P.; Lo, K.W.; Pettinger, A.; Foster, N.R.; Riess, J.W.; Agulnik, M.; Chang, A.Y.C.; et al. Antitumor Activity of Nivolumab in Recurrent and Metastatic Nasopharyngeal Carcinoma: An International, Multicenter Study of the Mayo Clinic Phase 2 Consortium (NCI-9742). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Lee, S.-H.; Ejadi, S.; Even, C.; Cohen, R.B.; Le Tourneau, C.; Mehnert, J.M.; Algazi, A.; van Brummelen, E.M.J.; Saraf, S. Safety and antitumor activity of pembrolizumab in patients with programmed death-ligand 1–positive nasopharyngeal carcinoma: Results of the KEYNOTE-028 study. J. Clin. Oncol. 2017, 35, 4050–4056. [Google Scholar] [CrossRef]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.S.; Kang, Y.; Hsu, D.; Balko, J.M. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti–PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef]
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and inflammasome inhibition for the treatment of inflammatory skin diseases. Front. Pharmacol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Ridker, P.; Lorenzatti, A.; Krum, H.; Varigos, J. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-C.; Xu, K.; Martinek, J.; Young, R.R.; Banchereau, R.; George, J.; Turner, J.; Kim, K.I.; Zurawski, S.; Wang, X. IL1 receptor antagonist controls transcriptional signature of inflammation in patients with metastatic breast cancer. Cancer Res. 2018, 78, 5243–5258. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Young, R.R.; Levin, M.K.; Baisch, J.; Timis, R.; Muniz, L.S.; Turner, J.; Pascual, V.; Palucka, K. Safety and immunologic activity of anakinra in HER2-negative metastatic breast cancer (MBC). Am. Soc. Clin. Oncol. 2016, 34, e14565. [Google Scholar] [CrossRef]
- Vajaitu, C.; Draghici, C.C.; Solomon, I.; Lisievici, C.V.; Popa, A.V.; Lupu, M.; Caruntu, C.; Constantin, M.M.; Voiculescu, V.M. The central role of inflammation associated with checkpoint inhibitor treatments. J. Immunol. Res. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stanam, A.; Gibson-Corley, K.N.; Love-Homan, L.; Ihejirika, N.; Simons, A.L. Interleukin-1 blockade overcomes erlotinib resistance in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 76087–76100. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, R.; Peng, Z.; Dong, D.; Wei, G.; Wang, Y. Inhibition of IL-18-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy against osteosarcoma cancer. J. Bone Oncol. 2017, 9, 59–64. [Google Scholar] [CrossRef]
- Chen, L.; Huang, C.-F.; Li, Y.-C.; Deng, W.-W.; Mao, L.; Wu, L.; Zhang, W.-F.; Zhang, L.; Sun, Z.-J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar] [CrossRef]
- Hu, H.; Tang, K.F.; Chua, Y.N.; Lu, J.; Feng, P.; Chew, C.T.; Chan, S.H. Expression of interleukin-18 by nasopharyngeal carcinoma cells: A factor that possibly initiates the massive leukocyte infiltration. Hum. Pathol. 2004, 35, 722–728. [Google Scholar] [CrossRef]
- Li, F.; Xu, M.; Wang, M.; Wang, L.; Wang, H.; Zhang, H.; Chen, Y.; Gong, J.; Zhang, J.J.; Adcock, I.M. Roles of mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced lung inflammation and emphysema. Respir. Res. 2018, 19, 230. [Google Scholar] [CrossRef]
- Loher, F.; Bauer, C.; Landauer, N.; Schmall, K.; Siegmund, B.; Lehr, H.A.; Dauer, M.; Schoenharting, M.; Endres, S.; Eigler, A. The interleukin-1 beta-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J. Pharmacol. Exp. Ther. 2004, 308, 583–590. [Google Scholar] [CrossRef]
- Palumbo, A.; Facon, T.; Sonneveld, P.; Blade, J.; Offidani, M.; Gay, F.; Moreau, P.; Waage, A.; Spencer, A.; Ludwig, H. Thalidomide for treatment of multiple myeloma: 10 years later. Blood 2008, 111, 3968–3977. [Google Scholar] [CrossRef] [PubMed]
- Sztiller-Sikorska, M.; Czyz, M. Parthenolide as Cooperating Agent for Anti-Cancer Treatment of Various Malignancies. Pharmaceuticals 2020, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Kim, S.L.; Park, Y.R.; Lee, S.T.; Kim, S.W. Parthenolide promotes apoptotic cell death and inhibits the migration and invasion of SW620 cells. Intest. Res. 2017, 15, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lin, Z.-N.; Yang, C.-F.; Shi, X.; Ong, C.-N.; Shen, H.-M. Suppressed NF-κB and sustained JNK activation contribute to the sensitization effect of parthenolide to TNF-α-induced apoptosis in human cancer cells. Carcinogenesis 2004, 25, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Xia, B.; Zhuang, Q.Y.; Hou, M.J.; Zhang, Y.J.; Luo, B.; Qiu, Y.; Gao, Y.F.; Li, X.J.; Chen, H.F.; et al. Parthenolide inhibits cancer stem-like side population of nasopharyngeal carcinoma cells via suppression of the NF-kappaB/COX-2 pathway. Theranostics 2015, 5, 302–321. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Fu, M.; Yao, Q.; Zhuo, H.; Lu, Q.; Niu, X.; Zhang, P.; Pei, Y.; Zhang, K. Parthenolide induces apoptosis and lytic cytotoxicity in Epstein-Barr virus-positive Burkitt lymphoma. Mol. Med. Rep. 2012, 6, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, S.L.; Park, Y.R.; Liu, Y.C.; Seo, S.Y.; Kim, S.H.; Kim, I.H.; Lee, S.O.; Lee, S.T.; Kim, S.W. Balsalazide Potentiates Parthenolide-Mediated Inhibition of Nuclear Factor-kappaB Signaling in HCT116 Human Colorectal Cancer Cells. Intest. Res. 2015, 13, 233–241. [Google Scholar] [CrossRef]
- Kim, S.-L.; Kim, S.H.; Park, Y.R.; Liu, Y.-C.; Kim, E.-M.; Jeong, H.-J.; Kim, Y.N.; Seo, S.Y.; Kim, I.H.; Lee, S.O. Combined parthenolide and balsalazide have enhanced antitumor efficacy through blockade of NF-κB activation. Mol. Cancer Res. 2017, 15, 141–151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mishra, K.P. Andrographolide Ameliorates Inflammation through Inhibition of NLRP3 Inflammasome Activation in Intestinal Epithelial Cells. Jpn. J. Gastroenterol. Hepatol. 2019, 2, 1–9. [Google Scholar]
- Peng, T.; Hu, M.; Wu, T.-T.; Zhang, C.; Chen, Z.; Huang, S.; Zhou, X.-H. Andrographolide suppresses proliferation of nasopharyngeal carcinoma cells via attenuating NF-B pathway. BioMed Res. Int. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumour Types | Roles of Inflammasomes | Inflammasomes and Signalling Molecules | Results | Reference | |
|---|---|---|---|---|---|
| NPC | Tumour suppressor | NLRP3, IL-1β | Upregulation tumour inflammasome-derived IL-1β actively induced the influx of TANs to the tumour site and positively correlated to patient survival. | [44] | |
| Tumour promoter | Caspase-1, IL-1β, IL-18 | Constitutive activation of caspase-1 facilitated EBV genome persistence and immune evasion. | [103] | ||
| Multiple myeloma | Tumour promoter | NLRP1, IL-18 | NLRP1-mediated IL-18 induced immunosuppression through MDSC recruitment. | [21] | |
| OSCC | Tumour promoter | NLRP3 | Knockdown of NLRP3 increased E-cadherin expression and suppressed proliferation, invasion, and migration of OSCC cells. | [136] | |
| Melanoma | Tumour promoter | NLRP3, IL-1β | NLRP3 activation increased IL-1β secretion, resulting in auto-inflammation and enhanced tumour growth. | [137] | |
| Breast cancer | Tumour promoter | NLRP1, IL-1β, IL-18 | NLRP1 promoted tumour progression by inducing EMT, both in vivo and in vitro. | [138] | |
| Tumour promoter | NLRP3, IL-1β | NLRP3 activation and IL-1β promoted tumour growth and lung metastasis via increased infiltration of MDSCs and TAMs. | [139] | ||
| Tumour suppressor | Caspase-1 | Downregulation of caspase-1 increased breast cancer cell proliferation and invasion. | [140] | ||
| Lung cancer | Tumour promoter | NLRP3 | NLRP3 activation decreased E-cadherin expression and promoted proliferation and migration of lung cancer cells. | [141] | |
| Tumour suppressor | NLRP3, Caspase-1 | Impairment of NLRP3/caspase-1 activation promoted immunosuppression in the lung microenvironment. | [142] | ||
| CRC | Tumour promoter | NLRP3 | Knockdown of NLRP3 reversed mesenchymal phenotypes of tumour cells and diminished tumour migration and invasion. | [143] | |
| Tumour suppressor | NLRP3, IL-18 | NLRP3-derived IL-18 enhanced NK cell priming and trigger FasL-induced apoptosis of CRC cells. | [144] | ||
| Tumour suppressor | NLRP3, IL-18 | NLRP3 activation enhanced maturation and tumouricidal activity of NK cells as well as suppressed metastatic growth of CRC in liver. | [49,145] | ||
| HNSCC | Tumour promoter | NLRP3, IL-1β | Upregulation of NLRP3 activity positively correlated to carcinogenesis and CSC markers. | [146] | |
| - | NLRP3, P2X7R | Blockade of NLRP3 activation and P2X7R significantly reduced invasiveness and viability of HNSCC cells. | [147] | ||
| Pancreatic cancer | Tumour promoter | NLRP3, caspase-1, IL-1β | NLRP3 activation promoted tumour survival via upregulation of caspase-1 activity and IL-1β production. | [148] | |
| - | NLRP3, ASC, caspase-1 | Inhibition of NLRP3, ASC, or caspase-1 activation restored innate and adaptive antitumour immune response. | [149] | ||
| CAC | Tumour suppressor | NLRP1, NLRP3, IL-1β, IL-18 | Loss of inflammasome-secreted IL-1β and IL-18 increased susceptibility of mice to CAC. | [150,151] | |
| - | NLRP3, IL-18 | Defective NLRP3 activation decreased IL-18 production and led to the overgrowth of commensal, massive infiltration of leucocytes, and overproduction of chemokines in the colon, which ultimately, resulted in increased mortality rates. | [152] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Looi, C.K.; Hii, L.-W.; Chung, F.F.-L.; Mai, C.-W.; Lim, W.-M.; Leong, C.-O. Roles of Inflammasomes in Epstein–Barr Virus-Associated Nasopharyngeal Cancer. Cancers 2021, 13, 1786. https://doi.org/10.3390/cancers13081786
Looi CK, Hii L-W, Chung FF-L, Mai C-W, Lim W-M, Leong C-O. Roles of Inflammasomes in Epstein–Barr Virus-Associated Nasopharyngeal Cancer. Cancers. 2021; 13(8):1786. https://doi.org/10.3390/cancers13081786
Chicago/Turabian StyleLooi, Chin King, Ling-Wei Hii, Felicia Fei-Lei Chung, Chun-Wai Mai, Wei-Meng Lim, and Chee-Onn Leong. 2021. "Roles of Inflammasomes in Epstein–Barr Virus-Associated Nasopharyngeal Cancer" Cancers 13, no. 8: 1786. https://doi.org/10.3390/cancers13081786
APA StyleLooi, C. K., Hii, L.-W., Chung, F. F.-L., Mai, C.-W., Lim, W.-M., & Leong, C.-O. (2021). Roles of Inflammasomes in Epstein–Barr Virus-Associated Nasopharyngeal Cancer. Cancers, 13(8), 1786. https://doi.org/10.3390/cancers13081786