
Systematic Analysis of the Transcriptome Profiles and Co-Expression Networks of Tumour Endothelial Cells Identifies Several Tumour-Associated Modules and Potential Therapeutic Targets in Hepatocellular Carcinoma

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
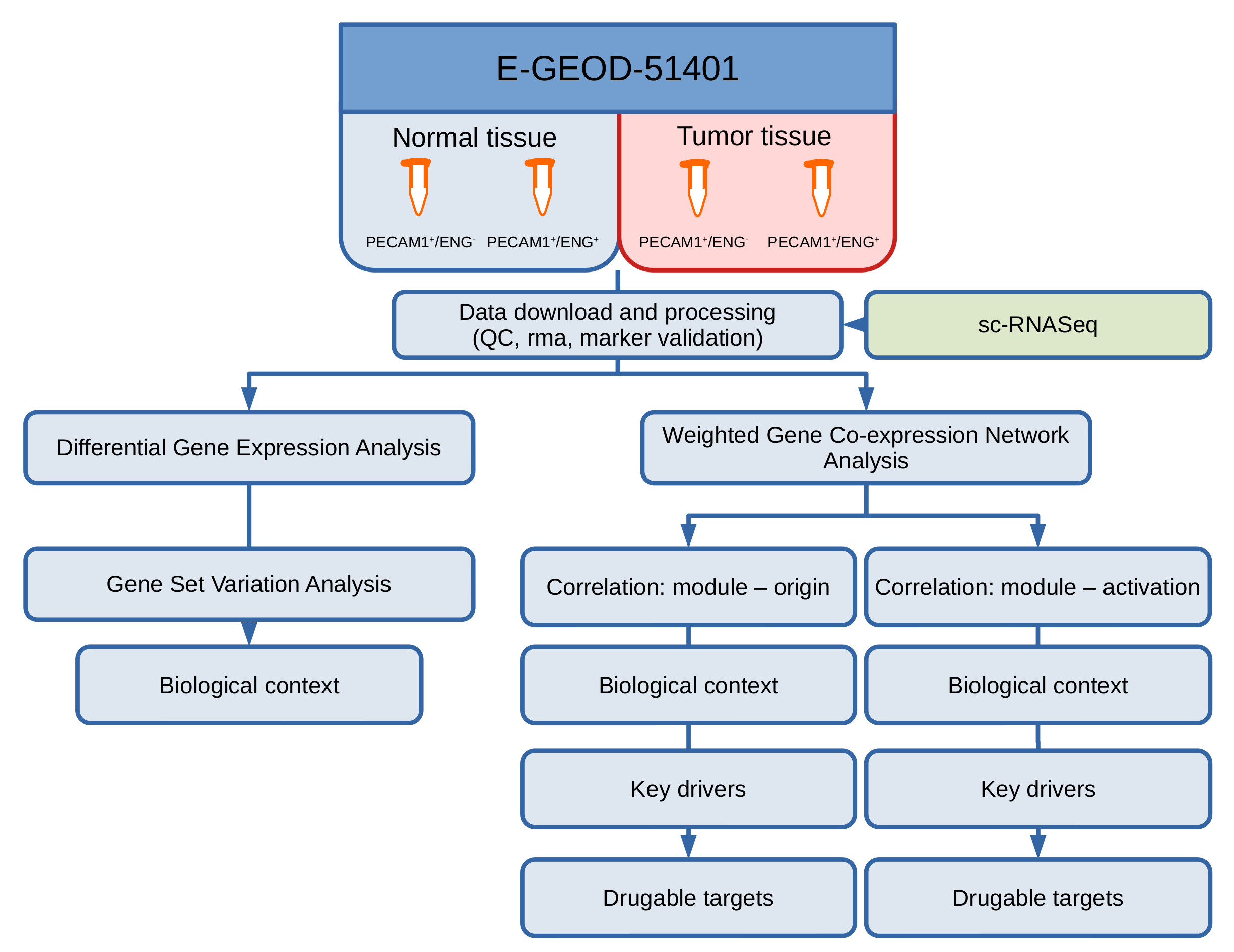
2. Materials and Methods
2.1. Data Generation and Access
2.2. Data Preprocessing and QC
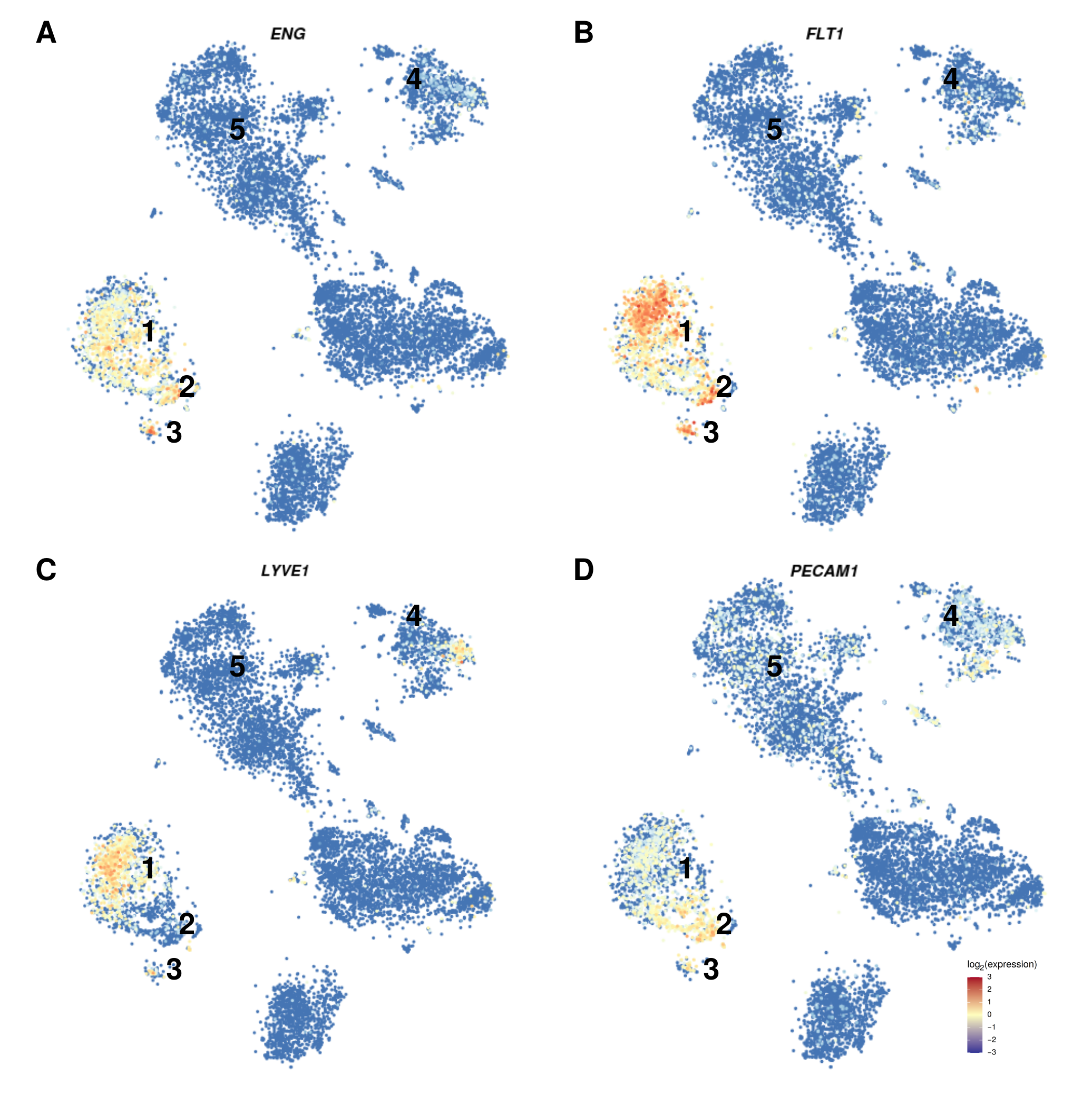
2.3. Single-Cell RNASeq
2.4. Differential Gene and Pathway Expression Analysis
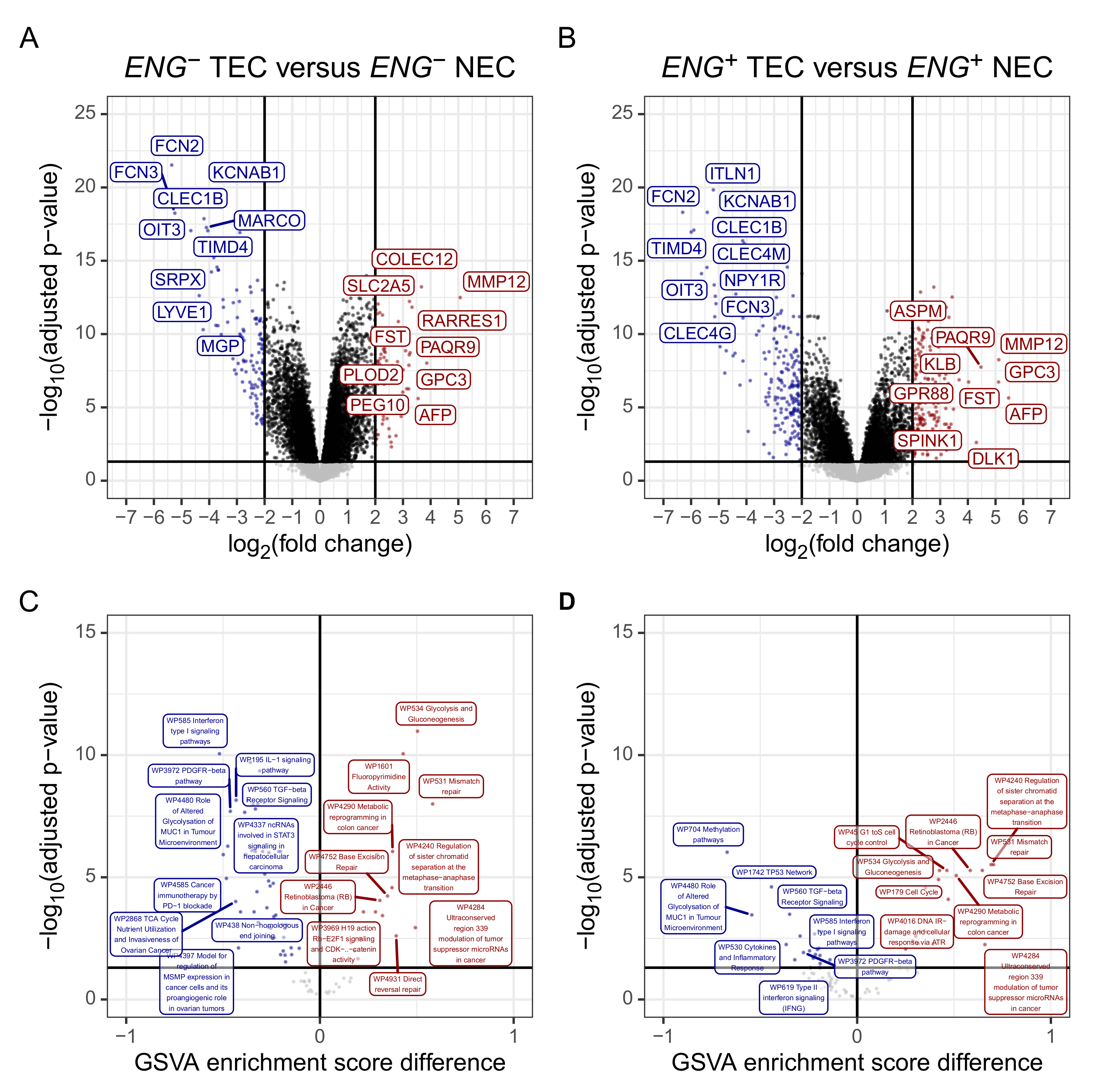
2.5. Gene Set Variation Analysis
2.6. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.7. Term Enrichment Analysis for Detected Modules
2.8. Statistics
3. Results
3.1. Cohort Characteristics
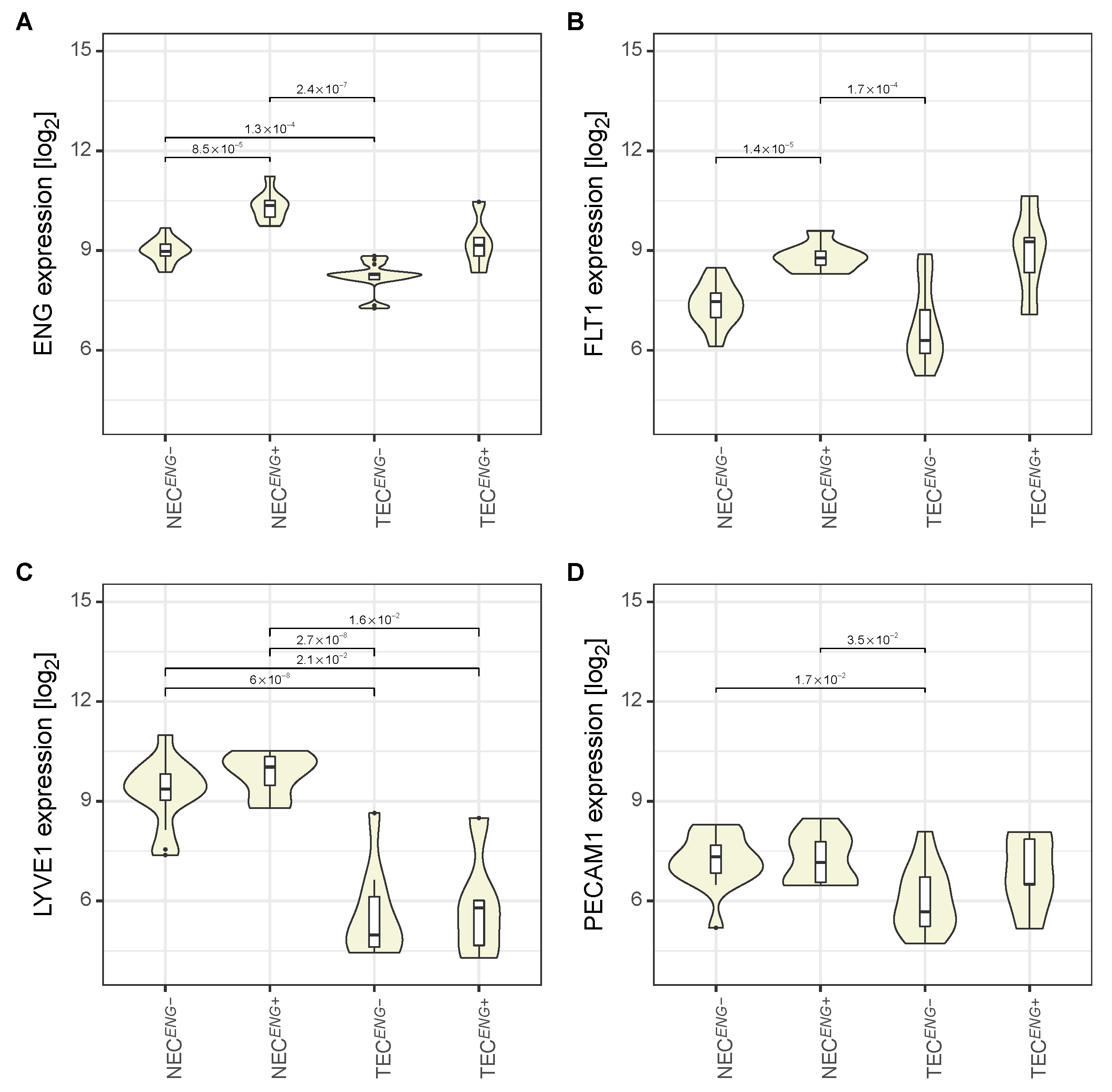
3.2. The Expression Pattern of Endothelial Markers Is in Accordance with That of Liver Endothelial Cells
3.3. The Gene Expression Profile of Tumour Endothelial Cells Is Characterised by Upregulated Evasion from Growth Suppressors, Downregulated Immune and Inflammation-Related Pathways, and Resistance to Apoptosis
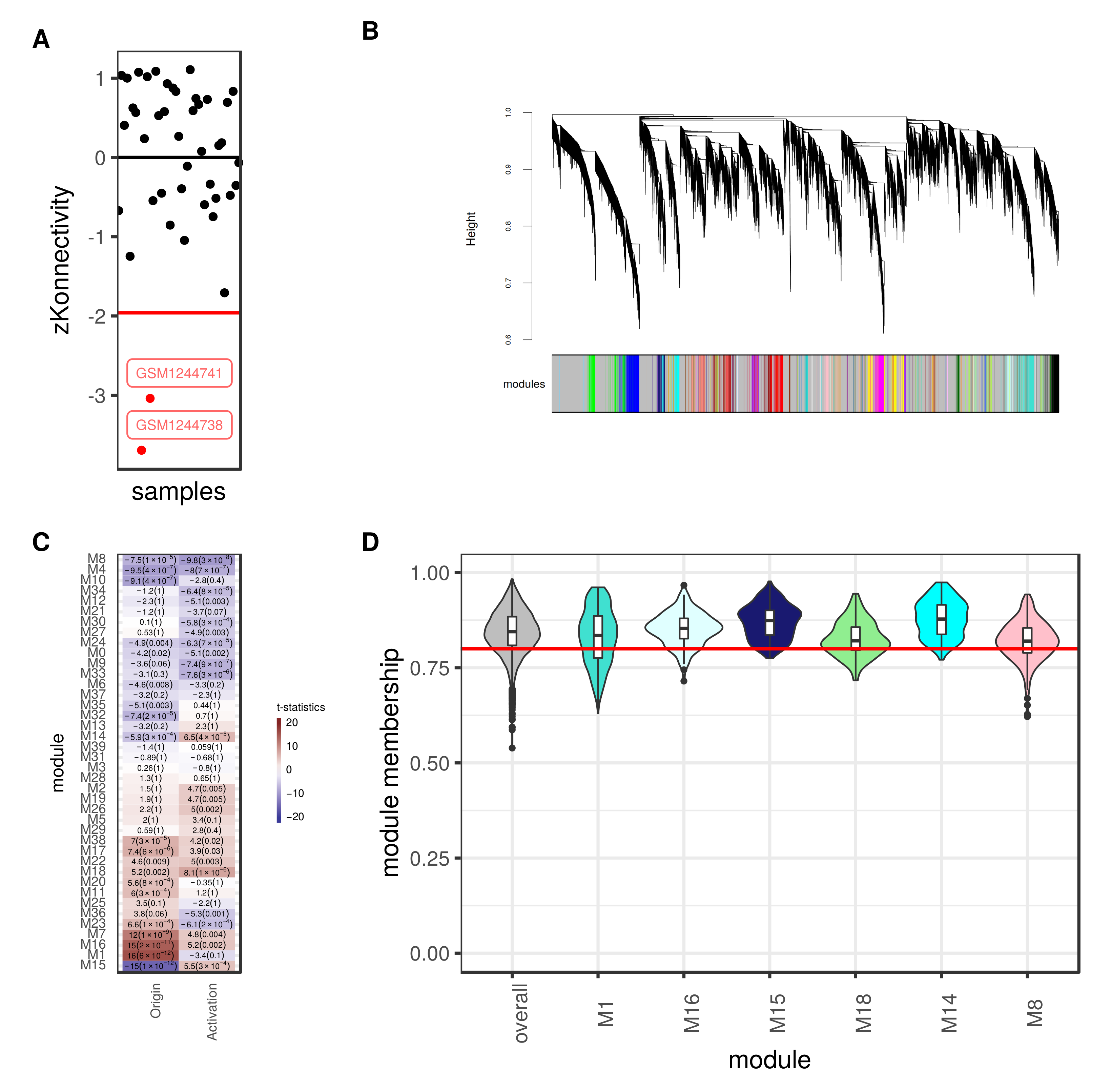
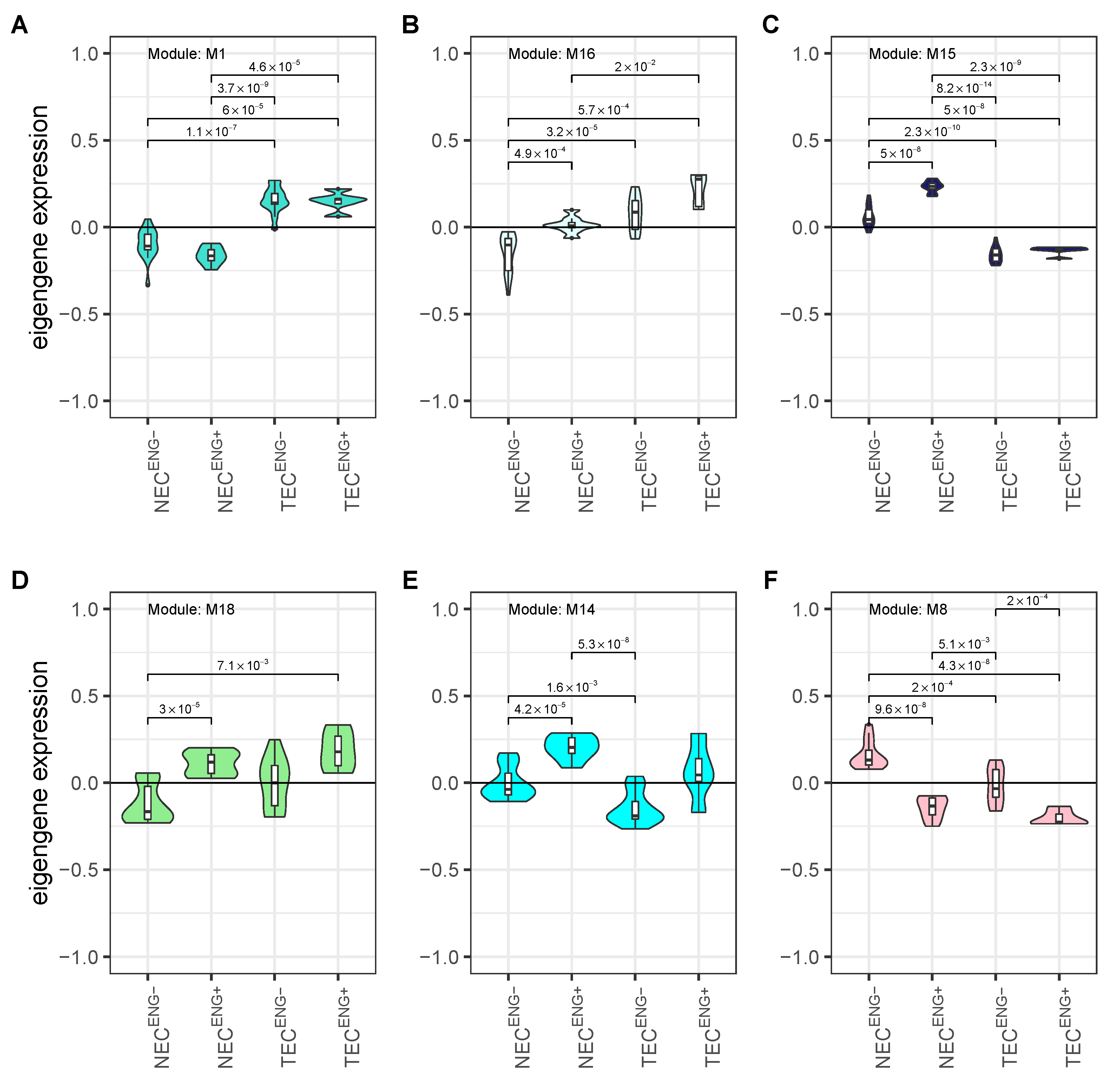
3.4. Weighted Gene Co-Expression Network Analysis Reveals Several Gene Modules Associated Strongly with Cell Origin and Angiogenic Activation
3.5. Cell Origin Related Modules Are Positively Associated with Cell Growth and Survival and Negatively Associated with Immune Functions
3.6. Activation Related Modules Are Associated with Angiogenesis, Cell Adhesion and Immunologic Functions
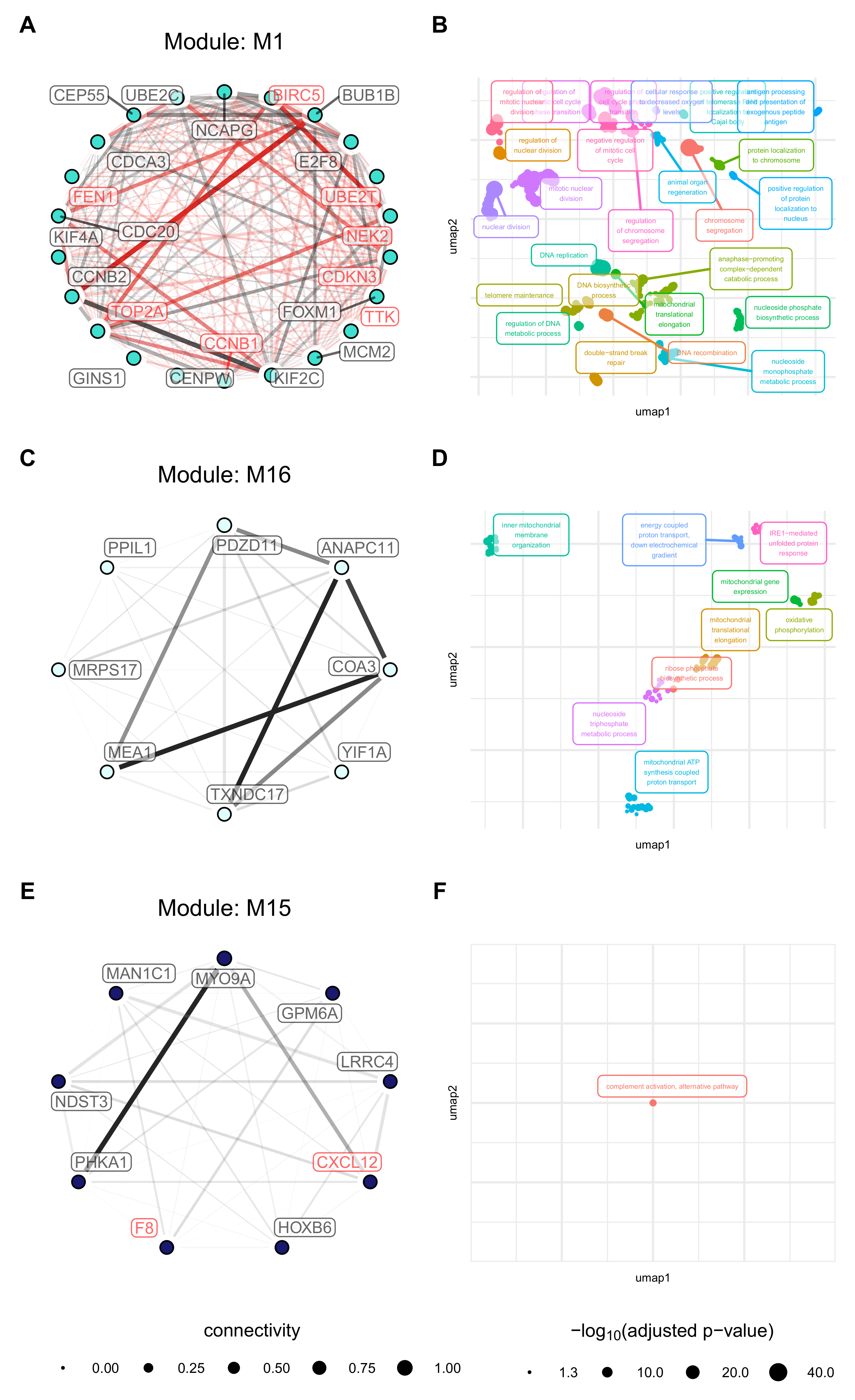
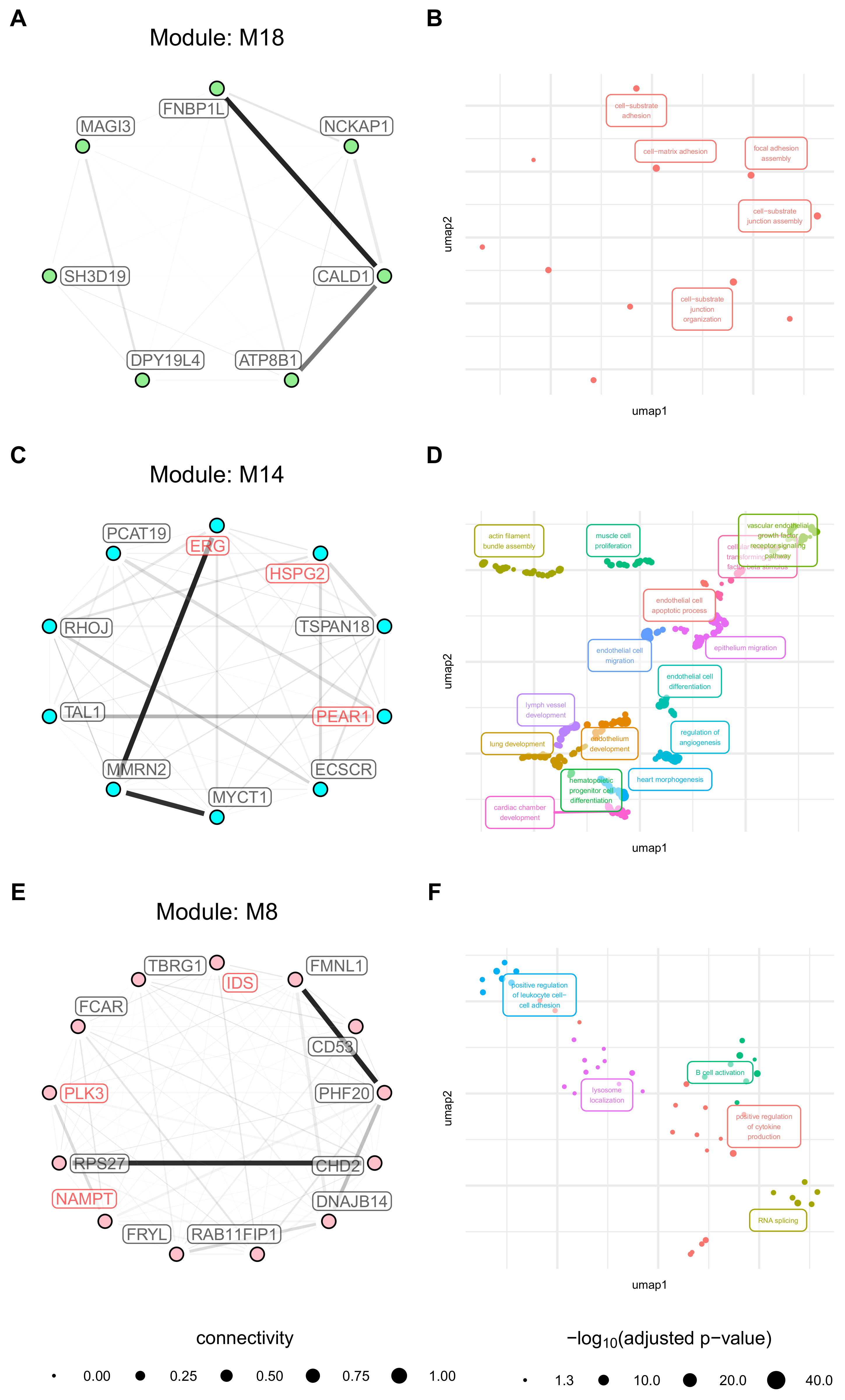
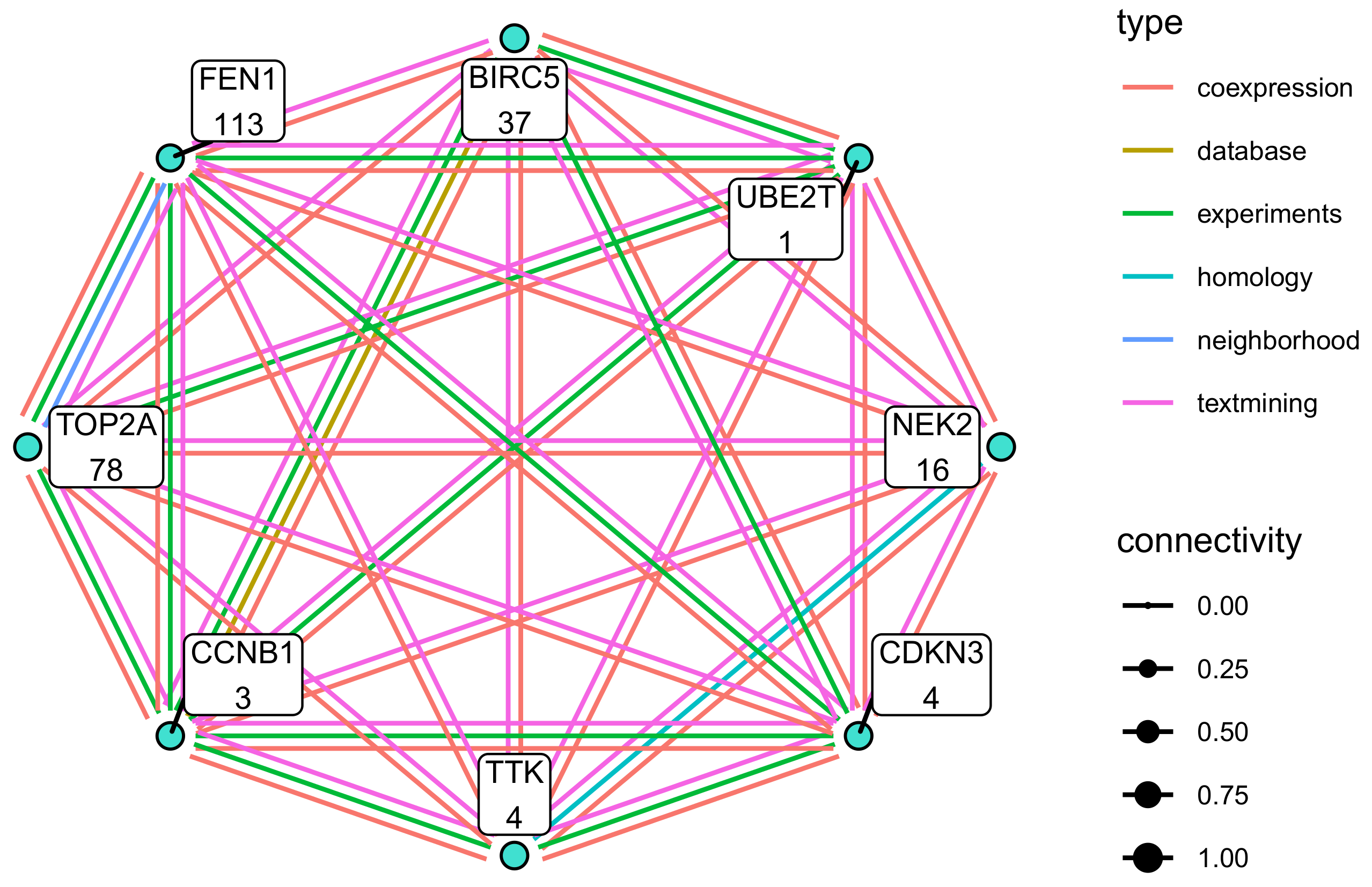
3.7. Modules with a Strong Positive Association with Cell Origin and Angiogenic Activation Contain Several Potentially Druggable Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DGIdb | Drug–Gene Interaction database |
| NEC | endothelial cells derived from non-tumour tissue (liver sinusoidal, macrovascular, and other endothelial cells) |
| TEC | endothelial cells derived from tumour tissue (liver sinusoidal, macrovascular endothelial cells, and other endothelial cells) |
| LSEC | Liver sinusoidal endothelial cells |
| LEC | Liver endothelial cells (liver sinusoidal, macrovascular, and other endothelial cells) |
| HCC | Hepatocellular carcinoma |
| log2FC | logarithm of the fold change to the base 2 |
| WGCNA | Weighted Gene Network Analysis |
| GSEA | Gene Set Enrichment Analysis |
| GOTEA | GO Term Enrichment Analysis |
| GSVA | Gene Set Variation Analysis |
| GOTSSA | GO Term Semantic Similarity Analysis |
| FI | Functional interactions |
| DEG | differentially expressed gene |
| 5FU | 5-fluorouracil |
References
- Wong, M.C.S.; Jiang, J.Y.; Goggins, W.B.; Liang, M.; Fang, Y.; Fung, F.D.H.; Leung, C.; Wang, H.H.X.; Wong, G.L.H.; Wong, V.W.S.; et al. International Incidence and Mortality Trends of Liver Cancer: A Global Profile. Sci. Rep. 2017, 7, srep45846. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M. Early HCC: Diagnosis and Molecular Markers. J. Gastroenterol. 2009, 44, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Bertuccio, P.; Turati, F.; Carioli, G.; Rodriguez, T.; La Vecchia, C.; Malvezzi, M.; Negri, E. Global Trends and Predictions in Hepatocellular Carcinoma Mortality. J. Hepatol. 2017, 67, 302–309. [Google Scholar] [CrossRef]
- Kumar, V.; Rahman, M.; Gahtori, P.; Al-Abbasi, F.; Anwar, F.; Kim, H.S. Current Status and Future Directions of Hepatocellular Carcinoma-Targeted Nanoparticles and Nanomedicine. Expert Opin. Drug Deliv. 2020, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tabrizian, P.; Jibara, G.; Shrager, B.; Schwartz, M.; Roayaie, S. Recurrence of Hepatocellular Cancer After Resection: Patterns, Treatments, and Prognosis. Ann. Surg. 2015, 261, 947–955. [Google Scholar] [CrossRef]
- Maslak, E.; Gregorius, A.; Chlopicki, S. Liver Sinusoidal Endothelial Cells (LSECs) Function and NAFLD; NO-Based Therapy Targeted to the Liver. Pharmacol. Rep. 2015, 67, 689–694. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver Sinusoidal Endothelial Cells: Physiology and Role in Liver Diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef]
- Augustin, H.G.; Kozian, D.H.; Johnson, R.C. Differentiation of Endothelial Cells: Analysis of the Constitutive and Activated Endothelial Cell Phenotypes. Bioessays News Rev. Mol. Cell. Dev. Biol. 1994, 16, 901–906. [Google Scholar] [CrossRef]
- Géraud, C.; Evdokimov, K.; Straub, B.K.; Peitsch, W.K.; Demory, A.; Dörflinger, Y.; Schledzewski, K.; Schmieder, A.; Schemmer, P.; Augustin, H.G.; et al. Unique Cell Type-Specific Junctional Complexes in Vascular Endothelium of Human and Rat Liver Sinusoids. PLoS ONE 2012, 7, e34206. [Google Scholar] [CrossRef]
- Strauss, O.; Phillips, A.; Ruggiero, K.; Bartlett, A.; Dunbar, P.R. Immunofluorescence Identifies Distinct Subsets of Endothelial Cells in the Human Liver. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver Sinusoidal Endothelial Cells — Gatekeepers of Hepatic Immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor Endothelial Cells Acquire Drug Resistance by MDR1 Up-Regulation via VEGF Signaling in Tumor Microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated Tumor Endothelial Cells Maintain Specific Character during Long-Term Culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef] [PubMed]
- St. Croix, B.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes Expressed in Human Tumor Endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef]
- Wu, L.Q.; Zhang, W.J.; Niu, J.X.; Ye, L.Y.; Yang, Z.H.; Grau, G.E.; Lou, J.N. Phenotypic and Functional Differences between Human Liver Cancer Endothelial Cells and Liver Sinusoidal Endothelial Cells. J. Vasc. Res. 2008, 45, 78–86. [Google Scholar] [CrossRef]
- Semela, D.; Dufour, J.F. Angiogenesis and Hepatocellular Carcinoma. J. Hepatol. 2004, 41, 864–880. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Li, Y.; Zhai, Z.; Liu, D.; Zhong, X.; Meng, X.; Yang, Q.; Liu, J.; Li, H. CD105 Promotes Hepatocarcinoma Cell Invasion and Metastasis through VEGF. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Hida, K. Tumor Endothelial Cells Accelerate Tumor Metastasis. Cancer Sci. 2017, 108, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Q.; Sun, H.C.; Zhang, W.; Zhu, X.D.; Zhuang, P.Y.; Zhang, J.B.; Wang, L.; Wu, W.Z.; Qin, L.X.; Tang, Z.Y. Human Hepatocellular Carcinoma Tumor–Derived Endothelial Cells Manifest Increased Angiogenesis Capability and Drug Resistance Compared with Normal Endothelial Cells. Clin. Cancer Res. 2009, 15, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, J. Feature Selection with the Fisher Score Followed by the Maximal Clique Centrality Algorithm Can Accurately Identify the Hub Genes of Hepatocellular Carcinoma. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Presson, A.P.; Yoon, N.K.; Bagryanova, L.; Mah, V.; Alavi, M.; Maresh, E.L.; Rajasekaran, A.K.; Goodglick, L.; Chia, D.; Horvath, S. Protein Expression Based Multimarker Analysis of Breast Cancer Samples. BMC Cancer 2011, 11, 230. [Google Scholar] [CrossRef] [PubMed]
- Tian, A.; Pu, K.; Li, B.; Li, M.; Liu, X.; Gao, L.; Mao, X. Weighted Gene Coexpression Network Analysis Reveals Hub Genes Involved in Cholangiocarcinoma Progression and Prognosis. Hepatol. Res. 2019, 49, 1195–1206. [Google Scholar] [CrossRef]
- Wu, H.; Chen, S.; Yu, J.; Li, Y.; Zhang, X.Y.; Yang, L.; Zhang, H.; Hou, Q.; Jiang, M.; Brunicardi, F.C.; et al. Single-Cell Transcriptome Analyses Reveal Molecular Signals to Intrinsic and Acquired Paclitaxel Resistance in Esophageal Squamous Cancer Cells. Cancer Lett. 2018, 420, 156–167. [Google Scholar] [CrossRef]
- Horvath, S. Weighted Network Analysis: Applications in Genomics and Systems Biology; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Jeong, H.; Mason, S.P.; Barabási, A.L.; Oltvai, Z.N. Lethality and Centrality in Protein Networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef]
- Tolios, A.; De Las Rivas, J.; Hovig, E.; Trouillas, P.; Scorilas, A.; Mohr, T. Computational Approaches in Cancer Multidrug Resistance Research: Identification of Potential Biomarkers, Drug Targets and Drug-Target Interactions. Drug Resist. Updat. 2019, 48, 100662. [Google Scholar] [CrossRef]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin Promotes Endothelial Cell Proliferation and TGF-β/ALK1 Signal Transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin Prevents Vascular Malformation by Regulating Flow-Induced Cell Migration and Specification through VEGFR2 Signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef]
- Schoonderwoerd, M.J.A.; Goumans, M.J.T.H.; Hawinkels, L.J.A.C. Endoglin: Beyond the Endothelium. Biomolecules 2020, 10, 289. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Gautier, L.; Cope, L.M.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2009, 20, 307–315. [Google Scholar] [CrossRef]
- Kauffmann, A.; Gentleman, R.; Huber, W. arrayQualityMetrics—A Bioconductor Package for Quality Assessment of Microarray Data. Bioinformatics 2009, 25, 415–416. [Google Scholar] [CrossRef]
- Gentleman, R.; Carey, V.; Huber, W.; Hahne, F. Genefilter: Genefilter: Methods for Filtering Genes from Microarray Experiments. R Package Version 1.72.1, 2021. [Google Scholar]
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A Human Liver Cell Atlas Reveals Heterogeneity and Epithelial Progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.S.; Sagar; Grün, D. FateID Infers Cell Fate Bias in Multipotent Progenitors from Single-Cell RNA-Seq Data. Nat. Methods 2018, 15, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl. Acids Res. 2015, 43, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, Y.; Benjamani, Y. More Powerful Procedures for Multiple Significance Testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Mélius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. WikiPathways: Capturing the Full Diversity of Pathway Knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Greenfest-Allen, E.; Cartailler, J.P.; Magnuson, M.A.; Stoeckert, C.J. iterativeWGCNA: Iterative Refinement to Improve Module Detection from WGCNA Co-Expression Networks. bioRxiv 2017, 234062. [Google Scholar] [CrossRef]
- Benaglia, T.; Chauveau, D.; Hunter, D.R.; Young, D.S. Mixtools: An R Package for Analyzing Finite Mixture Models. J. Stat. Softw. 2009, 32, 1–29. [Google Scholar] [CrossRef]
- Oldham, M.C.; Langfelder, P.; Horvath, S. Network Methods for Describing Sample Relationships in Genomic Datasets: Application to Huntington’s Disease. BMC Syst. Biol. 2012, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67. [Google Scholar] [CrossRef]
- Li, J.; Zhou, D.; Qiu, W.; Shi, Y.; Yang, J.J.; Chen, S.; Wang, Q.; Pan, H. Application of Weighted Gene Co-Expression Network Analysis for Data from Paired Design. Sci. Rep. 2018, 8, 622. [Google Scholar] [CrossRef]
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 1–26. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A Redesign and Expansion of the Drug–Gene Interaction Database. Nucleic Acids Res. 2018, 46, D1068–D1073. [Google Scholar] [CrossRef]
- Wagner, A.H.; Coffman, A.C.; Ainscough, B.J.; Spies, N.C.; Skidmore, Z.L.; Campbell, K.M.; Krysiak, K.; Pan, D.; McMichael, J.F.; Eldred, J.M.; et al. DGIdb 2.0: Mining Clinically Relevant Drug–Gene Interactions. Nucleic Acids Res. 2016, 44, D1036–D1044. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Li, F.; Qin, Y.; Bo, X.; Wu, Y.; Wang, S. GOSemSim: An R Package for Measuring Semantic Similarity among GO Terms and Gene Products. Bioinformatics 2010, 26, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Du, Z.; Payattakool, R.; Yu, P.S.; Chen, C.F. A New Method to Measure the Semantic Similarity of GO Terms. Bioinformatics 2007, 23, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2020, arXiv:1802.03426. [Google Scholar]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, KDD’96; AAAI Press: Portland, OR, USA, 1996; pp. 226–231. [Google Scholar]
- Sayols, S. Rrvgo: A Bioconductor Package to Reduce and Visualize Gene Ontology Terms. 2020. Available online: https://ssayols.github.io/rrvgo (accessed on 31 March 2021).
- Seaman, S.; Stevens, J.; Yang, M.Y.; Logsdon, D.; Graff-Cherry, C.; St. Croix, B. Genes That Distinguish Physiological and Pathological Angiogenesis. Cancer Cell 2007, 11, 539–554. [Google Scholar] [CrossRef]
- Li, J.J. A New Bioinformatics Tool to Recover Missing Gene Expression in Single-Cell RNA Sequencing Data. J. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Damen, C.A.; Blijham, G.H.; Groenewegen, G. Tumor Angiogenesis Is Accompanied by a Decreased Inflammatory Response of Tumor-Associated Endothelium. Blood 1996, 88, 667–673. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. [Google Scholar] [CrossRef]
- Li, F.; Aljahdali, I.; Ling, X. Cancer Therapeutics Using Survivin BIRC5 as a Target: What Can We Do after over Two Decades of Study? J. Exp. Clin. Cancer Res. 2019, 38. [Google Scholar] [CrossRef]
- Chen, B.; Garmire, L.; Calvisi, D.F.; Chua, M.S.; Kelley, R.K.; Chen, X. Harnessing Big ‘omics’ Data and AI for Drug Discovery in Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 238–251. [Google Scholar] [CrossRef]
- Kaur, H.; Dhall, A.; Kumar, R.; Raghava, G.P.S. Identification of Platform-Independent Diagnostic Biomarker Panel for Hepatocellular Carcinoma Using Large-Scale Transcriptomics Data. bioRxiv 2019, 758250. [Google Scholar] [CrossRef]
- Liu, J.; Hua, P.; Hui, L.; Zhang, L.L.; Hu, Z.; Zhu, Y.W. Identification of Hub Genes and Pathways Associated with Hepatocellular Carcinoma Based on Network Strategy. Exp. Ther. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Goveia, J.; Rohlenova, K.; Taverna, F.; Treps, L.; Conradi, L.C.; Pircher, A.; Geldhof, V.; de Rooij, L.P.M.H.; Kalucka, J.; Sokol, L.; et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 2020, 37, 21–36.e13. [Google Scholar] [CrossRef]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al. Heterogeneity of Tumor Endothelial Cells. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef]
- Chavakis, E.; Dimmeler, S. Regulation of Endothelial Cell Survival and Apoptosis During Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Addison, C.L.; Daniel, T.O.; Burdick, M.D.; Liu, H.; Ehlert, J.E.; Xue, Y.Y.; Buechi, L.; Walz, A.; Richmond, A.; Strieter, R.M. The CXC Chemokine Receptor 2, CXCR2, Is the Putative Receptor for ELR+ CXC Chemokine-Induced Angiogenic Activity. J. Immunol. 2000, 165, 5269–5277. [Google Scholar] [CrossRef]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The Role of CXC Chemokines and Their Receptors in Cancer. Cancer Lett. 2008, 267, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.; Zabel, B.A. Support of Tumor Endothelial Cells by Chemokine Receptors. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Xie, H.; Zhou, L.; Zhou, W.; Zhang, W.; Ding, S.; Wei, B.; Yu, X.; Su, R.; Zheng, S. Cyclin-Dependent Kinase Inhibitor 3 Is Overexpressed in Hepatocellular Carcinoma and Promotes Tumor Cell Proliferation. Biochem. Biophys. Res. Commun. 2012, 420, 29–35. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, Y.; Shao, S.; Sun, Y.; Lin, Z. Identification of Hub Genes and Biological Pathways in Hepatocellular Carcinoma by Integrated Bioinformatics Analysis. PeerJ 2021, 9, e10594. [Google Scholar] [CrossRef]
- Zhao, X.; Weng, W.; Jin, M.; Li, S.; Chen, Q.; Li, B.; Zhou, Z.; Lan, C.; Yang, Y. Identification of Biomarkers Based on Bioinformatics Analysis: The Expression of Ubiquitin-Conjugating Enzyme E2T (UBE2T) in the Carcinogenesis and Progression of Hepatocellular Carcinoma. Med Sci. Monit. Int. Med J. Exp. Clin. Res. 2021, 27, e929023-1–e929023-11. [Google Scholar] [CrossRef]
- Dudley, A.C. Tumor Endothelial Cells. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fu, G.; Dai, T.; Huang, H. Migration of Endothelial Progenitor Cells Mediated by Stromal Cell-Derived Factor-1α/CXCR4 via PI3K/Akt/eNOS Signal Transduction Pathway. J. Cardiovasc. Pharmacol. 2007, 50, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Cheng, I.C.; Chen, B.C.; Shuai, H.H.; Chien, F.C.; Chen, P.; Hsieh, T.S. Wuho Is a New Member in Maintaining Genome Stability through Its Interaction with Flap Endonuclease 1. PLoS Biol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Kathera, C.; Zhang, J.; Janardhan, A.; Sun, H.; Ali, W.; Zhou, X.; He, L.; Guo, Z. Interacting Partners of FEN1 and Its Role in the Development of Anticancer Therapeutics. Oncotarget 2017, 8, 27593–27602. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, L.; Luo, L.; Zhu, H.; Yang, H.; Zhang, Y.; Wu, H.; Sun, H.; Jiang, F.; Kathera, C.S.; Liu, L.; et al. FEN1 Promotes Tumor Progression and Confers Cisplatin Resistance in Non-Small-Cell Lung Cancer. Mol. Oncol. 2017, 11, 640–654. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, Y.; Sun, H.; Jiang, F.; Yang, H.; Wu, H.; Zhou, T.; Hu, S.; Kathera, C.S.; Wang, X.; et al. Targeting DNA Flap Endonuclease 1 to Impede Breast Cancer Progression. EBioMedicine 2016, 14, 32–43. [Google Scholar] [CrossRef]
- Pu, J.; Wang, J.; Qin, Z.; Wang, A.; Zhang, Y.; Wu, X.; Wu, Y.; Li, W.; Xu, Z.; Lu, Y.; et al. IGF2BP2 Promotes Liver Cancer Growth Through an m6A-FEN1-Dependent Mechanism. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Aspe, J.R.; Asumen, M.G.; Almaguel, F.; Odumosu, O.; Acevedo-Martinez, S.; De Leon, M.; Langridge, W.H.R.; Wall, N.R. Extracellular, Cell-Permeable Survivin Inhibits Apoptosis While Promoting Proliferative and Metastatic Potential. Br. J. Cancer 2009, 100, 1073–1086. [Google Scholar] [CrossRef]
- Shin, S.; Sung, B.J.; Cho, Y.S.; Kim, H.J.; Ha, N.C.; Hwang, J.I.; Chung, C.W.; Jung, Y.K.; Oh, B.H. An Anti-Apoptotic Protein Human Survivin Is a Direct Inhibitor of Caspase-3 and -7. Biochemistry 2001, 40, 1117–1123. [Google Scholar] [CrossRef]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.J.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. SEARCH: A Phase III, Randomized, Double-Blind, Placebo-Controlled Trial of Sorafenib plus Erlotinib in Patients with Advanced Hepatocellular Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef]
- Xia, W.; Bisi, J.; Strum, J.; Liu, L.; Carrick, K.; Graham, K.M.; Treece, A.L.; Hardwicke, M.A.; Dush, M.; Liao, Q.; et al. Regulation of Survivin by ErbB2 Signaling: Therapeutic Implications for ErbB2-Overexpressing Breast Cancers. Cancer Res. 2006, 66, 1640–1647. [Google Scholar] [CrossRef]
- Collins, D.C.; Cocchiglia, S.; Tibbitts, P.; Solon, G.; Bane, F.T.; McBryan, J.; Treumann, A.; Eustace, A.; Hennessy, B.; Hill, A.D.; et al. Growth Factor Receptor/Steroid Receptor Cross Talk in Trastuzumab-Treated Breast Cancer. Oncogene 2015, 34, 525–530. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Bhola, N.E.; Sutton, C.; Ghosh, R.; Kuba, M.G.; Dave, B.; Chang, J.C.; Arteaga, C.L. Trastuzumab-Resistant Cells Rely on a HER2-PI3K-FoxO-Survivin Axis and Are Sensitive to PI3K Inhibitors. Cancer Res. 2013, 73, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Guo, W.Z.; Jin, Y.; Zhang, H.P.; Pang, C.; Li, J.; Line, P.D.; Zhang, S.J. Recognition of HER2 Expression in Hepatocellular Carcinoma and Its Significance in Postoperative Tumor Recurrence. Cancer Med. 2019, 8, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; et al. Rational Design of Shepherdin, a Novel Anticancer Agent. Cancer Cell 2005, 7, 457–468. [Google Scholar] [CrossRef]
- Oikawa, T.; Unno, Y.; Matsuno, K.; Sawada, J.I.; Ogo, N.; Tanaka, K.; Asai, A. Identification of a Small-Molecule Inhibitor of the Interaction between Survivin and Smac/DIABLO. Biochem. Biophys. Res. Commun. 2010, 393, 253–258. [Google Scholar] [CrossRef]
- Wadegaonkar, V.P.; Wadegaonkar, P.A. Withanone as an Inhibitor of Survivin: A Potential Drug Candidate for Cancer Therapy. J. Biotechnol. 2013, 168, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Sattarinezhad, E.; Bordbar, A.K.; Fani, N. Piperine Derivatives as Potential Inhibitors of Survivin: An in Silico Molecular Docking. Comput. Biol. Med. 2015, 63, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Shin, I.; Park, S.H.; Kim, N.D.; Shin, I. An Inhibitor of the Interaction of Survivin with Smac in Mitochondria Promotes Apoptosis. Chem. Asian J. 2019, 14, 4035–4041. [Google Scholar] [CrossRef]
- Hida, K.; Akiyama, K.; Ohga, N.; Maishi, N.; Hida, Y. Tumour Endothelial Cells Acquire Drug Resistance in a Tumour Microenvironment. J. Biochem. 2013, 153, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Sun, J.; Chen, X.; Liu, L.; Wu, D. Nek2 Augments Sorafenib Resistance by Regulating the Ubiquitination and Localization of β-Catenin in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 316. [Google Scholar] [CrossRef] [PubMed]
- Kokuryo, T.; Yokoyama, Y.; Yamaguchi, J.; Tsunoda, N.; Ebata, T.; Nagino, M. NEK2 Is an Effective Target for Cancer Therapy With Potential to Induce Regression of Multiple Human Malignancies. Anticancer. Res. 2019, 39, 2251–2258. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhang, X. Targeting NEK2 as a Promising Therapeutic Approach for Cancer Treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Zhu, J.; Guo, X.E.; Chen, W.; Qiu, X.L.; Ngo, B.; Chien, R.; Wang, Y.V.; Tsai, C.Y.; Wu, G.; et al. Novel Small Molecules Disrupting Hec1/Nek2 Interaction Ablate Tumor Progression by Triggering Nek2 Degradation through a Death-Trap Mechanism. Oncogene 2015, 34, 1220–1230. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | n | Median Age (Years) | Non-Tumour Endothelial Cells (NEC) | Tumour Endothelial Cells (TEC) | ||
|---|---|---|---|---|---|---|
| ENG− | ENG+ | ENG− | ENG+ | |||
| F | 3 | 64 | 3/3 | 2/2 | 2/3 1 | 2/2 |
| M | 13 | 52 | 12/13 2 | 6/6 | 12/13 3 | 4/6 4 |
| Total | 16 | 53 | 15/16 | 8/8 | 14/16 | 6/8 |
| Gene Symbol | Gene Name | Drug | PMIDs |
|---|---|---|---|
| Module: M1 | |||
| BIRC5 | baculoviral IAP repeat containing 5 | UCN-01, STAUROSPORINE, ROMIDEPSIN, VALDECOXIB, ERLOTINIB, OPRELVEKIN, VORINOSTAT, IMATINIB, TRASTUZUMAB, PACLITAXEL, FLUTAMIDE, CALCITONIN, GATAPARSEN, DEXAMETHASONE, ROFECOXIB, PLICAMYCIN, IRINOTECAN HYDROCHLORIDE, REGRAMOSTIM, EPIRUBICIN, TRETINOIN, SULINDAC, CARBOPLATIN, AMMONIUM TRICHLOROTELLURATE, DOCETAXEL, RESVERATROL, OMACETAXINE MEPESUCCINATE, METHOTREXATE, DOXORUBICIN, EPOETIN ALFA, LAPATINIB, INDOMETHACIN, CAMPTOTHECIN, PRASTERONE, GENISTEIN, FLUOROURACIL, RESERPINE, BERBERINE | 15255949, 17255535, 14767553, 16707021, 17047074, 14734714, 16951239, 16254145, 23204226, 16452223, 15347474, 16211241, 15735703, 16222118, 16787583, 14627349, 17124180, 15956246, 16403261, 16608080, 14587026, 16950207, 16889755, 14729643, 15837718, 17270149, 15854289, 15670151, 16211302, 17112829, 16461558, 17375591, 15067352, 17968851 |
| CCNB1 | cyclin B1 | KENPAULLONE, PROTOAPIGENONE, SELICICLIB | 21080703 |
| CDKN3 | cyclin-dependent kinase inhibitor 3 | PHA-793887, RONICICLIB, AZD-5438, AT-7519 | |
| FEN1 | flap structure-specific endonuclease 1 | TYRPHOSTIN 23, APOMORPHINE HYDROCHLORIDE HEMIHYDRATE, STREPTONIGRIN, ISOTHYMONIN, CALMIDAZOLIUM CHLORIDE, PIRARUBICIN, MYRICETIN, PROTOPORPHYRIN DISODIUM, CIANIDANOL, DEOXIEPINEPHRINE, TRANSPLATIN, SLAZINIC ACID, OXIDOPAMINE HYDROCHLORIDE, PENTABROMOPHENOL, AMINODIMETHOXYQUINAZOLINYLPIPERAZINE, 4-CHLOROMERCURIBENZOIC ACID, ELLIPTECINE, PINAFIDE, HYDROXYZINE PAMOATE, CLINAFLOXACIN, SENNOSIDE B, 3-O-METHYLQUERCETIN, PHENYLSTIBONIC ACID, PURPUROGALLIN, IDARUBICIN, CLOSANTEL, OXOPURPUREINE, THUNBERGINOL B, DIOTYROSINE, TYRPHOSTIN 51, DAPHNETIN, TETRAIDOFLUORESCEIN, EBSELEN, MITONAFIDE, QUINACRINE, LEVODOPA, STICTIC ACID, DEMECLOCYCLINE, DOXYCYCLINE, ATHRAQUINONES A, AMARANTH, THIMEROSAL, CEPHALOCHROMIN, HAEMATOXYLIN, METHOXSALEN, COUMESTROL, AURINTRICARBOXYLIC ACID, GW305074X, FERROUS FUMARATE, FERROUS GLYCINE SULFATE, GOSSYPOL, NOREPINEPHRINE, CETYLPYRIDINIUM BROMIDE, BENSERAZIDE HYDROCHLORIDE, METHACYCLINE HYDROCHLORIDE, SURAMIN, QUERCETIN, ASTERRIC ACID, METHYLENE BLUE, EMODIN, LUTEOLIN, SANGUINARINE SULFATE, RHEIN, METHYLDOPA (RACEMIC), ELLAGIC ACID, HOMIDIUM, DEPHOSTATIN, HOMIDIUM BROMIDE, EPINEPHRINE, AMINACRINE HYDROCHLORIDE, DITHIAZANINE, ACRIFLAVINE, CARMINE, AMENTOFLAVONE, DEQUALINIUM, LAVENDUSTIN C, TOLONIUM CHLORIDE, PROTOPORPHYRIN, HEXAMETHYL PARAROSANILINE, TAXIFOLIN, ALEXIDINE HYDROCHLORIDE, PICEATANNOL, METHYLTHIONINIUM CHLORIDE, OXOGLAUCINE, FAGARONINE, ACID BLUE 129, MORIN, DAUNORUBICIN HYDROCHLORIDE, CEFSULODIN SODIUM, HISPIDIN, METHYLDOPA, PAMOIC ACID, FRAXETIN, LOMOFUNGIN, ISOKAEMPFERIDE, NORDIHYDROGUAIARETIC ACID, EUPAFOLIN, FURAMIDINE, SANGUINARIUM CHLORIDE, MITOXANTRONE, HYCANTHONE, PYRONIN Y, CHARTREUSIN, CARMINIC ACID, 2-METHOXY-1, 4-NAPHTHOQUINONE, ERBSTATIN, MITOXANTRONE HYDROCHLORIDE, BAICALEIN, CATECHOL, PURPURIN, OXYTETRACYCLINE, DOPAMINE, INDOCYANINE GREEN | 3319774, 20622253 |
| NEK2 | NIMA related kinase 2 | ADAVOSERTIB, HESPERADIN, R-406, DACTOLISIB, PAZOPANIB, DOVITINIB, FOSTAMATINIB, GW441756X, TAE-684, CENISERTIB, GW843682X, ILORASERTIB, PALBOCICLIB, CYC-116, SP-600125, GSK-579289A | 19035792, 26516587 |
| TOP2A | topoisomerase (DNA) II alpha | DOXORUBICIN, BECATECARIN, C-1311, CARINATIN G, ETOPOSIDE, AMSACRINE, PODOFILOX, VALRUBICIN, EPIRUBICIN, IDRONOXIL, AMONAFIDE, 13-DEOXYDOXORUBICIN, AMRUBICIN HYDROCHLORIDE, HYDROQUINONE, DIAZIRINE, LUPEOL, DOXORUBICIN HYDROCHLORIDE, UNGEREMINE, DAUNORUBICIN HYDROCHLORIDE, SPARFLOXACIN, ANNAMYCIN, ELSAMITRUCIN, LOMEFLOXACIN, NORFLOXACIN, DACTINOMYCIN, FLEROXACIN, LUCANTHONE, AMRUBICIN, VOSAROXIN, TENIPOSIDE, DAUNORUBICIN, MITOXANTRONE, DAUNORUBICIN CITRATE, DEXRAZOXANE, BETULIN, IDARUBICIN, KAEMPFERITRIN, GANCOTAMAB, BERUBICIN HYDROCHLORIDE, ELINAFIDE, ETOPOSIDE PHOSPHATE, MYRICETIN, DEMETHYLZEYLASTERONE, FRANGULIN B, IDARUBICIN HYDROCHLORIDE, PACLITAXEL, FLUOROURACIL, SECAUBRYOLIDE, MITOXANTRONE HYDROCHLORIDE, GENISTEIN, QUERCETIN, HURATOXIN, FISETIN, DIGITOXIN, MOXIFLOXACIN, PEFLOXACIN, TROVAFLOXACIN, ENOXACIN, DECLOPRAMIDE, CIPROFLOXACIN, OFLOXACIN, ELLIPTECINE, ADRIAMYCIN, 4’-O-ACETYLPATENTIFLORIN B, MAKALUVAMINE E, DIPHYLLIN, SIMOCYCLINONE D8, BANOXANTRONE, FINAFLOXACIN, MAKALUVAMINE C TFA SALT, CAMPTOTHECIN, MAKALUVAMINE A, MAKALUVAMINE F, ALDOXORUBICIN, LYCOBETAINE, VINCRISTINE, TRICITRINOL B, OLEANDEROLIDE | 26211460, 17089011, 11752352, 17578914, 17010609, 20170164, 22276998, 21388138, 17351394, 17016621, 16377807, 16309315, 16271071, 8823806, 8870683, 23711769, 22867019, 25466187, 23968711, 23920485, 11678653, 25003995, 23566520, 21644529, 18258442, 25941559, 20863598, 22364746, 25922181, 23353750, 24931277, 24012683, 23360284, 20006518, 26216018, 17361331, 17514873, 9426516, 9485461, 9494516, 16759114, 25945730, 19691293, 24326278, 22620261, 26264845, 19783445, 26291037, 19386396, 24334150, 21435753, 25815139, 22867097, 25799376, 25800514, 25240702, 24775914, 18816045, 26292628, 24507920, 24095018, 1311390, 11473732, 17911018, 19155103, 25626146, 22537681, 19725581, 11006484, 11716434, 10691026, 1322791, 8519659, 8632768, 1845848, 10783066, 16061385, 1331331, 1334447, 16019763, 16234514, 17639997, 14728934, 17658777, 24054489, 15833037, 11754608, 22014547, 7756657, 3015015, 1323952, 9169823, 10487533, 17628745, 16480143, 8702194, 22260166, 1963303, 6380596, 11004693, 9631585, 10451375, 11278845, 18687447, 11046078, 12911317, 10194547, 17115008, 11179439, 17652819, 11984069, 11332155, 10203104, 12034365, 11836027, 8036155, 10523799, 22014228, 21880496, 7769390, 8759170, 20561793, 17139284, 17016423, 20596674, 20802486, 18471102, 10089819, 2847647, 22119124, 8691207, 17340571, 25808831, 11205246, 25312684, 23266176, 21761866 |
| TTK | TTK protein kinase | HESPERADIN, BAY-1217389, FOSTAMATINIB, BAY-1161909 | 19035792, 26516587 |
| UBE2T | ubiquitin conjugating enzyme E2T | MK-2206 | |
| Module: M14 | |||
| HSPG2 | heparan sulfate proteoglycan 2 | HALOPERIDOL, PALIFERMIN, CYCLOSPORINE | 27023437, 10593896, 16989989, 14753849, 9788974, 14974815 |
| PEAR1 | platelet endothelial aggregation receptor 1 | ASPIRIN, CLOPIDOGREL, PRASUGREL, TICAGRELOR | 23392654, 23859572, 28820077, 26962983, 27937053 |
| ERG | v-ets avian erythroblastosis virus E26 oncogene homolog | DOFETILIDE, SOTALOL HYDROCHLORIDE, AZD1305, NERISPIRDINE, IDARUBICIN HYDROCHLORIDE, MITONAFIDE, N-ACETYLASPARTIC ACID, GUANIDINE HYDROCHLORIDE, DALFAMPRIDINE, AZD7009, AMIODARONE HYDROCHLORIDE, MEDROXYPROGESTERONE ACETATE, HOMIDIUM BROMIDE, DAUNORUBICIN HYDROCHLORIDE, PERGOLIDE MESYLATE, 1, 4-DIMETHOXYANTHRAQUINONE, TEDISAMIL | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohr, T.; Katz, S.; Paulitschke, V.; Aizarani, N.; Tolios, A. Systematic Analysis of the Transcriptome Profiles and Co-Expression Networks of Tumour Endothelial Cells Identifies Several Tumour-Associated Modules and Potential Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2021, 13, 1768. https://doi.org/10.3390/cancers13081768
Mohr T, Katz S, Paulitschke V, Aizarani N, Tolios A. Systematic Analysis of the Transcriptome Profiles and Co-Expression Networks of Tumour Endothelial Cells Identifies Several Tumour-Associated Modules and Potential Therapeutic Targets in Hepatocellular Carcinoma. Cancers. 2021; 13(8):1768. https://doi.org/10.3390/cancers13081768
Chicago/Turabian StyleMohr, Thomas, Sonja Katz, Verena Paulitschke, Nadim Aizarani, and Alexander Tolios. 2021. "Systematic Analysis of the Transcriptome Profiles and Co-Expression Networks of Tumour Endothelial Cells Identifies Several Tumour-Associated Modules and Potential Therapeutic Targets in Hepatocellular Carcinoma" Cancers 13, no. 8: 1768. https://doi.org/10.3390/cancers13081768
APA StyleMohr, T., Katz, S., Paulitschke, V., Aizarani, N., & Tolios, A. (2021). Systematic Analysis of the Transcriptome Profiles and Co-Expression Networks of Tumour Endothelial Cells Identifies Several Tumour-Associated Modules and Potential Therapeutic Targets in Hepatocellular Carcinoma. Cancers, 13(8), 1768. https://doi.org/10.3390/cancers13081768