Multi-Omic Biomarkers as Potential Tools for the Characterisation of Pancreatic Cystic Lesions and Cancer: Innovative Patient Data Integration

 ,
,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Current Management of PCLs
3. Identification of Biomarkers in PCLs and PC Using Omics
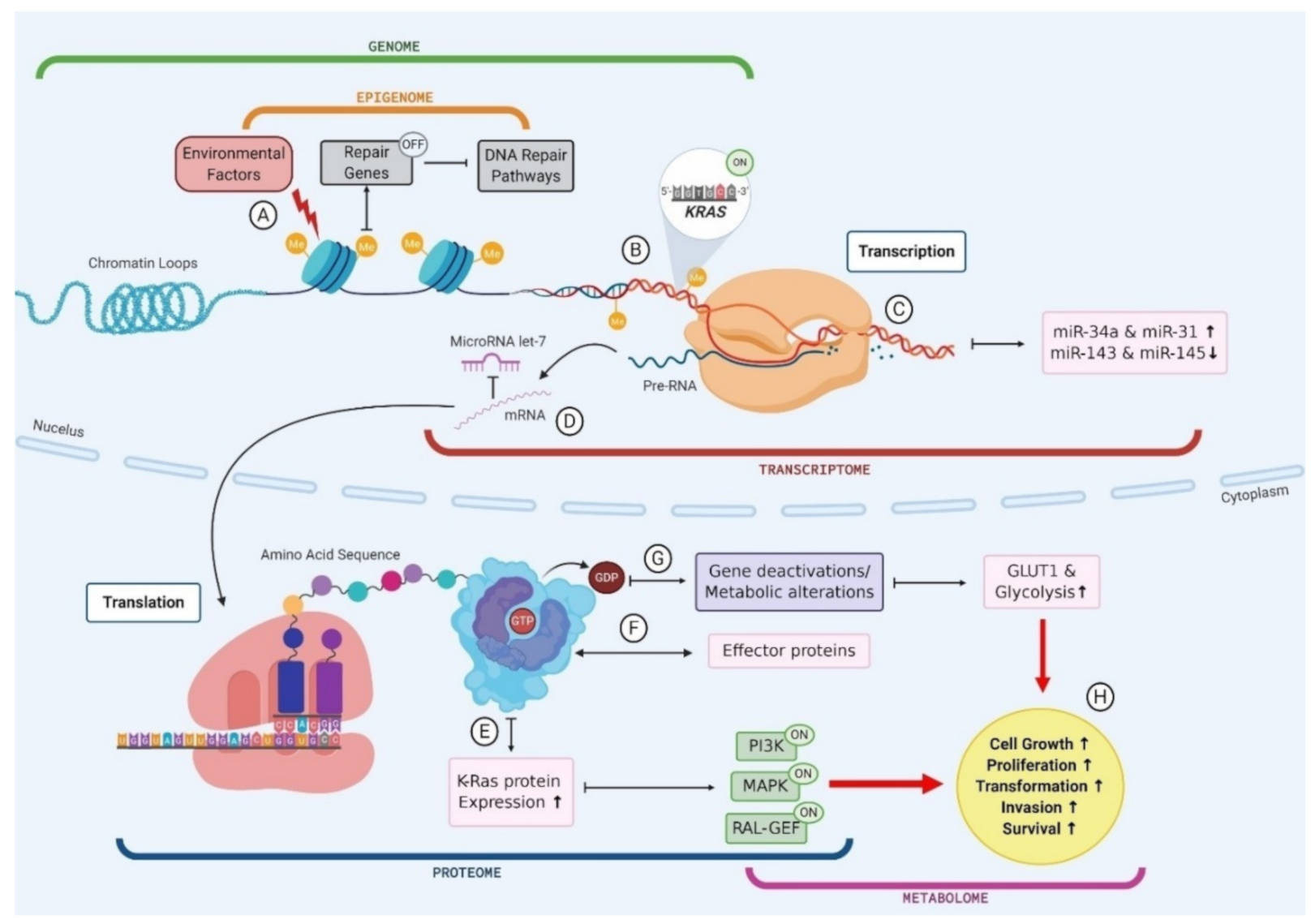
3.1. Genomics
3.2. Transcriptomics
3.3. Epigenomics
3.4. Proteomics
3.5. Metabolomics
4. Multi-Omics as the Key to Biomarker Identification
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Park, J.; Han, D.; Do, M.; Woo, J.; Wang, J.I.; Han, Y.; Kwon, W.; Kim, S.W.; Jang, J.Y.; Kim, Y. Proteome characterization of human pancreatic cyst fluid from intraductal papillary mucinous neoplasm by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2017, 31, 1761–1772. [Google Scholar] [CrossRef]
- Soufi, M.; Yip-Schneider, M.T.; Carr, R.A.; Roch, A.M.; Wu, H.H.; Schmidt, C.M. Multifocal High-Grade Pancreatic Precursor Lesions: A Case Series and Management Recommendations. J. Pancreat. Cancer 2019, 5, 8–11. [Google Scholar] [CrossRef]
- Matthaei, H.; Schulick, R.D.; Hruban, R.H.; Maitra, A. Cystic precursors to invasive pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846. [Google Scholar] [CrossRef] [PubMed]
- Midha, S.; Chawla, S.; Garg, P.K. Modifiable and non-modifiable risk factors for pancreatic cancer: A review. Cancer Lett. 2016, 381, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.; Donahue, T.R.; Reber, H.A.; Hines, O.J. Pancreatic cyst disease: A review. JAMA 2016, 315, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Tersmette, A.C.; Petersen, G.M.; Offerhaus, G.J.A.; Falatko, F.C.; Brune, K.A.; Goggins, M.; Rozenblum, E.; Wilentz, R.E.; Yeo, C.J.; Cameron, J.L. Increased risk of incident pancreatic cancer among first-degree relatives of patients with familial pancreatic cancer. Clin. Cancer Res. 2001, 7, 738–744. [Google Scholar] [PubMed]
- Greer, J.B.; Whitcomb, D.C. Role of BRCA1 and BRCA2 mutations in pancreatic cancer. Gut 2007, 56, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Sadowski, S.M.; Linehan, W.M.; Libutti, S.K.; Patel, D.; Nilubol, N.; Kebebew, E. Association of VHL genotype with pancreatic neuroendocrine tumor phenotype in patients with von Hippel–Lindau disease. JAMA Oncol. 2018, 4, 124–126. [Google Scholar] [CrossRef]
- Kromrey, M.-L.; Bülow, R.; Hübner, J.; Paperlein, C.; Lerch, M.M.; Ittermann, T.; Völzke, H.; Mayerle, J.; Kühn, J.-P. Prospective study on the incidence, prevalence and 5-year pancreatic-related mortality of pancreatic cysts in a population-based study. Gut 2018, 67, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Torisu, Y.; Takakura, K.; Kinoshita, Y.; Tomita, Y.; Nakano, M.; Saruta, M. Pancreatic cancer screening in patients with presumed branch-duct intraductal papillary mucinous neoplasms. World J. Clin. Oncol. 2019, 10, 67. [Google Scholar] [CrossRef]
- Wong, M.C.; Jiang, J.Y.; Liang, M.; Fang, Y.; Yeung, M.S.; Sung, J.J. Global temporal patterns of pancreatic cancer and association with socioeconomic development. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Brugge, W.R. Diagnosis and management of cystic lesions of the pancreas. J. Gastrointest. Oncol. 2015, 6, 375. [Google Scholar] [PubMed]
- Maggi, G.; Guarneri, G.; Gasparini, G.; Fogliati, A.; Partelli, S.; Falconi, M.; Crippa, S. Pancreatic cystic neoplasms: What is the most cost-effective follow-up strategy? Endosc. Ultrasound 2018, 7, 319. [Google Scholar]
- de Jong, K.; Nio, C.Y.; Hermans, J.J.; Dijkgraaf, M.G.; Gouma, D.J.; van Eijck, C.H.; van Heel, E.; Klass, G.; Fockens, P.; Bruno, M.J. High prevalence of pancreatic cysts detected by screening magnetic resonance imaging examinations. Clin. Gastroenterol. Hepatol. 2010, 8, 806–811. [Google Scholar] [CrossRef]
- Chang, Y.R.; Park, J.K.; Jang, J.-Y.; Kwon, W.; Yoon, J.H.; Kim, S.-W. Incidental pancreatic cystic neoplasms in an asymptomatic healthy population of 21,745 individuals: Large-scale, single-center cohort study. Medicine 2016, 95, e5535. [Google Scholar] [CrossRef]
- Sureka, B.; Varshney, V. Pancreatic Incidentalomas: Review and Current Management Recommendations. Ann. Natl. Acad. Med. Sci. 2019, 55, 006–013. [Google Scholar] [CrossRef]
- Sakhdari, A.; Moghaddam, P.A.; Ok, C.Y.; Walter, O.; Tomaszewicz, K.; Caporelli, M.-L.; Meng, X.; LaFemina, J.; Whalen, G.; Belkin, E.; et al. Somatic molecular analysis augments cytologic evaluation of pancreatic cyst fluids as a diagnostic tool. Oncotarget 2018, 10, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fernández-del Castillo, C.; Adsay, V.; Chari, S.; Falconi, M.; Jang, J.-Y.; Kimura, W.; Levy, P.; Pitman, M.B.; Schmidt, C.M. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology 2012, 12, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Hanania, A.N.; Bantis, L.E.; Feng, Z.; Wang, H.; Tamm, E.P.; Katz, M.H.; Maitra, A.; Koay, E.J. Quantitative imaging to evaluate malignant potential of IPMNs. Oncotarget 2016, 7, 85776. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fernández-del, C.; Kamisawa, T.; Jang, J.Y.; Levy, P.; Ohtsuka, T.; Salvia, R.; Shimizu, Y.; Tada, M.; Wolfgang, C.L. Revisions of international consensus Fukuoka guidelines for the management of IPMN of the pancreas. Pancreatology 2017, 17, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Vege, S.S.; Ziring, B.; Jain, R.; Moayyedi, P.; Adams, M.A.; Dorn, S.D.; Dudley-Brown, S.L.; Flamm, S.L.; Gellad, Z.F.; Gruss, C.B. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015, 148, 819–822. [Google Scholar] [CrossRef] [PubMed]
- European Study Group on Cystic Tumours of the Pancreas, European evidence-based guidelines on pancreatic cystic neoplasms. Gut 2018, 67, 789–804. [CrossRef] [PubMed]
- Maker, A.V.; Carrara, S.; Jamieson, N.B.; Pelaez-Luna, M.; Lennon, A.M.; Molin, M.D.; Scarpa, A.; Frulloni, L.; Brugge, W.R. Cyst fluid biomarkers for intraductal papillary mucinous neoplasms of the pancreas: A critical review from the international expert meeting on pancreatic branch-duct-intraductal papillary mucinous neoplasms. J. Am. Coll. Surg. 2015, 220, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M. Intraductal papillary mucinous neoplasm of the pancreas as the main focus for early detection of pancreatic adenocarcinoma. Pancreas 2018, 47, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Kadiyala, V.; Lee, L.S. Evaluation of AGA and Fukuoka Guidelines for EUS and surgical resection of incidental pancreatic cysts. Endosc. Int. Open 2017, 5, E116–E122. [Google Scholar] [CrossRef] [PubMed]
- Valsangkar, N.P.; Morales-Oyarvide, V.; Thayer, S.P.; Ferrone, C.R.; Wargo, J.A.; Warshaw, A.L.; Castillo, C.F.-d. 851 resected cystic tumors of the pancreas: A 33-year experience at the Massachusetts General Hospital. Surgery 2012, 152, S4–S12. [Google Scholar] [CrossRef]
- Fritz, S.; Klauss, M.; Bergmann, F.; Strobel, O.; Schneider, L.; Werner, J.; Hackert, T.; Büchler, M.W. Pancreatic main-duct involvement in branch-duct IPMNs: An underestimated risk. Ann. Surg. 2014, 260, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Meyer, J.E.; Waters, J.A.; Al-Haddad, M.; DeWitt, J.; Sherman, S.; Lillemoe, K.D.; Schmidt, C.M. Outcome of the pancreatic remnant following segmental pancreatectomy for non-invasive intraductal papillary mucinous neoplasm. HPB 2011, 13, 759–766. [Google Scholar] [CrossRef]
- Tan, M.C.; Basturk, O.; Brannon, A.R.; Bhanot, U.; Scott, S.N.; Bouvier, N.; LaFemina, J.; Jarnagin, W.R.; Berger, M.F.; Klimstra, D. GNAS and KRAS mutations define separate progression pathways in intraductal papillary mucinous neoplasm-associated carcinoma. J. Am. Coll. Surg. 2015, 220, 845–854.e1. [Google Scholar] [CrossRef]
- Maker, A.V.; Lee, L.S.; Raut, C.P.; Clancy, T.E.; Swanson, R.S. Cytology from pancreatic cysts has marginal utility in surgical decision-making. Ann. of Surg. Oncol. 2008, 15, 3187–3192. [Google Scholar] [CrossRef]
- Le Borgne, J.; de Calan, L.; Partensky, C.; Association, F.S. Cystadenomas and cystadenocarcinomas of the pancreas: A multiinstitutional retrospective study of 398 cases. Ann. Surg. 1999, 230, 152. [Google Scholar] [CrossRef]
- Thornton, G.; McPhail, M.; Nayagam, S.; Hewitt, M.; Vlavianos, P.; Monahan, K. Endoscopic ultrasound guided fine needle aspiration for the diagnosis of pancreatic cystic neoplasms: A meta-analysis. Pancreatology 2013, 13, 48–57. [Google Scholar] [CrossRef]
- Hasan, S.; Jacob, R.; Manne, U.; Paluri, R. Advances in pancreatic cancer biomarkers. Oncol. Rev. 2019, 13, 410. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Singhi, A.D.; Nikiforova, M.N.; Fasanella, K.E.; McGrath, K.M.; Pai, R.K.; Ohori, N.P.; Bartholow, T.L.; Brand, R.E.; Chennat, J.S.; Lu, X. Preoperative GNAS and KRAS testing in the diagnosis of pancreatic mucinous cysts. Clin. Cancer Res. 2014, 20, 4381–4389. [Google Scholar] [CrossRef] [PubMed]
- Kadayifci, A.; Atar, M.; Wang, J.L.; Forcione, D.G.; Casey, B.W.; Pitman, M.B.; Brugge, W.R. Value of adding GNAS testing to pancreatic cyst fluid KRAS and carcinoembryonic antigen analysis for the diagnosis of intraductal papillary mucinous neoplasms. Dig. Endosc. 2017, 29, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; McGrath, K.; Brand, R.E.; Khalid, A.; Zeh, H.J.; Chennat, J.S.; Fasanella, K.E.; Papachristou, G.I.; Slivka, A.; Bartlett, D.L. Preoperative next-generation sequencing of pancreatic cyst fluid is highly accurate in cyst classification and detection of advanced neoplasia. Gut 2018, 67, 2131–2141. [Google Scholar] [CrossRef]
- Ding, J.; Li, Y.; Zhang, Y.; Fan, B.; Li, Q.; Zhang, J.; Zhang, J. Identification of key lncRNAs in the tumorigenesis of intraductal pancreatic mucinous neoplasm by coexpression network analysis. Cancer Med. 2020, 9, 3840–3851. [Google Scholar] [CrossRef] [PubMed]
- Park, W.G.; Wu, M.; Bowen, R.; Zheng, M.; Fitch, W.L.; Pai, R.K.; Wodziak, D.; Visser, B.C.; Poultsides, G.A.; Norton, J.A. Metabolomic-derived novel cyst fluid biomarkers for pancreatic cysts: Glucose and kynurenine. Gastrointest. Endosc. 2013, 78, 295–302.e2. [Google Scholar] [CrossRef]
- Carr, R.A.; Yip-Schneider, M.T.; Simpson, R.E.; Dolejs, S.; Schneider, J.G.; Wu, H.; Ceppa, E.P.; Park, W.; Schmidt, C.M. Pancreatic cyst fluid glucose: Rapid, inexpensive, and accurate diagnosis of mucinous pancreatic cysts. Surgery 2018, 163, 600–605. [Google Scholar] [CrossRef]
- Fahrmann, J.F.; Bantis, L.E.; Capello, M.; Scelo, G.; Dennison, J.B.; Patel, N.; Murage, E.; Vykoukal, J.; Kundnani, D.L.; Foretova, L. A plasma-derived protein-metabolite multiplexed panel for early-stage pancreatic cancer. J. Natl. Cancer Inst. 2019, 111, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Eissa, M.A.; Lerner, L.; Abdelfatah, E.; Shankar, N.; Canner, J.K.; Hasan, N.M.; Yaghoobi, V.; Huang, B.; Kerner, Z.; Takaesu, F. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin. Epigenetics 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Molin, M.D.; Hong, S.-M.; Tamura, K.; Suenaga, M.; Yu, J.; Sedogawa, H.; Weiss, M.J.; Wolfgang, C.L.; Lennon, A.M. Predicting the grade of dysplasia of pancreatic cystic neoplasms using cyst fluid DNA methylation markers. Clin. Cancer Res. 2017, 23, 3935–3944. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Taylor, W.R.; Yab, T.C.; Berger, C.K.; Dukek, B.A.; Cao, X.; Foote, P.H.; Wu, C.W.; Mahoney, D.W.; Aslanian, H.R. Novel Methylated DNA Markers Discriminate Advanced Neoplasia in Pancreatic Cysts: Marker Discovery, Tissue Validation, and Cyst Fluid Testing. Am. J. Gastroenterol. 2019, 114, 1539–1549. [Google Scholar] [CrossRef]
- Li, A.; Yu, J.; Kim, H.; Wolfgang, C.L.; Canto, M.I.; Hruban, R.H.; Goggins, M. MicroRNA array analysis finds elevated serum miR-1290 accurately distinguishes patients with low-stage pancreatic cancer from healthy and disease controls. Clin. Cancer Res. 2013, 19, 3600–3610. [Google Scholar] [CrossRef]
- Karasek, P.; Gablo, N.; Hlavsa, J.; Kiss, I.; Vychytilova-Faltejskova, P.; Hermanova, M.; Kala, Z.; Slaby, O.; Prochazka, V. Pre-operative plasma miR-21-5p is a sensitive biomarker and independent prognostic factor in patients with pancreatic ductal adenocarcinoma undergoing surgical resection. Cancer Genom. Proteom. 2018, 15, 321–327. [Google Scholar] [CrossRef]
- Wei, J.; Yang, L.; Wu, Y.-n.; Xu, J. Serum miR-1290 and miR-1246 as Potential Diagnostic Biomarkers of Human Pancreatic Cancer. J. Cancer 2020, 11, 1325–1333. [Google Scholar] [CrossRef]
- Matthaei, H.; Wylie, D.; Lloyd, M.B.; Molin, M.D.; JKemppainen Mayo, S.C.; Wolfgang, C.L.; Schulick, R.D.; Langfield, L.; Andruss, B.F. miRNA biomarkers in cyst fluid augment the diagnosis and management of pancreatic cysts. Clin. Cancer Res. 2012, 18, 4713–4724. [Google Scholar] [CrossRef]
- Xie, Z.; Yin, X.; Gong, B.; Nie, W.; Wu, B.; Zhang, X.; Huang, J.; Zhang, P.; Zhou, Z.; Li, Z. Salivary microRNAs show potential as a noninvasive biomarker for detecting resectable pancreatic cancer. Cancer Prev. Res. 2015, 8, 165–173. [Google Scholar] [CrossRef]
- Brand, R.E.; Nolen, B.M.; Zeh, H.J.; Allen, P.J.; Eloubeidi, M.A.; Goldberg, M.; Elton, E.; Arnoletti, J.P.; Christein, J.D.; Vickers, S.M. Serum biomarker panels for the detection of pancreatic cancer. Clin. Cancer Res. 2011, 17, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Prassas, I.; Dimitromanolakis, A.; Brand, R.E.; Serra, S.; Diamandis, E.P.; Blasutig, I.M. Validation of biomarkers that complement CA19. 9 in detecting early pancreatic cancer. Clin. Cancer Res. 2014, 20, 5787–5795. [Google Scholar] [PubMed]
- Azadeh, A.; Felix, R.; Krause, T.; Bernhardt, M.; Jo, P.; König, A.; Mathias, K.; Andreas, L.; Ghadimi, M.; Jochen, G. CA19-9 for detecting recurrence of pancreatic cancer. Sci. Rep. 2020, 10, 1–10. [Google Scholar]
- Carr, R.A.; Yip-Schneider, M.T.; Dolejs, S.; Hancock, B.A.; Wu, H.; Radovich, M.; Schmidt, C.M. Pancreatic cyst fluid vascular endothelial growth factor A and carcinoembryonic antigen: A highly accurate test for the diagnosis of serous cystic neoplasm. J. Am. Coll. Surg. 2017, 225, 93–100. [Google Scholar] [CrossRef]
- Sinha, J.; Cao, Z.; Dai, J.; Tang, H.; Partyka, K.; Hostetter, G.; Simeone, D.M.; Feng, Z.; Allen, P.J.; Brand, R.E. A gastric glycoform of MUC5AC is a biomarker of mucinous cysts of the pancreas. PLoS ONE 2016, 11, e0167070. [Google Scholar] [CrossRef]
- Cao, Z.; Maupin, K.; Curnutte, B.; Fallon, B.; Feasley, C.L.; Brouhard, E.; Kwon, R.; West, C.M.; Cunningham, J.; Brand, R. Specific glycoforms of MUC5AC and endorepellin accurately distinguish mucinous from nonmucinous pancreatic cysts. Mol. Cell. Proteom. 2013, 12, 2724–2734. [Google Scholar] [CrossRef]
- Rebours, V.; le Faouder, J.; Laouirem, S.; Mebarki, M.; Albuquerque, M.; Camadro, J.-M.; Léger, T.; Ruszniewski, P.; Lévy, P.; Paradis, V. In situ proteomic analysis by MALDI imaging identifies ubiquitin and thymosin-β4 as markers of malignant intraductal pancreatic mucinous neoplasms. Pancreatology 2014, 14, 117–124. [Google Scholar] [CrossRef]
- Kung, J.S.; Lopez, O.A.; McCoy, E.E.; Reicher, S.; Eysselein, V.E. Fluid genetic analyses predict the biological behavior of pancreatic cysts: Three-year experience. JOP J. Pancreas 2014, 15, 427–432. [Google Scholar] [CrossRef]
- Wu, J.; Matthaei, H.; Maitra, A.; Molin, M.D.; Wood, L.D.; Eshleman, J.R.; Goggins, M.; Canto, M.I.; Schulick, R.D.; Edil, B.H. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 2011, 3, 92ra66. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef]
- Lu, S.; Ahmed, T.; Du, P.; Wang, Y. Genomic variations in pancreatic cancer and potential opportunities for development of new approaches for diagnosis and treatment. Int. J. Mol. Sci. 2017, 18, 1201. [Google Scholar] [CrossRef]
- Macgregor-Das, A.M.; Iacobuzio-Donahue, C.A. Molecular pathways in pancreatic carcinogenesis. J. Surg. Oncol. 2013, 107, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Khalid, A.; Zahid, M.; Finkelstein, S.D.; LeBlanc, J.K.; Kaushik, N.; Ahmad, N.; Brugge, W.R.; Edmundowicz, S.A.; Hawes, R.H.; McGrath, K.M. Pancreatic cyst fluid DNA analysis in evaluating pancreatic cysts: A report of the PANDA study. Gastrointest. Endosc. 2009, 69, 1095–1102. [Google Scholar] [CrossRef]
- Reid, M.D.; Saka, B.; Balci, S.; Goldblum, A.S.; Adsay, N.V. Molecular genetics of pancreatic neoplasms and their morphologic correlates: An update on recent advances and potential diagnostic applications. Am. J. Clin. Pathol. 2014, 141, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Tulla, K.A.; Maker, A.V. Can we better predict the biologic behavior of incidental IPMN? A comprehensive analysis of molecular diagnostics and biomarkers in intraductal papillary mucinous neoplasms of the pancreas. Langenbeck’s Arch. Surg. 2018, 403, 151–194. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, Y.; Choi, J.-W.; Kim, Y.-S. KRAS, GNAS, and RNF43 mutations in intraductal papillary mucinous neoplasm of the pancreas: A meta-analysis. Springerplus 2016, 5, 1172. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, W.; Sasaki, E.; Murakami, Y.; Yamao, K.; Shimizu, Y.; Yatabe, Y. GNAS mutation is a frequent event in pancreatic intraductal papillary mucinous neoplasms and associated adenocarcinomas. Virchows Arch. 2015, 466, 665–674. [Google Scholar] [CrossRef]
- Omori, Y.; Ono, Y.; Tanino, M.; Karasaki, H.; Yamaguchi, H.; Furukawa, T.; Enomoto, K.; Ueda, J.; Sumi, A.; Katayama, J. Pathways of progression from intraductal papillary mucinous neoplasm to pancreatic ductal adenocarcinoma based on molecular features. Gastroenterology 2019, 156, 647–661. [Google Scholar] [CrossRef]
- Liang, W.S.; Craig, D.W.; Carpten, J.; Borad, M.J.; Demeure, M.J.; Weiss, G.J.; Izatt, T.; Sinari, S.; Christoforides, A.; Aldrich, J. Genome-wide characterization of pancreatic adenocarcinoma patients using next generation sequencing. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chaudhary, K.; Garmire, L.X. More is better: Recent progress in multi-omics data integration methods. Front. Genet. 2017, 8, 84. [Google Scholar] [CrossRef]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, Y.G.; Lucas, A.L. MicroRNA in pancreatic ductal adenocarcinoma and its precursor lesions. World J. Gastrointest. Oncol. 2016, 8, 18. [Google Scholar] [CrossRef]
- Lee, L.S.; Szafranska-Schwarzbach, A.E.; Wylie, D.; Doyle, L.A.; Bellizzi, A.M.; Kadiyala, V.; Suleiman, S.; Banks, P.A.; Andruss, B.F.; Conwell, D.L. Investigating microRNA expression profiles in pancreatic cystic neoplasms. Clin. Transl. Gastroenterol. 2014, 5, e47. [Google Scholar] [CrossRef] [PubMed]
- Vila-Navarro, E.; Vila-Casadesús, M.; Moreira, L.; Duran-Sanchon, S.; Sinha, R.; Ginés, À.; Fernández-Esparrach, G.; Miquel, R.; Cuatrecasas, M.; Castells, A. MicroRNAs for detection of pancreatic neoplasia: Biomarker discovery by next-generation sequencing and validation in 2 independent cohorts. Ann. Surg. 2017, 265, 1226. [Google Scholar] [CrossRef]
- Humeau, M.; Vignolle-Vidoni, A.; Sicard, F.; Martins, F.; Bournet, B.; Buscail, L.; Torrisani, J.; Cordelier, P. Salivary microRNA in pancreatic cancer patients. PLoS ONE 2015, 10, e0130996. [Google Scholar] [CrossRef]
- Rapado-González, Ó.; Martínez-Reglero, C.; Salgado-Barreira, Á.; Takkouche, B.; López-López, R.; Suárez-Cunqueiro, M.M.; Muinelo-Romay, L. Salivary biomarkers for cancer diagnosis: A meta-analysis. Ann. Med. 2020, 52, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef]
- Wang, Z.; Monteiro, C.D.; Jagodnik, K.M.; Fernandez, N.F.; Gundersen, G.W.; Rouillard, A.D.; Jenkins, S.L.; Feldmann, A.S.; Hu, K.S.; McDermott, M.G. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Toll, A.D.; Dasgupta, A.; Potoczek, M.; Yeo, C.J.; Kleer, C.G.; Brody, J.R.; Witkiewicz, A.K. Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Hum. Pathol. 2010, 41, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xie, D.; Li, W.Y.; Cheung, C.M.; Yao, H.; Chan, C.Y.; Chan, C.-y.; Xu, F.-P.; Liu, Y.-H.; Sung, J.J. RNAi targeting EZH2 inhibits tumor growth and liver metastasis of pancreatic cancer in vivo. Cancer Lett. 2010, 297, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yang, C.; Fan, P.; Xiao, J.; Zhang, W.; Zhan, S.; Liu, T.; Wang, D.; Wu, H. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J. Biol. Chem. 2017, 292, 6269–6280. [Google Scholar] [CrossRef]
- Rahman, M.M.; Brane, A.C.; Tollefsbol, T.O. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells 2019, 8, 1214. [Google Scholar] [CrossRef]
- Fujiyama, Y.; Kumamoto, Y.; Nishizawa, N.; Nakamoto, S.; Harada, H.; Yokota, K.; Tanaka, Y.; Igarashi, K.; Oiki, H.; Okuwaki, K. Promoter DNA Hypermethylation of the Cysteine Dioxygenase 1 (CDO1) Gene in Intraductal Papillary Mucinous Neoplasm (IPMN). Ann. Surg. Oncol. 2020, 27, 4007–4016. [Google Scholar] [CrossRef]
- Meissner, A. What can epigenomics do for you? Genome Biol. 2012, 13, 420. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Briefings Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Yu, K.-H.; Snyder, M. Omics Profiling in Precision Oncology. Mol. Cell. Proteom. 2016, 15, 2525–2536. [Google Scholar] [CrossRef]
- Kwon, R.S.; Simeone, D.M. The Use of Protein-Based Biomarkers for the Diagnosis of Cystic Tumors of the Pancreas. Int. J. Proteom. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ngamruengphong, S.; Bartel, M.J.; Raimondo, M. Cyst carcinoembryonic antigen in differentiating pancreatic cysts: A meta-analysis. Dig. Liver Dis. 2013, 45, 920–926. [Google Scholar] [CrossRef]
- Al-Haddad, M.; DeWitt, J.; Sherman, S.; Schmidt, C.M.; LeBlanc, J.K.; McHenry, L.; Coté, G.; el Chafic, A.H.; Luz, L.; Stuart, J.S. Performance characteristics of molecular (DNA) analysis for the diagnosis of mucinous pancreatic cysts. Gastrointest. Endosc. 2014, 79, 79–87. [Google Scholar] [CrossRef]
- Kurita, Y.; Kuwahara, T.; Hara, K.; Mizuno, N.; Okuno, N.; Matsumoto, S.; Obata, M.; Koda, H.; Tajika, M.; Shimizu, Y. Diagnostic ability of artificial intelligence using deep learning analysis of cyst fluid in differentiating malignant from benign pancreatic cystic lesions. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Okusaka, T.; Felix, K.; Nakamori, S.; Sata, N.; Nagai, H.; Ioka, T.; Tsuchida, A.; Shimahara, T.; Shimahara, M. Altered plasma apolipoprotein modifications in patients with pancreatic cancer: Protein characterization and multi-institutional validation. PLoS ONE 2012, 7, e46908. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Kobayashi, M.; Okusaka, T.; Rinaudo, J.A.; Huang, Y.; Marsh, T.; Sanada, M.; Sasajima, Y.; Nakamori, S.; Shimahara, M. Plasma biomarker for detection of early stage pancreatic cancer and risk factors for pancreatic malignancy using antibodies for apolipoprotein-AII isoforms. Sci. Rep. 2015, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Katzke, V.A.; Hüsing, A.; Okaya, S.; Shoji, H.; Onidani, K.; Olsen, A.; Tjønneland, A.; Overvad, K.; Weiderpass, E. CA19-9 and apolipoprotein-A2 isoforms as detection markers for pancreatic cancer: A prospective evaluation. Int. J. Cancer 2019, 144, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Porterfield, M.; Zhao, P.; Han, H.; Cunningham, J.; Aoki, K.; von Hoff, D.D.; Demeure, M.J.; Pierce, J.M.; Tiemeyer, M.; Wells, L. Discrimination between adenocarcinoma and normal pancreatic ductal fluid by proteomic and glycomic analysis. J. Proteome Res. 2014, 13, 395–407. [Google Scholar] [CrossRef]
- de Oliveira, G.; Freire, P.P.; Cury, S.S.; de Moraes, D.; Oliveira, J.S.; Dal-Pai-Silva, M.; Reis, P.P.; Carvalho, R.F. An Integrated Meta-Analysis of Secretome and Proteome Identify Potential Biomarkers of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 716. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Horinouchi, M.; Saitou, M.; Higashi, M.; Nomoto, M.; Goto, M.; Yonezawa, S. Mucin expression profile in pancreatic cancer and the precursor lesions. J. Hepato BiliaryPancreat. Surg. 2007, 14, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Carrara, S.; Cangi, M.G.; Arcidiacono, P.G.; Perri, F.; Petrone, M.C.; Mezzi, G.; Boemo, C.; Talarico, A.; Cin, E.D.; Grassini, G. Mucin expression pattern in pancreatic diseases: Findings from EUS-guided fine-needle aspiration biopsies. Am. J. Gastroenterol. 2011, 106, 1359–1363. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Wu, H.; Dumas, R.P.; Hancock, B.A.; Agaram, N.; Radovich, M.; Schmidt, C.M. Vascular endothelial growth factor, a novel and highly accurate pancreatic fluid biomarker for serous pancreatic cysts. J. Am. Coll. Surg. 2014, 218, 608–617. [Google Scholar] [CrossRef]
- Malaker, S.A.; Pedram, K.; Ferracane, M.J.; Bensing, B.A.; Krishnan, V.; Pett, C.; Yu, J.; Woods, E.C.; Kramer, J.R.; Westerlind, U. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc. Natl. Acad. Sci. USA 2019, 116, 7278–7287. [Google Scholar] [CrossRef]
- Rudnick, P.A.; Markey, S.P.; Roth, J.; Mirokhin, Y.; Yan, X.; Tchekhovskoi, D.V.; Edwards, N.J.; Thangudu, R.R.; Ketchum, K.A.; Kinsinger, C.R. A description of the clinical proteomic tumor analysis consortium (CPTAC) common data analysis pipeline. J. Proteome Res. 2016, 15, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Côté, R.G.; Griss, J.; Dianes, J.A.; Wang, R.; Wright, J.C.; van den Toorn, H.W.; van Breukelen, B.; Heck, A.J.; Hulstaert, N.; Martens, L. The PRoteomics IDEntification (PRIDE) Converter 2 framework: An improved suite of tools to facilitate data submission to the PRIDE database and the ProteomeXchange consortium. Mol. Cell. Proteom. 2012, 11, 1682–1689. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Mayerle, J.; Kalthoff, H.; Reszka, R.; Kamlage, B.; Peter, E.; Schniewind, B.; Maldonado, S.G.; Pilarsky, C.; Heidecke, C.-D.; Schatz, P. Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut 2018, 67, 128–137. [Google Scholar] [CrossRef]
- Zhang, G.; He, P.; Tan, H.; Budhu, A.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.G.; Lee, D.H.; Maitra, A. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin. Cancer Res. 2013, 19, 4983–4993. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef] [PubMed]
- Springer, S.; Masica, D.L.; Molin, M.D.; Douville, C.; Thoburn, C.J.; Afsari, B.; Li, L.; Cohen, J.D.; Thompson, E.; Allen, P.J. A multimodality test to guide the management of patients with a pancreatic cyst. Sci. Transl. Med. 2019, 11, eaav4772. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Calanzani, N.; Druce, P.E.; Snudden, C.; Milley, K.M.; Boscott, R.; Behiyat, D.; Saji, S.; Martinez-Gutierrez, J.; Oberoi, J.; Funston, G. Identifying Novel Biomarkers Ready for Evaluation in Low-Prevalence Populations for the Early Detection of Upper Gastrointestinal Cancers: A Systematic Review. Adv. Ther. 2020, 1–42. [Google Scholar]
- Springer, S.; Wang, Y.; Molin, M.D.; Masica, D.L.; Jiao, Y.; Kinde, I.; Blackford, A.; Raman, S.P.; Wolfgang, C.L.; Tomita, T. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology 2015, 149, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Natale, F.; Vivo, M.; Falco, G.; Angrisano, T. Deciphering DNA methylation signatures of pancreatic cancer and pancreatitis. Clin. Epigenetics 2019, 11, 132. [Google Scholar] [CrossRef]
- Kent, O.A.; Chivukula, R.R.; Mullendore, M.; Wentzel, E.A.; Feldmann, G.; Lee, K.H.; Liu, S.; Leach, S.D.; Maitra, A.; Mendell, J.T. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010, 24, 2754–2759. [Google Scholar] [CrossRef]
- Kent, O.A.; Mendell, J.T.; Rottapel, R. Transcriptional regulation of miR-31 by oncogenic KRAS mediates metastatic phenotypes by repressing RASA1. Mol. Cancer Res. 2016, 14, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Rachagani, S.; Macha, M.A.; Heimann, N.; Seshacharyulu, P.; Haridas, D.; Chugh, S.; Batra, S.K. Clinical implications of miRNAs in the pathogenesis, diagnosis and therapy of pancreatic cancer. Adv. Drug Deliv. Rev. 2015, 81, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-driven metabolic rewiring reveals novel actionable targets in cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Kong, L.; Liu, P.; Zheng, M.; Xue, B.; Liang, K.; Tan, X. Multi-omics analysis based on integrated genomics, epigenomics and transcriptomics in pancreatic cancer. Epigenomics 2020, 12, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Monkman, J.; Blick, T.; Duijf, P.H.; Nagaraj, S.H.; Thompson, E.W. Multi-omics characterization of the spontaneous mesenchymal–epithelial transition in the PMC42 breast cancer cell lines. J. Clin. Med. 2019, 8, 1253. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, B.; Liu, Z.; Ren, X.; Zhao, W.; Li, Z.; You, L.; Zhao, Y. Molecular subtyping of pancreatic cancer: Translating genomics and transcriptomics into the clinic. J. Cancer 2017, 8, 513. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Biomarker Name | Biomarker Type | Single or Multi-Study Validated | Platform | Sample Type | Sample Size (Total Number Patients) | Sensitivity (95% Confidence Interval) | Specificity (95% Confidence Interval) | P-Value | Purpose | References |
|---|---|---|---|---|---|---|---|---|---|---|
| KRAS and/OR GNAS | Genetic mutation panel | Multi | PCR | Cyst fluid | 91 | 65% | 100% (83–100) | N/A | MCN vs. non-MCN | [38] |
| (52–76) | 98% | |||||||||
| 84% | (86–100) | N/A | IPMN vs. non-IPMN | |||||||
| (70–92) | ||||||||||
| 95.50% | ||||||||||
| PCR using NGS | Cyst fluid | 197 | 68.50% | (N/A) | N/A | IPMN vs. non-IPMN | [39] | |||
| (N/A) | ||||||||||
| 100% | ||||||||||
| NGS | Cyst fluid | 595 | 89% | (88–100) | N/A | MCN vs. non-MCN | [40] | |||
| (79–95) | 100% | |||||||||
| Sanger sequencing | Cyst fluid | 159 | 65% | (N/A) | N/A | MCN vs. non-MCN | ||||
| (N/A) | ||||||||||
| lncRNA-TFG | Long noncoding RNA | Single | Affymetrix human exon 1.0 ST | Tissue | 28 | N/A | N/A | 6.23 × 10−8 | Positive correlation with tumorigenesis in IPMNs | [41] |
| CTD-2033D15.2 | Long non-coding RNA | Single | Affymetrix human exon 1.0 ST | Tissue | 28 | N/A | N/A | 1.47 × 10−4 | Negative correlation with tumorigenesis in IPMNs | [41] |
| HAND2-AS1 | Long non-coding RNA | Single | Affymetrix human exon 1.0 ST | Tissue | 28 | N/A | N/A | 2.66 × 10−3 | Negative correlation with tumorigenesis in IPMNs | [41] |
| Glucose | Metabolite | Multi | Liquid chromatography | Cyst fluid | 19 | 94% | 64% | 0.004 | Glucose ≤ 66 mg/dL in MCNs vs. non-MCNs | [42] |
| (N/A) | (N/A) | |||||||||
| Glucometer | Glucose ≤ 50 mg/dL in MCNs vs. non-MCNs | |||||||||
| Cyst fluid | 153 | 92% | 87% | N/A | [43] | |||||
| (N/A) | (N/A) | |||||||||
| Kynurenine | Metabolite | Single | Liquid chromatography | Cyst fluid | 19 | 90% | 100% | 0.002 | Lower in MCNs vs. non-MCNs | [42] |
| (N/A) | (N/A) | |||||||||
| AcSperm and | Metabolite panel | Single | Mass spectrometry | Blood plasma | 121 | 66.70% | 95% | N/A | PDAC vs. N | [44] |
| DAS and | (N/A) | (N/A) | ||||||||
| LPC(18: 0) and LPC(20: 3) and indole derivative | ||||||||||
| ADAMTS1 | Methylated gene | Single | Methylation on beads | Blood cfDNA | 39 | 87.20% | 95.80% | N/A | PDAC vs. N | [45] |
| (N/A) | (N/A) | |||||||||
| BNC1 | Methylated gene | Single | Methylation on beads | Blood cfDNA | 39 | 64.10% | 93.70% | N/A | PDAC vs. N | [45] |
| (N/A) | (N/A) | |||||||||
| SOX17 | Methylated gene | Single | Methylation-specific ddPCR | Cyst fluid | 154 | 78.40% | 85.60% | N/A | High-risk PCL vs. low-risk PCLs | [46] |
| (64.7–88.7) | (78.4–91.1) | |||||||||
| TBX15 and BMP3 | Methylated gene marker panel | Single | Whole-genome methylome discovery and qPCR | Cyst fluid | 134 | 90% | 92% | N/A | HGD/PC vs. LGD/N | [47] |
| (70–99) | (85–96) | |||||||||
| ADAMTS1 and/OR BNC1 | Methylated gene panel | Single | Methylation on beads | Blood cfDNA | 39 | 97.40% | 91.60% | N/A | PDAC vs. N | [45] |
| (N/A) | (N/A) | |||||||||
| FOXE1 and SLIT2 and | Methylated gene panel | Single | Methylation-specific ddPCR | Cyst fluid | 154 | 84.30% | 89.40% | N/A | High-risk PCL vs. low-risk PCLs | [46] |
| EYA4 and SFRP1 | (N/A) | (N/A) | ||||||||
| miR-1290 | MicroRNA | Multi | MicroRNA array analysis | Blood serum | 60 | 88% | 84% | N/A | PC vs. N | [48] |
| (N/A) | (N/A) | |||||||||
| 76 | 83% | 69% | N/A | PC vs. CP | ||||||
| (N/A) | (N/A) | |||||||||
| 95 | 83% | 78% | N/A | PC vs. CP and N | ||||||
| (N/A) | (N/A) | |||||||||
| qRT-PCR | Blood plasma | |||||||||
| 49 | N/A | N/A | 0.027 | PDAC vs. N | [49] | |||||
| Blood serum | ||||||||||
| qRT-PCR | ||||||||||
| 200 | 74.20% | 91.20% | N/A | PC vs. C | [50] | |||||
| (N/A) | (N/A) | |||||||||
| 9-miRNA model a | MicroRNA panel | Single | TaqMan miRNA Array | Tissue and | 33 and 50 | 89% | 100% | N/A | HG IPMNs, PanNETs and SPNs vs. LG IPMNs and SCAs | [51] |
| cyst fluid | (N/A) | (N/A) | ||||||||
| miR-3679-5p and miR-940 | MicroRNA panel | Single | qPCR | Saliva | 80 | 72.50% | 70.00% | N/A | PC vs. N | [52] |
| (N/A) | (N/A) | |||||||||
| 60 | 62.50% | 80.00% | N/A | PC vs. BPT | ||||||
| (N/A) | (N/A) | |||||||||
| 100 | 70.00% | 70.00% | N/A | PC vs. N and BPT | ||||||
| (N/A) | (N/A) | |||||||||
| CA19-9 | Protein- | Multi | Bead-based xMAP immunoassay | Blood serum | 267 | 57.20% | 90% | N/A | PDAC vs. N | [53] |
| associated | (N/A) | (N/A) | ||||||||
| ELISA | ||||||||||
| Blood plasma | ||||||||||
| 176 | 77.50% | 83.10% | N/A | CA19-9 >20.3 U/mL | [54] | |||||
| Retrospective clinical data | Blood serum | (N/A) | (N/A) | PDAC vs. C | ||||||
| 41 | 90% | 83.33% | N/A | 2.45 times elevated CA19-9 indicated recurrence of PC | [55] | |||||
| (N/A) | (N/A) | |||||||||
| CEA | Protein | Multi | Clinical data | Cyst fluid | 31 | 73% | 89% | N/A | CEA > 192 ;ng/mL for MCN | [42] |
| (N/A) | (N/A) | |||||||||
| ELISA | Cyst fluid | 149 | 95.50% | 81.50% | <0.0001 | CEA ≤ 10 ng/mL for SCN | [56] | |||
| (N/A) | (N/A) | |||||||||
| Enzyme-linked immunosorbent assay | Cyst fluid | 153 | 58% | 96% | N/A | CEA > 192 ng/mL for MC | [43] | |||
| (N/A) | (N/A) | |||||||||
| MUC5AC:WGA and MUC5AC:BGH and Endorepellin:WGA | Protein | Multi | Antibody-lectin sandwich microarray | Cyst fluid | 147 | 92% b | 94% b | N/A | Elevation in any two differentiates MCNs vs. non-MCNs | [57] |
| panel | (N/A) | (N/A) | ||||||||
| Antibody-lectin sandwich arrays | Elevation in any two differentiates MCNs vs. non-MCNs | |||||||||
| Cyst fluid | 22 | 87% | 100% | N/A | [58] | |||||
| (N/A) | (N/A) | |||||||||
| Thymosin- β4 | Protein | Single | MALDI imaging and mass spectrometry | Tissue | 45 | 70% | 71% | 0.011 | Overexpressed in IPMN with HGD | [59] |
| (N/A) | (N/A) | |||||||||
| Ubiquitin | Protein | Single | MALDI imaging and mass spectrometry | Tissue | 45 | 94% | 86% | 0.04 | Overexpressed in IPMN with HGD | [59] |
| (N/A) | (N/A) | |||||||||
| VEGF-A | Protein | Single | ELISA | Cyst fluid | 149 | 100% | 83.70% | <0.0001 | VEGF-A > 5000 pg/mL benign SCN | [56] |
| (N/A) | (N/A) | |||||||||
| VEGF-A and CEA | Protein panel | Single | ELISA | Cyst fluid | 149 | 95.50% | 100% | N/A | VEGF-A > 5000 pg/mL and CEA ≤ 10 ng/mL in benign SCN | [56] |
| (N/A) | (N/A) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kane, L.E.; Mellotte, G.S.; Conlon, K.C.; Ryan, B.M.; Maher, S.G. Multi-Omic Biomarkers as Potential Tools for the Characterisation of Pancreatic Cystic Lesions and Cancer: Innovative Patient Data Integration. Cancers 2021, 13, 769. https://doi.org/10.3390/cancers13040769
Kane LE, Mellotte GS, Conlon KC, Ryan BM, Maher SG. Multi-Omic Biomarkers as Potential Tools for the Characterisation of Pancreatic Cystic Lesions and Cancer: Innovative Patient Data Integration. Cancers. 2021; 13(4):769. https://doi.org/10.3390/cancers13040769
Chicago/Turabian StyleKane, Laura E., Gregory S. Mellotte, Kevin C. Conlon, Barbara M. Ryan, and Stephen G. Maher. 2021. "Multi-Omic Biomarkers as Potential Tools for the Characterisation of Pancreatic Cystic Lesions and Cancer: Innovative Patient Data Integration" Cancers 13, no. 4: 769. https://doi.org/10.3390/cancers13040769
APA StyleKane, L. E., Mellotte, G. S., Conlon, K. C., Ryan, B. M., & Maher, S. G. (2021). Multi-Omic Biomarkers as Potential Tools for the Characterisation of Pancreatic Cystic Lesions and Cancer: Innovative Patient Data Integration. Cancers, 13(4), 769. https://doi.org/10.3390/cancers13040769