
Microfluidic Organoids-on-a-Chip: Quantum Leap in Cancer Research

 , ,
, ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
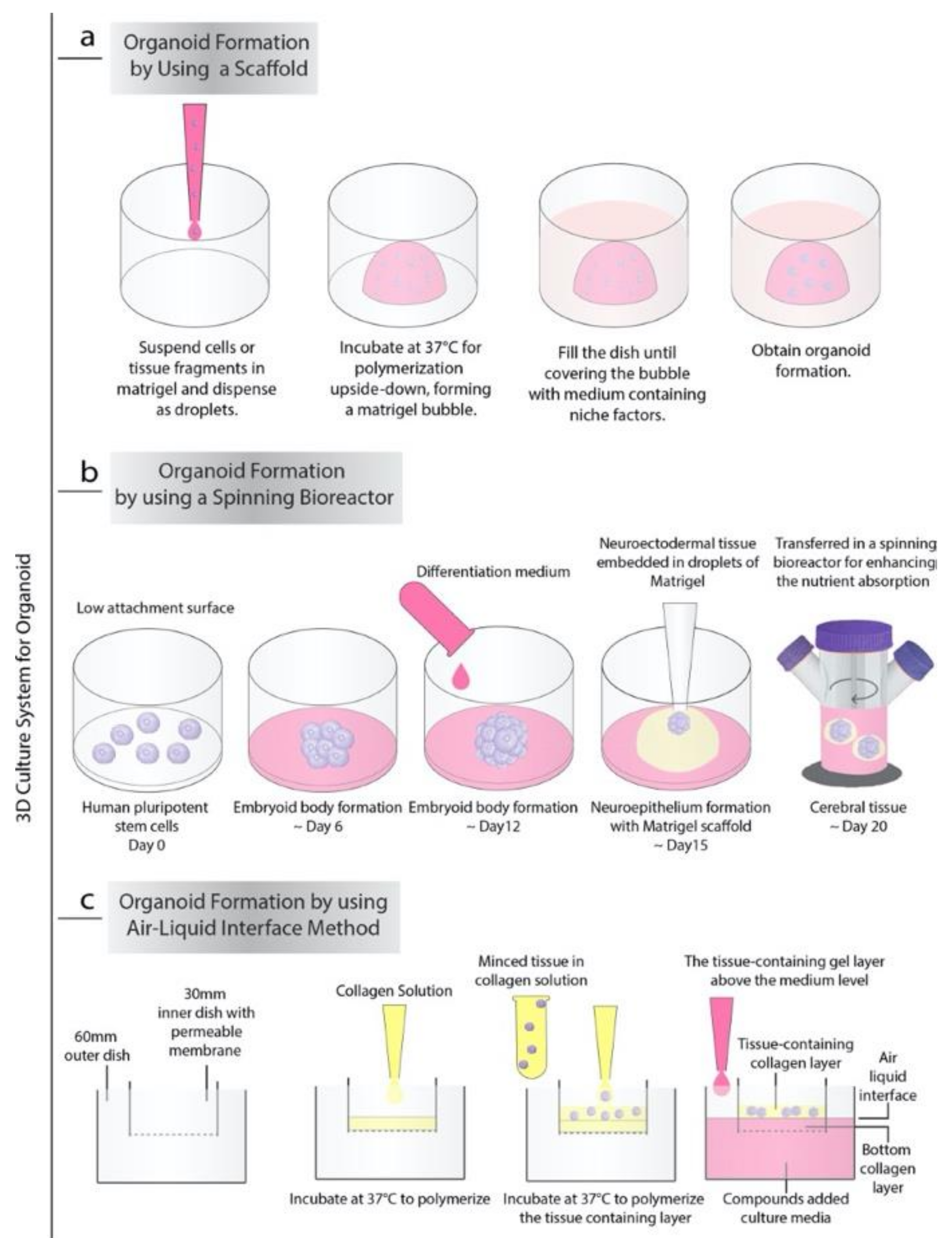
2. Approaches for Organoid Cultures
2.1. Niche Factors
2.1.1. Niche Factors for Organoids that Derived from Mammary Epithelial Cells
2.1.2. Niche Factors for Organoids that Derived from Gastrointestinal Tract
2.1.3. Niche Factors for Organoids Models to Study Liver Cancer
2.1.4. Niche Factors for Organoid Models to Study Brain Cancer
2.1.5. Niche Factors for Organoids Models to Study Ovarian Cancer
2.1.6. Synthetic Matrices for Organoids Models

{kind=link}
{kind=link}
{kind=link}
| Cancer-Derived Organoid Type | Niche Factor | ECM Component | References | |
|---|---|---|---|---|
| Proteins & Growth Factors | Small Molecules | |||
| Breast | EGF/Noggin/R-spondin Wnt-3A FGF7 FGF10 | A83-01 Y-27632 SB202190 Nicotinamide | Matrigel BME * Collagen-Matrigel | [22] [35] [36] |
| Colon/Colorectal | EGF/Noggin/R-spondin Wnt-3A FGF10 | Gastrin A83-01 Y-27632 SB202190 Nicotinamide Prostaglandin E2 | Matrigel Growth Factor Reduced-BME * | [16] [49] [50] |
| Gastrointestinal | EGF/Noggin/R-spondin BMP-4 | Gastrin A83-01 Y-27632 SB202190 Nicotinamide | Matrigel Growth Factor Reduced-Matrigel BME * Collagen Collagen-Matrigel PEG Hydrogel | [4] [46] [47] [70] |
| Glioblastoma | EGF/Noggin/R-spondin | Gastrin Nicotinamide A83-01 SB202190 Y-27632 Prostaglandin E2 CHIR 99021 | Matrigel | [59] [63] |
| Liver | EGF/R-spondin Wnt-3A FGF10 HGF | Nicotinamide Gastrin Forskolin | BME * PEG Hydrogel | [52] [53] [72] |
| Lung | EGF/Noggin/R-spondin Wnt-3A FGF4 FGF7 FGF10 | A83-01 | Matrigel | [39] [40] |
| Ovary | EGF/Noggin/R-spondin FGF10 Heregulinβ-1 BMP-4 | Nicotinamide A83-01 Y-27632 Forskolin | Growth Factor Reduced-BME * Matrigel | [25] [26] [68] |
| Prostate | EGF/Noggin/R-spondin Co-culture with stromal cells | EGF/Noggin/R-spondin 1 A83-01 Testosterone ROCK inhibitors | Matrigel | [21] [38] |
| Pancreas | EGF/Noggin/R-spondin FGF10 | Wnt-3A Retinoic acid A83-01 Nicotinamide Gastrin | Matrigel GFR Matrigel Collagen | [19] [37] [48] |
2.2. Co-Cultures of Organoids
2.3. Organoids and Viral Infections
2.4. Advantages of the Organoid Models
2.5. Limitation of the Organoid Models
3. Advantages of Microfluidic-Chip Technology for Evolving Current Organoid Models
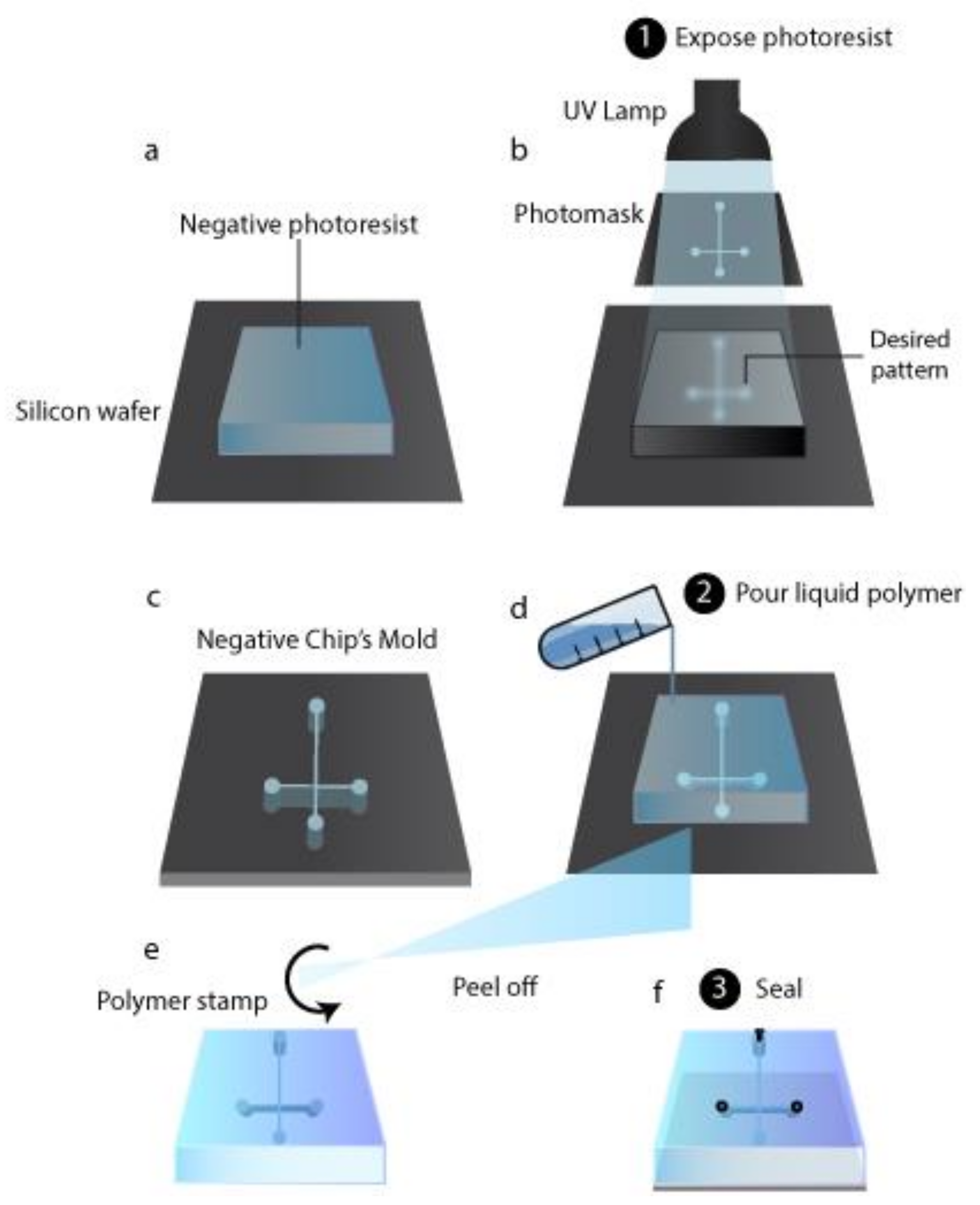
3.1. Chip Production
3.2. Flow Mechanisms
| PFM | Actuation | Recirculation |
|---|---|---|
| Gravity-driven flow | Using a rocking platform which allows the fluid to flow inside interconnected microchannels continuously [191] -Placing the fluid reservoir in a higher position than the chip [192] | Using the rocking platform, the medium continuously recirculate in the chip [191] |
| Hydrostatic pressure-driven flow | -Connecting liquid column (reservoirs) to the inlet and outlet of the chip, it can be controlled via their differential heights | Recirculation can be achieved by: -manually refilling the inlet reservoir with the medium collected at the outlet [193] -siphon-based auto-filled hydrostatic pressure-driven passive micropump [194] |
| Surface tension-driven flow | -Placing over the inlet port of a microchannel a drop with a smaller radius than the one placed at the outlet port [195] | It is not possible to recirculate the medium |
| Osmosis-driven flow | Using a membrane permeable only to solvent; the flow is obtained thanks to different concentrations across that layer [196] | It is not possible to recirculate the medium |
3.3. Monitoring Cell Parameters
| Parameter | Sensor | Functioning | Peculiarities |
|---|---|---|---|
| pH | Ion Sensitive Field-Effect Transistor (ISFET) | pH is determined through the current flowing between the terminals called source and drain. Indeed, the device’s potential is established in the solution using a detecting interface (specifically sensitive to H+) and a reference electrode (called gate potential). | Miniature size, durability, rapid response, and ion selectivity [197]. |
| Light addressable potentiometric sensor (LAPS) | LED or laser light is used to generate photocurrent, which intensity depends upon ion concentration in the analyzed solution affecting the potential establish between a detecting interface (specifically sensitive to H+) and the reference electrode. | Can be used as imaging devices allowing to visualize the ion concentration as a two-dimensional map [198] | |
| Potentiometric sensor | pH is determined from the voltage measured between a reference electrode and a sensitive electrode obtained exploiting metal oxides sensitivity to H+ | To make a space-saving device, screen printing has been used to produce thick film based potentiometric pH sensors by depositing on the same substrate both the reference and the sensitive electrodes [199]. | |
| Optical measure | pH is determined by measuring the absorbance of the culture medium containing pH indicator phenol red. The absorbance can be determined using a transmitted-light microscope and a digital camera with a CCD sensor. The generated light by a microscope lamp passes through (in-plane) the microfluidic device and reaches the CCD sensor where its intensity is measured, and therefore absorbance is calculated [200]. | ||
| Oxygen concentration | Amperometric sensor | Oxygen is determined using cathode and anode electrodes placed in the electrolyte solution. Indeed, the current generated by oxygen reduction at the cathode depends on its partial pressure. From oxygen partial pressure the gas concentration in the solution is determined since these two values are directly proportional. As a cathode electrode, it is possible to use a noble metal dipped into the medium, i.e., a direct amperometric sensor or a Clark’s type sensor in which a gas-permeable membrane separates the sensor from the electrolyte. | To improve the miniaturization of the amperometric sensor, it is possible to measure oxygen concentration through local pH changes due to oxygen reduction at the cathode with a pH-sensitive ISFET [201]. |
| Optical sensor | Oxygen concentration is measured dependently on the quenching of a dye fluorescence exerted by molecular oxygen. Dye molecules are excited by a particular electromagnetic radiation and emit light to return to the normal energy state. When oxygen is present in the sample, the emitted light is limited or altered for its interaction with the dye. The measured effects (fluorescence amplitude, phase shift, or fluorescence lifetime) are inversely proportional to the oxygen partial pressure. | The dye can be incorporated in a polymer to create a sensor site [202] or in a membrane coating the entire surface of the cell culture vessel to monitor spatial change in oxygen concentration [203]. Dye excitation is pursued by using LED, and the signal is registered with a photodetector. These three elements are placed together in commercially available sensor devices. However, it is preferable to separate the optical set up from the cell environment to avoid sterilization. Also, an inverted microscope can be used to read sensor spots. | |
| Metabolites | Functionalized sensors | Metabolites can be measured with electrochemical sensors based on enzymes able to react with these molecules. The enzyme can be immobilized in a membrane or directly on the electrode surface. By-products of the enzyme reactions are reduced or oxidized at a polarized electrode, and the reaction is electrochemically followed. Electrodes can also be functionalized with antibodies obtaining immunobiosensors sensitive to specific analytes. |
3.4. Bioprinted Organs-on-a-Chip
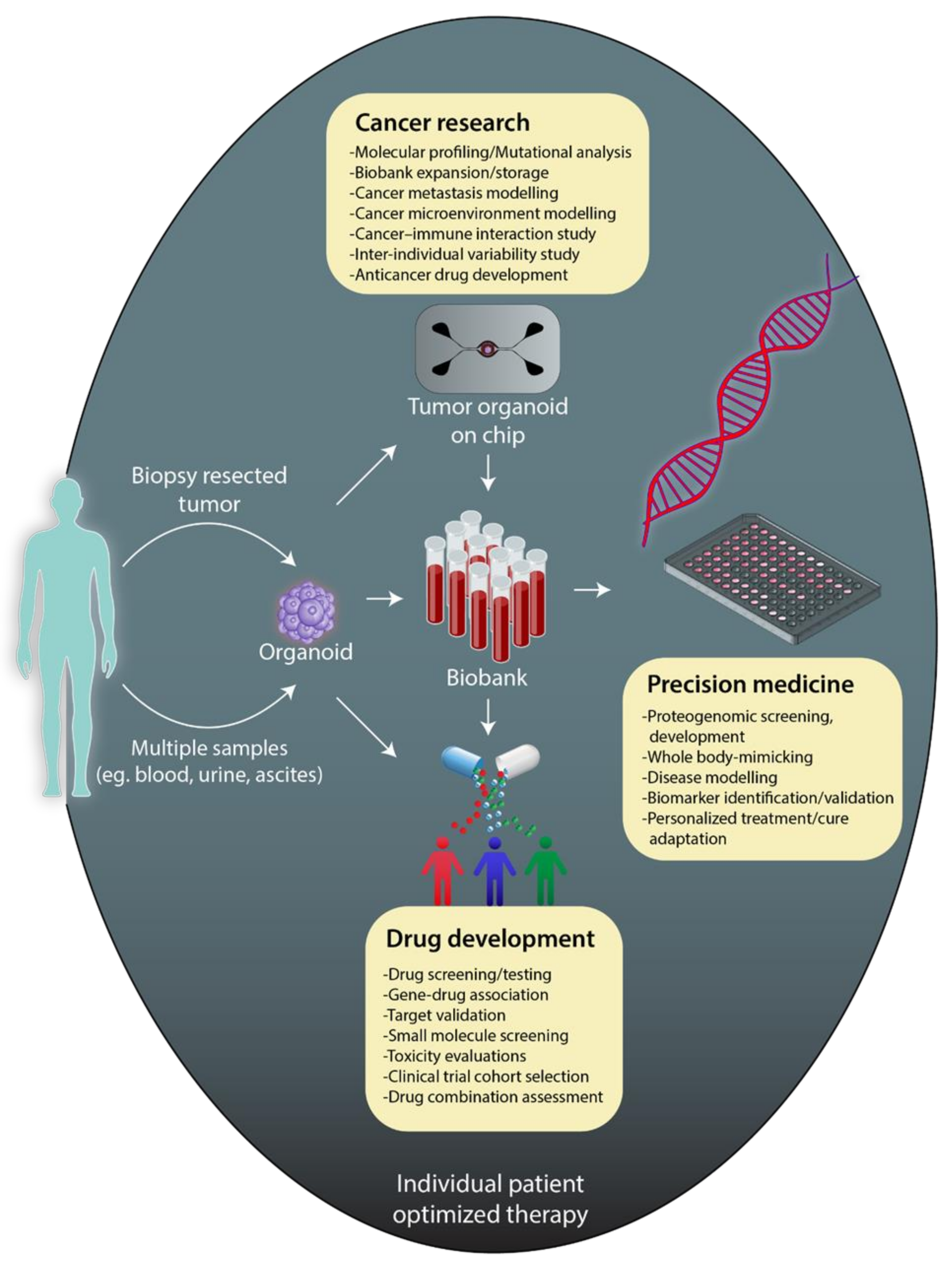
4. Patient-Derived Organoids to Improve Cancer Therapeutics
5. Possible Limitations of Current Organoids on-a-Chip Technologies
6. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension-how 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef]
- Yu, F.; Hunziker, W.; Choudhury, D. Engineering microfluidic organoid-on-a-chip platforms. Micromachines 2019, 10, 165. [Google Scholar] [CrossRef]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Tsukamoto, Y.; Kujala, P.; Peters, P.J.; Clevers, H. Intestinal epithelial organoids fuse to form self-organizing tubes in floating collagen gels. Dev. 2017, 144, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Jee, J.H.; Lee, D.H.; Ko, J.; Hahn, S.; Jeong, S.Y.; Kim, H.K.; Park, E.; Choi, S.Y.; Jeong, S.; Lee, J.W.; et al. Development of Collagen-Based 3D Matrix for Gastrointestinal Tract-Derived Organoid Culture. Stem Cells Int. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Staton, C.A.; Stribbling, S.M.; Tazzyman, S.; Hughes, R.; Brown, N.J.; Lewis, C.E. Current methods for assaying angiogenesis in vitro and in vivo. Int. J. Exp. Pathol. 2004, 85, 233–248. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Pohida, T.; Swaroop, A. Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors. Stem Cell Reports 2018, 10, 300–313. [Google Scholar] [CrossRef]
- Li, X.; Ootani, A.; Kuo, C. An air-liquid interface culture system for 3D organoid culture of diverse primary gastrointestinal tissues. Methods Mol. Biol. 2016, 1422, 33–40. [Google Scholar] [PubMed]
- Giandomenico, S.L.; Mierau, S.B.; Gibbons, G.M.; Wenger, L.M.D.; Masullo, L.; Sit, T.; Sutcliffe, M.; Boulanger, J.; Tripodi, M.; Derivery, E.; et al. Cerebral organoids at the air–liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 2019, 22, 669–679. [Google Scholar] [CrossRef]
- Chen, Y.W.; Huang, S.X.; De Carvalho, A.L.R.T.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Dedhia, P.H.; Bertaux-Skeirik, N.; Zavros, Y.; Spence, J.R. Organoid Models of Human Gastrointestinal Development and Disease. Gastroenterology 2016, 150, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.e11. [Google Scholar] [CrossRef] [PubMed]
- Cristobal, A.; van den Toorn, H.W.P.; van de Wetering, M.; Clevers, H.; Heck, A.J.R.; Mohammed, S. Personalized Proteome Profiles of Healthy and Tumor Human Colon Organoids Reveal Both Individual Diversity and Basic Features of Colorectal Cancer. Cell Rep. 2017, 18, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Van Houdt, W.; Van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Palazzolo, S.; Hadla, M.; Spena, C.R.; Caligiuri, I.; Rotondo, R.; Adeel, M.; Kumar, V.; Corona, G.; Canzonieri, V.; Toffoli, G.; et al. An effective multi-stage liposomal DNA origami nanosystem for in vivo cancer therapy. Cancers 2019, 11, 1997. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Phipson, B.; Er, P.X.; Combes, A.N.; Forbes, T.A.; Howden, S.E.; Zappia, L.; Yen, H.J.; Lawlor, K.T.; Hale, L.J.; Sun, J.; et al. Evaluation of variability in human kidney organoids. Nat. Methods 2019, 16, 79–87. [Google Scholar] [CrossRef]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; Van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.M.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed]
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef]
- Scattolin, T.; Bortolamiol, E.; Visentin, F.; Palazzolo, S.; Caligiuri, I.; Perin, T.; Canzonieri, V.; Demitri, N.; Rizzolio, F.; Togni, A. Palladium(II)-η3-Allyl Complexes Bearing N-Trifluoromethyl N-Heterocyclic Carbenes: A New Generation of Anticancer Agents that Restrain the Growth of High-Grade Serous Ovarian Cancer Tumoroids. Chem. A Eur. J. 2020, 26, 11868–11876. [Google Scholar] [CrossRef]
- Maru, Y.; Hippo, Y. Current Status of Patient-Derived Ovarian Cancer Models. Cells 2019, 8, 505. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vincan, E.; Schwab, R.H.M.; Flanagan, D.J.; Moselen, J.M.; Tran, B.M.; Barker, N.; Phesse, T.J. The Central Role of Wnt Signaling and Organoid Technology in Personalizing Anticancer Therapy. Prog. Mol. Biol. Transl. Sci. 2018, 153, 299–319. [Google Scholar] [PubMed]
- Plaks, V.; Brenot, A.; Lawson, D.A.; Linnemann, J.R.; Van Kappel, E.C.; Wong, K.C.; de Sauvage, F.; Klein, O.D.; Werb, Z. Lgr5-Expressing Cells Are Sufficient And Necessary for Postnatal Mammary Gland Organogenesis. Cell Rep. 2013, 3, 70–78. [Google Scholar] [CrossRef]
- Makarem, M.; Kannan, N.; Nguyen, L.V.; Knapp, D.J.H.F.; Balani, S.; Prater, M.D.; Stingl, J.; Raouf, A.; Nemirovsky, O.; Eirew, P.; et al. Developmental Changes in the in Vitro Activated Regenerative Activity of Primitive Mammary Epithelial Cells. PLoS Biol. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, J.R.; Miura, H.; Meixner, L.K.; Irmler, M.; Kloos, U.J.; Hirschi, B.; Bartsch, H.S.; Sass, S.; Beckers, J.; Theis, F.J.; et al. Quantification of regenerative potential in primary human mammary epithelial cells. Development 2015, 142, 3239–3251. [Google Scholar] [CrossRef] [PubMed]
- Nedvetsky, P.I.; Kwon, S.H.; Debnath, J.; Mostov, K.E. Cyclic AMP regulates formation of mammary epithelial acini in vitro. Mol. Biol. Cell 2012, 23, 2973–2981. [Google Scholar] [CrossRef] [PubMed]
- Goldhammer, N.; Kim, J.; Timmermans-Wielenga, V.; Petersen, O.W. Characterization of organoid cultured human breast cancer. Breast Cancer Res. 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.L.; Pegoraro, A.F.; Li, H.; Li, K.; Yuan, Y.; Xu, G.; Gu, Z.; Sun, J.; Hao, Y.; Gupta, S.K.; et al. Cell swelling, softening and invasion in a three-dimensional breast cancer model. Nat. Phys. 2020, 16, 101–108. [Google Scholar] [CrossRef]
- Huch, M.; Bonfanti, P.; Boj, S.F.; Sato, T.; Loomans, C.J.M.; Van De Wetering, M.; Sojoodi, M.; Li, V.S.W.; Schuijers, J.; Gracanin, A.; et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013, 32, 2708–2721. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Radulovich, N.; Ng, C.; Liu, N.; Notsuda, H.; Cabanero, M.; Martins-Filho, S.N.; Raghavan, V.; Li, Q.; Mer, A.S.; et al. Organoid cultures as preclinical models of non-small cell lung cancer. Clin. Cancer Res. 2020, 26, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Longmire, T.A.; Ikonomou, L.; Hawkins, F.; Christodoulou, C.; Cao, Y.; Jean, J.C.; Kwok, L.W.; Mou, H.; Rajagopal, J.; Shen, S.S.; et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell 2012, 10, 398–411. [Google Scholar] [CrossRef]
- Roberts, D.J. Molecular mechanisms of development of the gastrointestinal tract. Dev. Dyn. 2000, 219, 109–120. [Google Scholar] [CrossRef]
- Dessimoz, J.; Opoka, R.; Kordich, J.J.; Grapin-Botton, A.; Wells, J.M. FGF signaling is necessary for establishing gut tube domains along the anterior-posterior axis in vivo. Mech. Dev. 2006, 123, 42–55. [Google Scholar] [CrossRef]
- McLin, V.A.; Rankin, S.A.; Zorn, A.M. Repression of Wnt/β-catenin signaling in the anterior endoderm is essential for liver and pancreas development. Development 2007, 134, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.C.H.; Kranenburg, O.; Xiao, H.; Yu, J. Organoid models of gastrointestinal cancers in basic and translational research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Holtzinger, A.; Jagan, I.; Begora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell. 2016, 18, 827–838. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef]
- Gilbert, S.F.; Barresi, M.J.F. Development Biology, 11th ed.; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Petros, T.J.; Tyson, J.A.; Anderson, S.A. Pluripotent Stem Cells for the Study of CNS Development. Front. Mol. Neurosci. 2011, 4. [Google Scholar] [CrossRef]
- Huch, M.; Koo, B.K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [PubMed]
- Danjo, T.; Eiraku, M.; Muguruma, K.; Watanabe, K.; Kawada, M.; Yanagawa, Y.; Rubenstein, J.L.R.; Sasai, Y. Subregional specification of embryonic stem cell-derived ventral telencephalic tissues by timed and combinatory treatment with extrinsic signals. J. Neurosci. 2011, 31, 1919–1933. [Google Scholar] [CrossRef] [PubMed]
- Su, H.L.; Muguruma, K.; Matsuo-Takasaki, M.; Kengaku, M.; Watanabe, K.; Sasai, Y. Generation of cerebellar neuron precursors from embryonic stem cells. Dev. Biol. 2006, 290, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell. 2009, 4, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Van Den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.W.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; Van De Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Berger, H.; Kulbe, H.; Thillainadarasan, S.; Mollenkopf, H.; Zemojtel, T.; Taube, E.; Darb-Esfahani, S.; Mangler, M.; Sehouli, J.; et al. Stable expansion of high-grade serous ovarian cancer organoids requires a low-Wnt environment. EMBO J. 2020, 39. [Google Scholar] [CrossRef] [PubMed]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef]
- Hernandez-Gordillo, V.; Kassis, T.; Lampejo, A.; Choi, G.H.; Gamboa, M.E.; Gnecco, J.S.; Brown, A.; Breault, D.T.; Carrier, R.; Griffith, L.G. Fully synthetic matrices for in vitro culture of primary human intestinal enteroids and endometrial organoids. Biomaterials 2020, 254. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Sorrentino, G.; Rezakhani, S.; Yildiz, E.; Nuciforo, S.; Heim, M.H.; Lutolf, M.P.; Schoonjans, K. Mechano-modulatory synthetic niches for liver organoid derivation. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; Van Es, J.H.; Li, V.S.W.; Van De Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5 + liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef]
- Richards, Z.; McCray, T.; Marsili, J.; Zenner, M.L.; Manlucu, J.T.; Garcia, J.; Kajdacsy-Balla, A.; Murray, M.; Voisine, C.; Murphy, A.B.; et al. Prostate Stroma Increases the Viability and Maintains the Branching Phenotype of Human Prostate Organoids. iScience 2019, 12, 304–317. [Google Scholar] [CrossRef]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; De Sousa Lopes, S.M.C.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.J.; Henrique, R.; Costa, V.L.; Jerónimo, C.; Carvalho, F.; Bastos, M.L.; de Pinho, P.G.; Carvalho, M. A rapid and simple procedure for the establishment of human normal and cancer renal primary cell cultures from surgical specimens. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef]
- Garcez, P.P.; Loiola, E.C.; Da Costa, R.M.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus: Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Cugola, F.R.; Fernandes, I.R.; Russo, F.B.; Freitas, B.C.; Dias, J.L.M.; Guimarães, K.P.; Benazzato, C.; Almeida, N.; Pignatari, G.C.; Romero, S.; et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016, 534, 267–271. [Google Scholar] [CrossRef]
- Wells, M.F.; Salick, M.R.; Wiskow, O.; Ho, D.J.; Worringer, K.A.; Ihry, R.J.; Kommineni, S.; Bilican, B.; Klim, J.R.; Hill, E.J.; et al. Genetic Ablation of AXL Does Not Protect Human Neural Progenitor Cells and Cerebral Organoids from Zika Virus Infection. Cell Stem Cell 2016, 19, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Su, K.Y.; Balasubramaniam, V.R.M.T. Zika Virus as Oncolytic Therapy for Brain Cancer: Myth or Reality? Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Chavali, P.L.; Stojic, L.; Meredith, L.W.; Joseph, N.; Nahorski, M.S.; Sanford, T.J.; Sweeney, T.R.; Krishna, B.A.; Hosmillo, M.; Firth, A.E.; et al. Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science 2017, 357, 83–88. [Google Scholar] [CrossRef]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wu, J.; Ye, Q.; Ma, F.; Zhu, Q.; Wu, Y.; Shan, C.; Xie, X.; Li, D.; Zhan, X.; et al. Treatment of human glioblastoma with a live attenuated Zika virus vaccine candidate. MBio 2018, 9. [Google Scholar] [CrossRef]
- Jemal, A.; Simard, E.P.; Dorell, C.; Noone, A.M.; Markowitz, L.E.; Kohler, B.; Eheman, C.; Saraiya, M.; Bandi, P.; Saslow, D.; et al. Annual report to the nation on the status of cancer, 1975-2009, featuring the burden and trends in human papillomavirus (HPV)-associated cancers and HPV vaccination coverage levels. J. Natl. Cancer Inst. 2013, 105, 175–201. [Google Scholar] [CrossRef] [PubMed]
- Friesland, S.; Mellin, H.; Munck-Wikland, E.; Nilsson, A.; Lindholm, J.; Dalianis, T.; Lewensohn, R. Human papilloma virus (HPV) and p53 immunostaining in advanced tonsillar carcinoma - Relation to radiotherapy response and survival. Anticancer Res. 2001, 21, 529–534. [Google Scholar] [PubMed]
- Tanaka, N.; Osman, A.A.; Takahashi, Y.; Lindemann, A.; Patel, A.A.; Zhao, M.; Takahashi, H.; Myers, J.N. Head and neck cancer organoids established by modification of the CTOS method can be used to predict in vivo drug sensitivity. Oral Oncol. 2018, 87, 49–57. [Google Scholar] [CrossRef]
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Motavaf, M.; Safari, S.; Saffari Jourshari, M.; Alavian, S.M. Hepatitis B virus-induced hepatocellular carcinoma: The role of the virus x protein. Acta Virol. 2013, 57, 389–396. [Google Scholar] [CrossRef]
- Yang, P.; Markowitz, G.J.; Wang, X.F. The hepatitis B virus-associated tumor microenvironment in hepatocellular carcinoma. Natl. Sci. Rev. 2014, 1, 396–412. [Google Scholar] [CrossRef]
- Crignis, E.D.; Carofiglio, F.; Moulos, P.; Verstegen, M.M.A.; Romal, S.; Khalid, M.M.; Pourfarzad, F.; Koutsothanassis, C.; Gehart, H.; Kan, T.W.; et al. Human liver organoids; A patient-derived primary model for HBV Infection and Related Hepatocellular Carcinoma. bioRxiv 2019, 568147. [Google Scholar]
- Yang, H.; Sun, L.; Pang, Y.; Hu, D.; Xu, H.; Mao, S.; Peng, W.; Wang, Y.; Xu, Y.; Zheng, Y.-C.; et al. Three-dimensional bioprinted hepatorganoids prolong survival of mice with liver failure. Gut 2020, 70, 567–574. [Google Scholar] [PubMed]
- Xie, F.; Sun, L.; Pang, Y.; Xu, G.; Jin, B.; Xu, H.; Lu, X.; Xu, Y.; Du, S.; Wang, Y.; et al. Three-dimensional bio-printing of primary human hepatocellular carcinoma for personalized medicine. Biomaterials 2021, 265, 120416. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Quantius, J.; Schmoldt, C.; Vazquez-Armendariz, A.I.; Becker, C.; El Agha, E.; Wilhelm, J.; Morty, R.E.; Vadász, I.; Mayer, K.; Gattenloehner, S.; et al. Influenza Virus Infects Epithelial Stem/Progenitor Cells of the Distal Lung: Impact on Fgfr2b-Driven Epithelial Repair. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.; Sachs, N.; Chiu, M.C.; Wong, B.H.Y.; Chu, H.; Poon, V.K.M.; Wang, D.; Zhao, X.; Wen, L.; et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc. Natl. Acad. Sci. USA 2018, 115, 6822–6827. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Ferren, M.; Chen, Y.W.; Siu, Y.; Makhsous, N.; Rima, B.; Briese, T.; Greninger, A.L.; Snoeck, H.W.; Moscona, A. Authentic modeling of human respiratory virus infection in human pluripotent stem cell-derived lung organoids. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Biggerstaff, M.; Cauchemez, S.; Reed, C.; Gambhir, M.; Finelli, L. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: A systematic review of the literature. BMC Infect. Dis. 2014, 14. [Google Scholar] [CrossRef]
- Zhai, P.; Ding, Y.; Wu, X.; Long, J.; Zhong, Y.; Li, Y. The epidemiology, diagnosis and treatment of COVID-19. Int. J. Antimicrob. Agents 2020, 55. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.; Assou, S.; Fieldes, M.; Vachier, I.; Chanez, P.; De Vos, J.; Bourdin, A. Differentiation of Induced Pluripotent Stem Cells (iPSC) into human bronchial epithelium from healthy and severe COPD patients. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef]
- Zhao, B.; Ni, C.; Gao, R.; Wang, Y.; Yang, L.; Wei, J.; Lv, T.; Liang, J.; Zhang, Q.; Xu, W.; et al. Recapitulation of SARS-CoV-2 infection and cholangiocyte damage with human liver ductal organoids. Protein Cell 2020, 10, 771–775. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Paul van Schayck, J.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef]
- Zang, R.; Castro, M.F.G.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Liu, X.; Chiu, M.C.; Zhao, X.; Wang, D.; Wei, Y.; Lee, A.; Zhang, A.J.; Chu, H.; et al. Infection of bat and human intestinal organoids by SARS-CoV-2. Nat. Med. 2020, 26, 1077–1083. [Google Scholar] [CrossRef]
- Ciceri, F.; Castagna, A.; Rovere-Querini, P.; De Cobelli, F.; Ruggeri, A.; Galli, L.; Conte, C.; De Lorenzo, R.; Poli, A.; Ambrosio, A.; et al. Early predictors of clinical outcomes of COVID-19 outbreak in Milan, Italy. Clin. Immunol. 2020, 217. [Google Scholar] [CrossRef]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H.; et al. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef]
- Bersanelli, M. Controversies about COVID-19 and anticancer treatment with immune checkpoint inhibitors. Immunotherapy 2020, 12, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Berkenbrock, J.A.; Grecco-Machado, R.; Achenbach, S. Microfluidic devices for the detection of viruses: Aspects of emergency fabrication during the COVID-19 pandemic and other outbreaks. Proc. R. Soc. A Math. Phys. Eng. Sci. 2020, 476. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, J.; Banerjee, I.; Vaishya, R.; Ghosh, S. Bioengineered in Vitro Tissue Models to Study SARS-CoV-2 Pathogenesis and Therapeutic Validation. ACS Biomater. Sci. Eng. 2020, 6. [Google Scholar] [CrossRef]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef]
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Han, S.H.; Park, S. Long-Term Culture of Intestinal Organoids. Methods Mol. Biol. 2018, 1817, 123–135. [Google Scholar] [PubMed]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- McCauley, H.A.; Wells, J.M. Pluripotent stem cell-derived organoids: Using principles of developmental biology to grow human tissues in a dish. Development 2017, 144, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, X.; Tan, Z.; Su, Y.; Liu, J.; Chang, M.; Yan, F.; Chen, J.; Chen, T.; Li, C.; et al. Human ESC-derived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury. Cell Res. 2019, 29, 1009–1026. [Google Scholar] [CrossRef]
- Freedman, B.S. Modeling kidney disease with iPS cells. Biomark. Insights 2015, 2015, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Demirci, U.; Chen, P. Emerging organoid models: Leaping forward in cancer research. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Iakobachvili, N.; Peters, P.J. Humans in a dish: The potential of organoids in modeling immunity and infectious diseases. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, G.; Andreoli, L.; Corti, S.; Faravelli, I. iPSCs-Based Neural 3D Systems: A Multidimensional Approach for Disease Modeling and Drug Discovery. Cells 2019, 8, 1438. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar]
- Boretto, M.; Maenhoudt, N.; Luo, X.; Hennes, A.; Boeckx, B.; Bui, B.; Heremans, R.; Perneel, L.; Kobayashi, H.; Van Zundert, I.; et al. Patient-derived organoids from endometrial disease capture clinical heterogeneity and are amenable to drug screening. Nat. Cell Biol. 2019, 21, 1041–1051. [Google Scholar] [CrossRef]
- Namekawa, T.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Application of Prostate Cancer Models for Preclinical Study: Advantages and Limitations of Cell Lines, Patient-Derived Xenografts, and Three-Dimensional Culture of Patient-Derived Cells. Cells 2019, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Inoue, M. Application of Cancer Organoid Model for Drug Screening and Personalized Therapy. Cells 2019, 8, 470. [Google Scholar] [CrossRef]
- Langhans, S.A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Ergir, E.; Bachmann, B.; Redl, H.; Forte, G.; Ertl, P. Small force, big impact: Next generation organ-on-a-chip systems incorporating biomechanical cues. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- Li, X.; Larsson, P.; Ljuslinder, I.; Öhlund, D.; Myte, R.; Löfgren-Burström, A.; Zingmark, C.; Ling, A.; Edin, S.; Palmqvist, R. Ex vivo organoid cultures reveal the importance of the tumor microenvironment for maintenance of colorectal cancer stem cells. Cancers 2020, 12, 923. [Google Scholar] [CrossRef]
- Warren, S.M.; Sailon, A.M.; Allori, A.C.; Davidson, E.H.; Reformat, D.D.; Allen, R.J. A novel flow-perfusion bioreactor supports 3D dynamic cell culture. J. Biomed. Biotechnol. 2009, 2009. [Google Scholar]
- Wyss Institute Human Organs-on-Chips. Available online: https://wyss.harvard.edu/technology/human-organs-on-chips/ (accessed on 6 January 2020).
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Kim, H.J.; Fraser, J.P.; Shea, D.E.; Khan, M.; Bahinski, A.; Hamilton, G.A.; Ingber, D.E. Microfabrication of human organs-on-chips. Nat. Protoc. 2013, 8, 2135–2157. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, F.; Schilling, N.; Mader, K.; Gruchow, M.; Klotzbach, U.; Lindner, G.; Horland, R.; Wagner, I.; Lauster, R.; Howitz, S.; et al. Design and prototyping of a chip-based multi-micro-organoid culture system for substance testing, predictive to human (substance) exposure. J. Biotechnol. 2010, 148, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Stucki, A.O.; Stucki, J.D.; Hall, S.R.R.; Felder, M.; Mermoud, Y.; Schmid, R.A.; Geiser, T.; Guenat, O.T. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip 2015, 15, 1302–1310. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting Organ-Level Lung. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lesher Perez, S.C.; Choul C Kim, B.; Yamanishi, C.; Labuz, J.M.; Leung, B.; Takayama, S. Pharmacokinetic profile that reduces nephrotoxicity of gentamicin in a perfused kidney-on-a-chip. Biofabrication 2016, 8, 015021. [Google Scholar] [CrossRef]
- Pocock, K.; Delon, L.; Bala, V.; Rao, S.; Priest, C.; Prestidge, C.; Thierry, B. Intestine-on-A-Chip Microfluidic Model for Efficient in Vitro Screening of Oral Chemotherapeutic Uptake. ACS Biomater. Sci. Eng. 2017, 3, 951–959. [Google Scholar] [CrossRef]
- Wufuer, M.; Lee, G.H.; Hur, W.; Jeon, B.; Kim, B.J.; Choi, T.H.; Lee, S.H. Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Agarwal, A.; Goss, J.A.; Cho, A.; McCain, M.L.; Parker, K.K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip 2013, 13, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Huh, D. A human breathing lung-on-a-chip. Ann. Am. Thorac. Soc. 2015, 12, S42–S44. [Google Scholar] [CrossRef]
- Nakao, Y.; Kimura, H.; Sakai, Y.; Fujii, T. Bile canaliculi formation by aligning rat primary hepatocytes in a microfluidic device. Biomicrofluidics 2011, 5. [Google Scholar] [CrossRef]
- Nawroth, J.C.; Barrile, R.; Conegliano, D.; van Riet, S.; Hiemstra, P.S.; Villenave, R. Stem cell-based Lung-on-Chips: The best of both worlds? Adv. Drug Deliv. Rev. 2019, 140, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Eng, G.; Lee, B.W.; Protas, L.; Gagliardi, M.; Brown, K.; Kass, R.S.; Keller, G.; Robinson, R.B.; Vunjak-Novakovic, G. Autonomous beating rate adaptation in human stem cell-derived cardiomyocytes. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Marsano, A.; Conficconi, C.; Lemme, M.; Occhetta, P.; Gaudiello, E.; Votta, E.; Cerino, G.; Redaelli, A.; Rasponi, M. Beating heart on a chip: A novel microfluidic platform to generate functional 3D cardiac microtissues. Lab Chip 2016, 16, 599–610. [Google Scholar] [CrossRef]
- Bercovici, M.; Levenberg, S. Nanoliter Cell Culture Array with Tunable Chemical Gradients. Anal. Chem. 2018, 90, 7480–7488. [Google Scholar]
- Chiang, H.J.; Yeh, S.L.; Peng, C.C.; Liao, W.H.; Tung, Y.C. Polydimethylsiloxane-polycarbonate microfluidic devices for cell migration studies under perpendicular chemical and oxygen gradients. J. Vis. Exp. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Atencia, J.; Morrow, J.; Locascio, L.E. The microfluidic palette: A diffusive gradient generator with spatio-temporal control. Lab Chip 2009, 9, 2707–2714. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Wang, Y.; Sims, C.E.; Magness, S.T.; Allbritton, N.L. Optimizing Wnt-3a and R-spondin1 concentrations for stem cell renewal and differentiation in intestinal organoids using a gradient-forming microdevice. RSC Adv. 2015, 5, 74881–74891. [Google Scholar] [CrossRef]
- Spitz, S.; Zanetti, C.; Bolognin, S.; Muwanigwa, M.N.; Smits, L.; Berger, E.; Jordan, C.; Harasek, M.; Schwamborn, J.; Ertl, P. Cultivation and characterization of human midbrain organoids in sensor integrated microfluidic chips. bioRxiv 2019. [Google Scholar]
- Lee, K.K.; McCauley, H.A.; Broda, T.R.; Kofron, M.J.; Wells, J.M.; Hong, C.I. Human stomach-on-a-chip with luminal flow and peristaltic-like motility. Lab Chip 2018, 18, 3079–3085. [Google Scholar] [CrossRef]
- Soffe, R.; Baratchi, S.; Nasabi, M.; Tang, S.Y.; Boes, A.; McIntyre, P.; Mitchell, A.; Khoshmanesh, K. Lateral trapezoid microfluidic platform for investigating mechanotransduction of cells to spatial shear stress gradients. Sens. Actuators B Chem. 2017, 251, 963–975. [Google Scholar] [CrossRef]
- Wang, X.; Phan, D.T.T.; Sobrino, A.; George, S.C.; Hughes, C.C.W.; Lee, A.P. Engineering anastomosis between living capillary networks and endothelial cell-lined microfluidic channels. Lab Chip 2016, 16, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Utoh, R.; Ohashi, K.; Tatsumi, K.; Yamato, M.; Okano, T.; Seki, M. Controlled formation of heterotypic hepatic micro-organoids in anisotropic hydrogel microfibers for long-term preservation of liver-specific functions. Biomaterials 2012, 33, 8304–8315. [Google Scholar] [CrossRef]
- Sart, S.; Tomasi, R.F.X.; Barizien, A.; Amselem, G.; Cumano, A.; Baroud, C.N. Mapping the structure and biological functions within mesenchymal bodies using microfluidics. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.; Magliaro, C.; Paczia, N.; Monzel, A.S.; Antony, P.; Linster, C.L.; Bolognin, S.; Ahluwalia, A.; Schwamborn, J.C. Millifluidic culture improves human midbrain organoid vitality and differentiation. Lab Chip 2018, 18, 3172–3183. [Google Scholar] [CrossRef] [PubMed]
- Homan, K.A.; Gupta, N.; Kroll, K.T.; Kolesky, D.B.; Skylar-Scott, M.; Miyoshi, T.; Mau, D.; Valerius, M.T.; Ferrante, T.; Bonventre, J.V.; et al. Flow-enhanced vascularization and maturation of kidney organoids in vitro. Nat. Methods 2019, 16, 255–262. [Google Scholar] [CrossRef]
- Sung, J.H.; Wang, Y.I.; Narasimhan Sriram, N.; Jackson, M.; Long, C.; Hickman, J.J.; Shuler, M.L. Recent Advances in Body-on-a-Chip Systems. Anal. Chem. 2019, 91, 330–351. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Abdalkader, R.; Kamei, K.I. Multi-corneal barrier-on-a-chip to recapitulate eye blinking shear stress forces. Lab Chip 2020, 20, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- van der Helm, M.W.; Henry, O.Y.F.; Bein, A.; Hamkins-Indik, T.; Cronce, M.J.; Leineweber, W.D.; Odijk, M.; van der Meer, A.D.; Segerink, L.I.; Ingber, D.E. Non-invasive sensing of transepithelial barrier function and tissue differentiation in organs-on-chips using impedance spectroscopy. Lab Chip 2019, 19, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Yeste, J.; García-Ramírez, M.; Illa, X.; Guimerà, A.; Hernández, C.; Simó, R.; Villa, R. A compartmentalized microfluidic chip with crisscross microgrooves and electrophysiological electrodes for modeling the blood–retinal barrier. Lab Chip 2018, 18, 95–105. [Google Scholar] [CrossRef]
- Brown, J.A.; Pensabene, V.; Markov, D.A.; Allwardt, V.; Diana Neely, M.; Shi, M.; Britt, C.M.; Hoilett, O.S.; Yang, Q.; Brewer, B.M.; et al. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 2015, 9. [Google Scholar] [CrossRef]
- Arlk, Y.B.; Van Der Helm, M.W.; Odijk, M.; Segerink, L.I.; Passier, R.; Van Den Berg, A.; Van Der Meer, A.D. Barriers-on-chips: Measurement of barrier function of tissues in organs-on-chips. Biomicrofluidics 2018, 12. [Google Scholar]
- Nge, P.N.; Rogers, C.I.; Woolley, A.T. Advances in microfluidic materials, functions, integration, and applications. Chem. Rev. 2013, 113, 2550–2583. [Google Scholar] [CrossRef]
- Tsao, C.W. Polymer microfluidics: Simple, low-cost fabrication process bridging academic lab research to commercialized production. Micromachines 2016, 7, 225. [Google Scholar] [CrossRef] [PubMed]
- Alrifaiy, A.; Lindahl, O.A.; Ramser, K. Polymer-based microfluidic devices for pharmacy, biology and tissue engineering. Polymers 2012, 4, 1349–1398. [Google Scholar] [CrossRef]
- ELVE Flow. Different Microfluidic Fabrication Techniques. Available online: https://www.elveflow.com/microfluidic-reviews/soft-lithography-microfabrication/soft-lithography-fabrication-technics/ (accessed on 6 January 2020).
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Publ. Gr. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Zhu, Y.; Qin, J. Human brain organoid-on-a-chip to model prenatal nicotine exposure. Lab Chip 2018, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Workman, M.J.; Gleeson, J.P.; Troisi, E.J.; Estrada, H.Q.; Kerns, S.J.; Hinojosa, C.D.; Hamilton, G.A.; Targan, S.R.; Svendsen, C.N.; Barrett, R.J. Enhanced Utilization of Induced Pluripotent Stem Cell–Derived Human Intestinal Organoids Using Microengineered Chips. CMGH 2018, 5, 669–677. [Google Scholar] [CrossRef]
- Achberger, K.; Probst, C.; Haderspeck, J.C.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Deng, P.; Chen, W.; Guo, Y.; Tao, T.; Qin, J. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Arneri, A.; Bersini, S.; Shin, S.R.; Zhu, K.; Goli-Malekabadi, Z.; Aleman, J.; Colosi, C.; Busignani, F.; Dell’Erba, V.; et al. Bioprinting 3D microfibrous scaffolds for engineering endothelialized myocardium and heart-on-a-chip. Biomaterials 2016, 110, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, A.R.; Rajan, S.A.P.; Votanopoulos, K.I.; Hall, A.R.; Skardal, A. In vitro patient-derived 3D mesothelioma tumor organoids facilitate patient-centric therapeutic screening. Sci. Rep. 2018, 8, 2886. [Google Scholar] [CrossRef] [PubMed]
- Laperrousaz, B.; Porte, S.; Gerbaud, S.; Härmä, V.; Kermarrec, F.; Hourtane, V.; Bottausci, F.; Gidrol, X.; Picollet-D’Hahan, N. Direct transfection of clonal organoids in Matrigel microbeads: A promising approach toward organoid-based genetic screens. Nucleic Acids Res. 2018, 46, e70. [Google Scholar] [CrossRef] [PubMed]
- FLUIGENT. How to select the right microfluidic pump? Available online: https://www.fluigent.com/resources/microfluidic-expertise/what-is-microfluidic/system-comparison-for-microfluidic-applications/ (accessed on 6 January 2020).
- Behrens, M.R.; Fuller, H.C.; Swist, E.R.; Wu, J.; Islam, M.M.; Long, Z.; Ruder, W.C.; Steward, R. Open-source, 3D-printed Peristaltic Pumps for Small Volume Point-of-Care Liquid Handling. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Z.; Zhao, J.; Chao, Z.; You, Z.; Zhao, J. An air-chamber-based microfluidic stabilizer for attenuating syringe-pump-induced fluctuations. Microfluid. Nanofluidics 2019, 23, 26. [Google Scholar] [CrossRef]
- Gao, R.Z.; Hébert, M.; Huissoon, J.; Ren, C.L. µPump: An open-source pressure pump for precision fluid handling in microfluidics. HardwareX 2020, 7, e00096. [Google Scholar] [CrossRef]
- De Groot, T.E.; Veserat, K.S.; Berthier, E.; Beebe, D.J.; Theberge, A.B. Surface-tension driven open microfluidic platform for hanging droplet culture. Lab Chip 2016, 16, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Kim, J.; Lee, J.S.; Min, S.; Kim, S.; Ahn, D.H.; Kim, Y.G.; Cho, S.W. Vascularized Liver Organoids Generated Using Induced Hepatic Tissue and Dynamic Liver-Specific Microenvironment as a Drug Testing Platform. Adv. Funct. Mater. 2018, 28, 1–15. [Google Scholar] [CrossRef]
- Zhu, X.; Chu, L.Y.; Chueh, B.H.; Shen, M.; Hazarika, B.; Phadke, N.; Takayama, S. Arrays of horizontally-oriented mini-reservoirs generate steady microfluidic flows for continuous perfusion cell culture and gradient generation. Analyst 2004, 129, 1026–1031. [Google Scholar] [CrossRef]
- Ong, L.J.Y.; Chong, L.H.; Jin, L.; Singh, P.K.; Lee, P.S.; Yu, H.; Ananthanarayanan, A.; Leo, H.L.; Toh, Y.C. A pump-free microfluidic 3D perfusion platform for the efficient differentiation of human hepatocyte-like cells. Biotechnol. Bioeng. 2017, 114, 2360–2370. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, D.; Phan, D.T.T.; Liu, J.; Chen, X.; Yang, B.; Hughes, C.C.W.; Zhang, W.; Lee, A.P. A hydrostatic pressure-driven passive micropump enhanced with siphon-based autofill function. Lab Chip 2018, 18, 2167–2177. [Google Scholar] [CrossRef]
- Berthier, E.; Beebe, D.J. Flow rate analysis of a surface tension driven passive micropump. Lab Chip 2007, 7, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, S.K.; Woo, D.H.; Lee, E.J.; Kim, J.H.; Lee, S.H. Differentiation of neural progenitor cells in a microfluidic chip-generated cytokine gradient. Stem Cells 2009, 27, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.B.; DeRoller, N.; Zhu, Y.; Koley, G. Application of ion-sensitive field effect transistors for ion channel screening. Biosens. Bioelectron. 2014, 54, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Özsoylu, D.; Kizildag, S.; Schöning, M.J.; Wagner, T. Differential chemical imaging of extracellular acidification within microfluidic channels using a plasma-functionalized light-addressable potentiometric sensor (LAPS). Phys. Med. 2020, 10. [Google Scholar] [CrossRef]
- Manjakkal, L.; Szwagierczak, D.; Dahiya, R. Metal oxides based electrochemical pH sensors: Current progress and future perspectives. Prog. Mater. Sci. 2020, 109, 100635. [Google Scholar] [CrossRef]
- Magnusson, E.B.; Halldorsson, S.; Fleming, R.M.T.; Leosson, K. Real-time optical pH measurement in a standard microfluidic cell culture system. Biomed. Opt. Express 2013, 4, 1749. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Baumann, W.; Brischwein, M.; Gahle, H.J.; Freund, I.; Ehret, R.; Drechsler, S.; Palzer, H.; Kleintges, M.; Sieben, U.; et al. Simultaneous measurement of cellular respiration and acidification with a single CMOS ISFET. Biosens. Bioelectron. 2001, 16, 195–203. [Google Scholar] [CrossRef]
- Beckers, S.; Noor, F.; Müller-Vieira, U.; Mayer, M.; Strigun, A.; Heinzle, E. High throughput, non-invasive and dynamic toxicity screening on adherent cells using respiratory measurements. Toxicol. Vitr. 2010, 24, 686–694. [Google Scholar] [CrossRef]
- Thomas, P.C.; Halter, M.; Tona, A.; Raghavan, S.R.; Plant, A.L.; Forry, S.P. A noninvasive thin film sensor for monitoring oxygen tension during in vitro cell culture. Anal. Chem. 2009, 81, 9239–9246. [Google Scholar] [CrossRef]
- Welch, D.; Christen, J.B. Real-time feedback control of pH within microfluidics using integrated sensing and actuation. Lab Chip 2014, 14, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Gómez, P.; Fernández-Sánchez, C.; Baldi, A. Microfluidic Modules with Integrated Solid-State Sensors for Reconfigurable Miniaturized Analysis Systems. ACS Omega 2019, 4, 6192–6198. [Google Scholar] [CrossRef]
- Thedinga, E.; Kob, A.; Holst, H.; Keuer, A.; Drechsler, S.; Niendorf, R.; Baumann, W.; Freund, I.; Lehmann, M.; Ehret, R. Online monitoring of cell metabolism for studying pharmacodynamic effects. Toxicol. Appl. Pharmacol. 2007, 220, 33–44. [Google Scholar] [CrossRef]
- Brischwein, M.; Motrescu, E.R.; Cabala, E.; Otto, A.M.; Grothe, H.; Wolf, B. Functional cellular assays with multiparametric silicon sensor chips. Lab Chip 2003, 3, 234–240. [Google Scholar] [CrossRef]
- Makarychev-Mikhailov, S.; Shvarev, A.; Bakker, E. New trends in ion-selective electrodes. In Electrochemical Sensors, Biosensors and their Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2008; pp. 71–114. [Google Scholar]
- Liang, T.; Gu, C.; Gan, Y.; Wu, Q.; He, C.; Tu, J.; Pan, Y.; Qiu, Y.; Kong, L.B.; Wan, H.; et al. Microfluidic chip system integrated with light addressable potentiometric sensor (LAPS) for real-time extracellular acidification detection. Sensors Actuators, B Chem. 2019, 301, 127004. [Google Scholar] [CrossRef]
- Hu, N.; Wu, C.; Ha, D.; Wang, T.; Liu, Q.; Wang, P. A novel microphysiometer based on high sensitivity LAPS and microfluidic system for cellular metabolism study and rapid drug screening. Biosens. Bioelectron. 2013, 40, 167–173. [Google Scholar] [CrossRef]
- Bonk, S.M.; Stubbe, M.; Buehler, S.M.; Tautorat, C.; Baumann, W.; Klinkenberg, E.D.; Gimsa, J. Design and characterization of a sensorized microfluidic cell-culture system with electro-thermal micro-pumps and sensors for cell adhesion, oxygen, and pH on a glass chip. Biosensors 2015, 5, 513–536. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Saito, T.; Yasukawa, T.; Shiku, H.; Abe, H.; Hoshi, H.; Matsue, T. Microfluidic chip integrated with amperometric detector array for in situ estimating oxygen consumption characteristics of single bovine embryos. Sens. Actuators B Chem. 2007, 125, 680–687. [Google Scholar] [CrossRef]
- Shaegh, S.A.M.; De Ferrari, F.; Zhang, Y.S.; Nabavinia, M.; Mohammad, N.B.; Ryan, J.; Pourmand, A.; Laukaitis, E.; Sadeghian, R.B.; Nadhman, A.; et al. A microfluidic optical platform for real-time monitoring of pH and oxygen in microfluidic bioreactors and organ-on-chip devices. Biomicrofluidics 2016, 10. [Google Scholar]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Shaegh, S.A.M.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef]
- Dornhof, J.; Kieninger, J.; Maurer, J.; Urban, G.A.; Weltin, A. Next Generation Organ-on-Chip System for Directional Control of Culture Conditions and Metabolic Monitoring of Tumor Organoids. In Proceedings of the 2019 20th International Conference on Solid-State Sensors, Actuators and Microsystems and Eurosensors XXXIII, TRANSDUCERS 2019 and EUROSENSORS XXXIII, Berlin, Germany, 23–27 June 2019; pp. 1106–1108. [Google Scholar]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Hospodiuk, M.; Dey, M.; Sosnoski, D.; Ozbolat, I.T. The bioink: A comprehensive review on bioprintable materials. Biotechnol. Adv. 2017, 35, 217–239. [Google Scholar] [CrossRef]
- Grolman, J.M.; Zhang, D.; Smith, A.M.; Moore, J.S.; Kilian, K.A. Rapid 3D Extrusion of Synthetic Tumor Microenvironments. Adv. Mater. 2015, 27, 5512–5517. [Google Scholar] [CrossRef]
- Skardal, A.; Murphy, S.V.; Devarasetty, M.; Mead, I.; Kang, H.W.; Seol, Y.J.; Zhang, Y.S.; Shin, S.R.; Zhao, L.; Aleman, J.; et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Pang, Y.; Mao, S.S.; Yao, R.; He, J.Y.; Zhou, Z.Z.; Feng, L.; Zhang, K.T.; Cheng, S.J.; Sun, W. TGF-β induced epithelial-mesenchymal transition in an advanced cervical tumor model by 3D printing. Biofabrication 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Sarker, S.F.; Crabbé, A.; Nickerson, C.A.; Prestwich, G.D. The generation of 3-D tissue models based on hyaluronan hydrogel-coated microcarriers within a rotating wall vessel bioreactor. Biomaterials 2010, 31, 8426–8435. [Google Scholar] [CrossRef]
- Aleman, J.; Skardal, A. A multi-site metastasis-on-a-chip microphysiological system for assessing metastatic preference of cancer cells. Biotechnol. Bioeng. 2019, 116, 936–944. [Google Scholar] [CrossRef]
- Hao, S.; Ha, L.; Cheng, G.; Wan, Y.; Xia, Y.; Sosnoski, D.M.; Mastro, A.M.; Zheng, S.Y. A Spontaneous 3D Bone-On-a-Chip for Bone Metastasis Study of Breast Cancer Cells. Small 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, E.; Guo, Z.; Yu, R.; Hao, H.; Xu, Y.; Sun, Z.; Li, X.; Lyu, J.; Wang, Q. Design and Construction of a Multi-Organ Microfluidic Chip Mimicking the in vivo Microenvironment of Lung Cancer Metastasis. ACS Appl. Mater. Interfaces 2016, 8, 25840–25847. [Google Scholar] [CrossRef]
- Zhou, X.; Qu, M.; Tebon, P.; Jiang, X.; Wang, C.; Xue, Y.; Zhu, J.; Zhang, S.; Oklu, R.; Sengupta, S.; et al. Screening Cancer Immunotherapy: When Engineering Approaches Meet Artificial Intelligence. Adv. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Aung, A.; Kumar, V.; Theprungsirikul, J.; Davey, S.K.; Varghese, S. An Engineered Tumor-on-a-Chip Device with Breast Cancer–Immune Cell Interactions for Assessing T-cell Recruitment. Cancer Res. 2020, 80, 263–275. [Google Scholar] [CrossRef]
- Hassell, B.A.; Goyal, G.; Lee, E.; Sontheimer-Phelps, A.; Levy, O.; Chen, C.S.; Ingber, D.E. Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In Vitro. Cell Rep. 2017, 21, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Schuster, B.; Junkin, M.; Kashaf, S.S.; Romero-Calvo, I.; Kirby, K.; Matthews, J.; Weber, C.R.; Rzhetsky, A.; White, K.P.; Tay, S. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Herland, A.; Maoz, B.M.; Das, D.; Somayaji, M.R.; Prantil-Baun, R.; Novak, R.; Cronce, M.; Huffstater, T.; Jeanty, S.S.F.; Ingram, M.; et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat. Biomed. Eng. 2020, 4, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.J.; Shin, T.H.; Kim, M.; Sung, C.O.; Jang, S.J.; Jeong, G.S. A one-stop microfluidic-based lung cancer organoid culture platform for testing drug sensitivity. Lab Chip 2019, 19, 2854–2865. [Google Scholar] [CrossRef] [PubMed]
- Shirure, V.S.; Bi, Y.; Curtis, M.B.; Lezia, A.; Goedegebuure, M.M.; Goedegebuure, S.P.; Aft, R.; Fields, R.C.; George, S.C. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 2018, 18, 3687–3702. [Google Scholar] [CrossRef]



| Features | Peristaltic Pump [186,187] | Syringe Pump [186,187,188] | Pressure Driven Pump [186,189] |
|---|---|---|---|
| Flow Rate Range | 2 μL/min–10 L/min | 0.012 nL/min–0.3 L/min | nL/min–mL/min |
| Working characteristics | Periodically compression of a flexible tubing | Controlled motion of the syringe piston | Pressure is applied to a sealed reservoir pushing liquid through tubes |
| Flow characteristics | -Pulsatile flow -Low stability -Flow recirculation is possible -Large volumes can be dispensed -Only the mean flow rate can be controlled | -Constant flow -Different stability levels according to device quality -Only a fixed volume of liquid can be dispensed | -Complex flow patterns can be performed -High stability -Flow recirculation is possible -Both pressure and flow rate can be controlled |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duzagac, F.; Saorin, G.; Memeo, L.; Canzonieri, V.; Rizzolio, F. Microfluidic Organoids-on-a-Chip: Quantum Leap in Cancer Research. Cancers 2021, 13, 737. https://doi.org/10.3390/cancers13040737
Duzagac F, Saorin G, Memeo L, Canzonieri V, Rizzolio F. Microfluidic Organoids-on-a-Chip: Quantum Leap in Cancer Research. Cancers. 2021; 13(4):737. https://doi.org/10.3390/cancers13040737
Chicago/Turabian StyleDuzagac, Fahriye, Gloria Saorin, Lorenzo Memeo, Vincenzo Canzonieri, and Flavio Rizzolio. 2021. "Microfluidic Organoids-on-a-Chip: Quantum Leap in Cancer Research" Cancers 13, no. 4: 737. https://doi.org/10.3390/cancers13040737
APA StyleDuzagac, F., Saorin, G., Memeo, L., Canzonieri, V., & Rizzolio, F. (2021). Microfluidic Organoids-on-a-Chip: Quantum Leap in Cancer Research. Cancers, 13(4), 737. https://doi.org/10.3390/cancers13040737