
Molecular Classification and Tumor Microenvironment Characterization of Gallbladder Cancer by Comprehensive Genomic and Transcriptomic Analysis

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
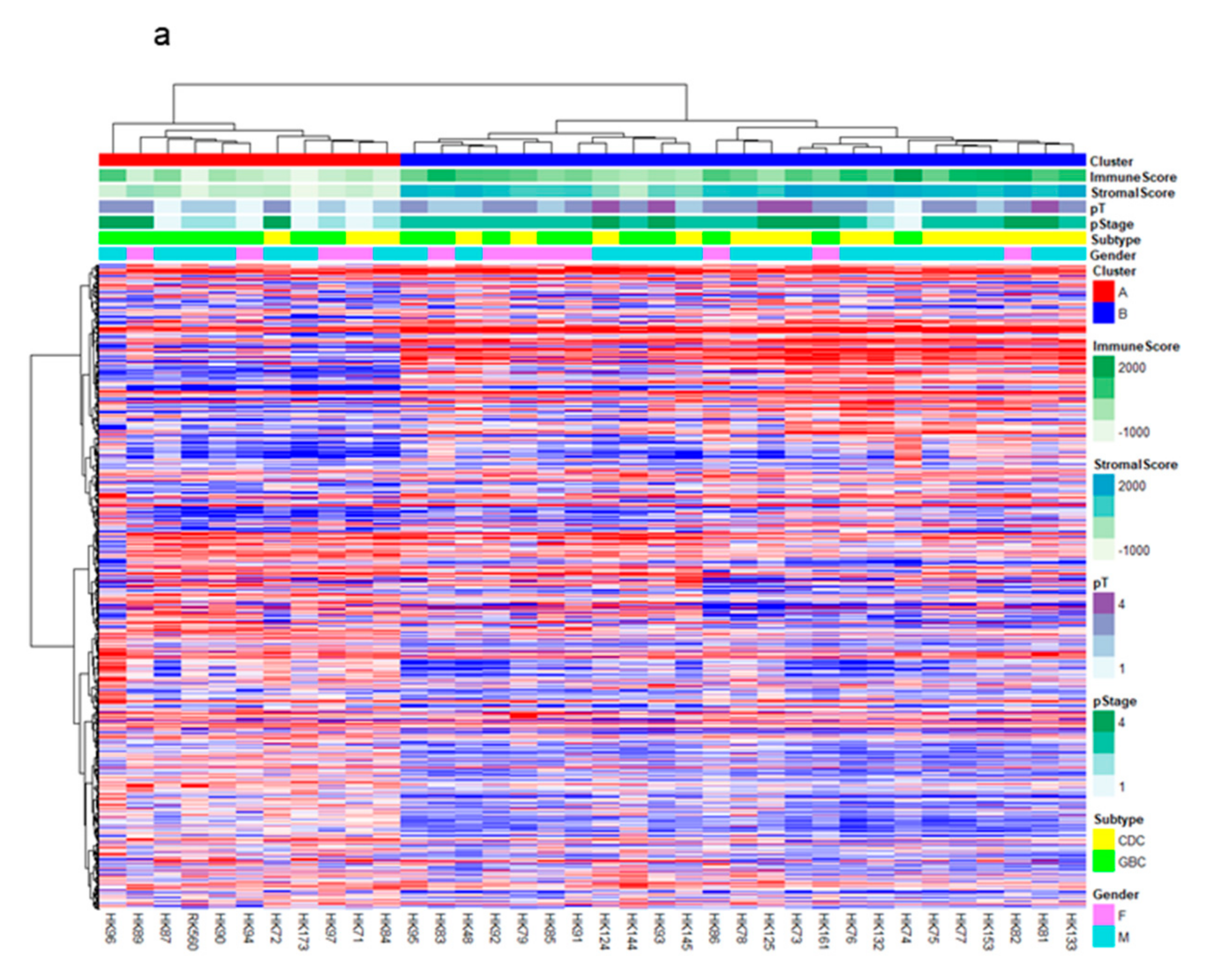
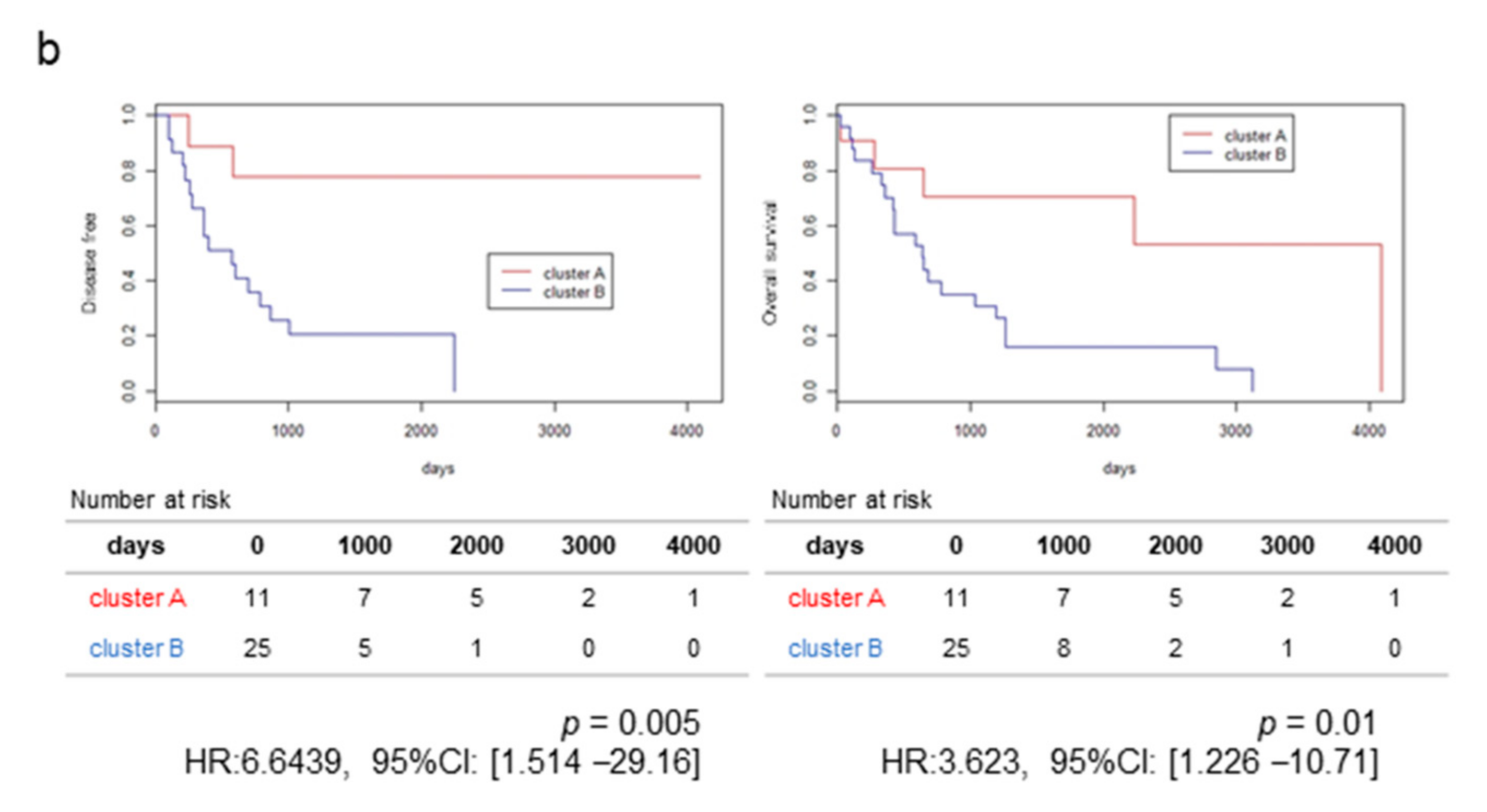
2.1. RNA-Sequencing (RNA-Seq) and Clustering Analysis Revealed Two Subclasses of GBCs
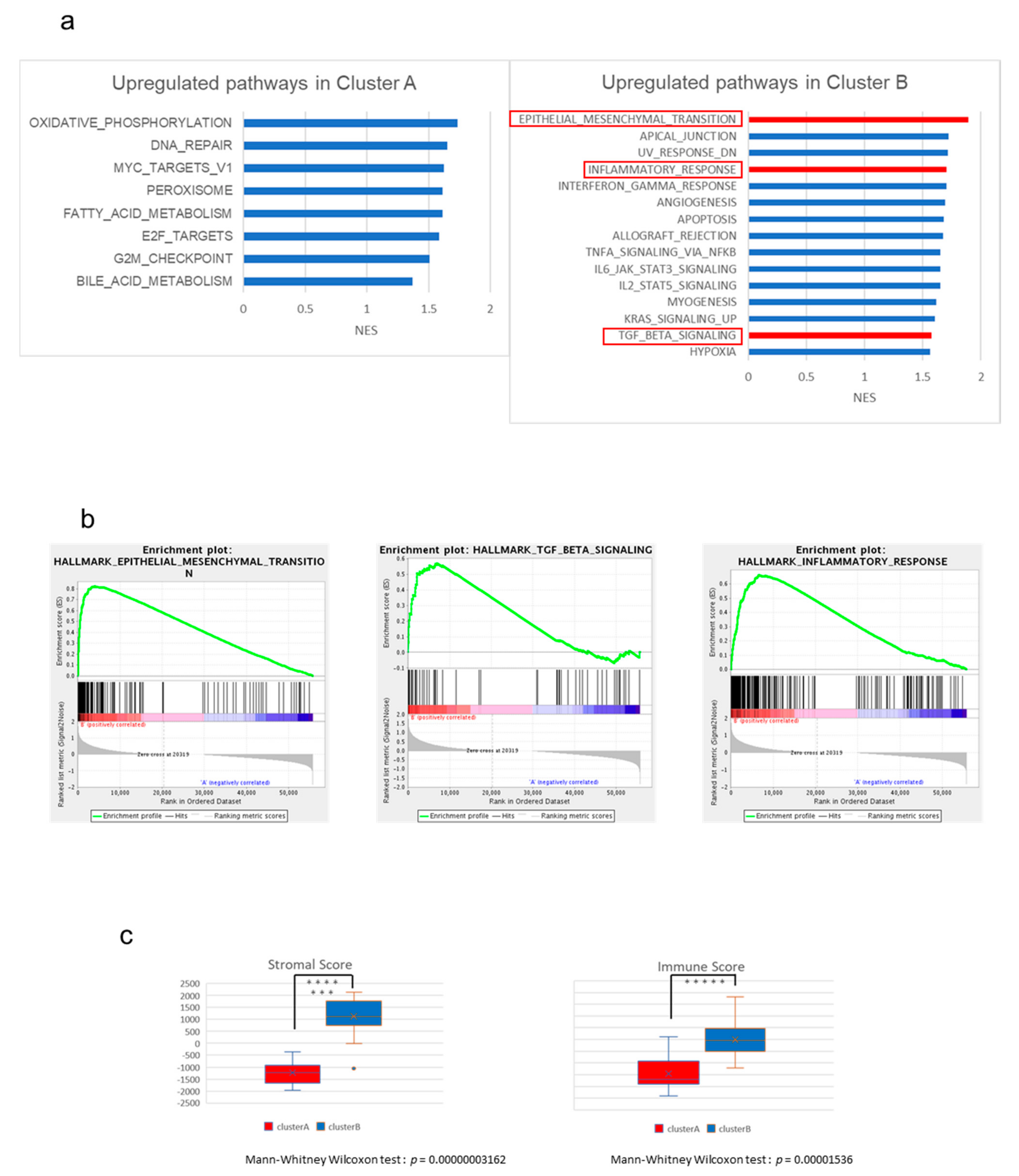
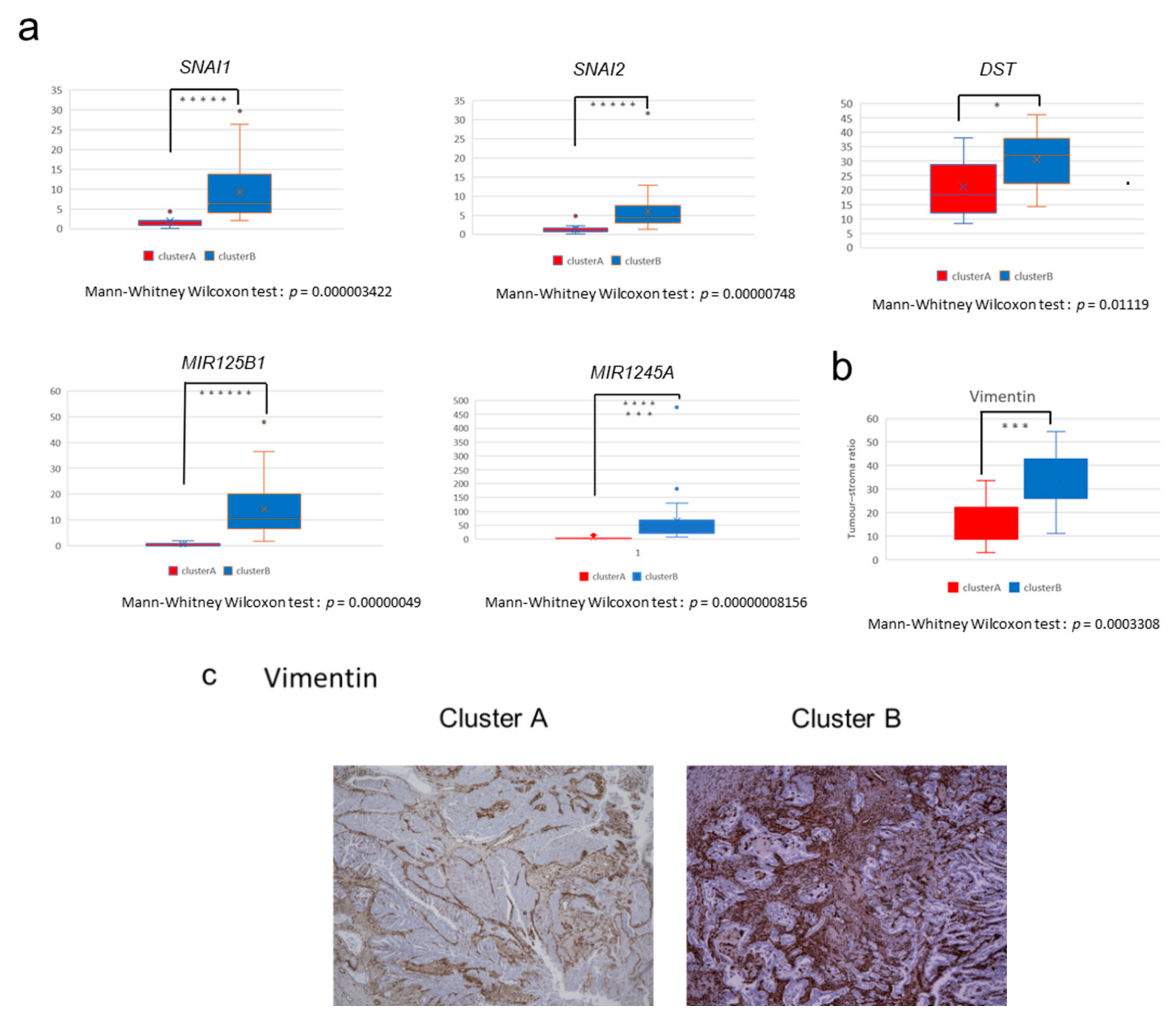
2.2. The High-Stromal Cluster Showed Upregulated Expression of Genes Involved in EMT and Inflammation
2.3. The High-Immune Core Cluster Showed Increased Expression of Immune Checkpoints
2.4. Immunostaining of Stromal Markers and Immune Cells in GBCs
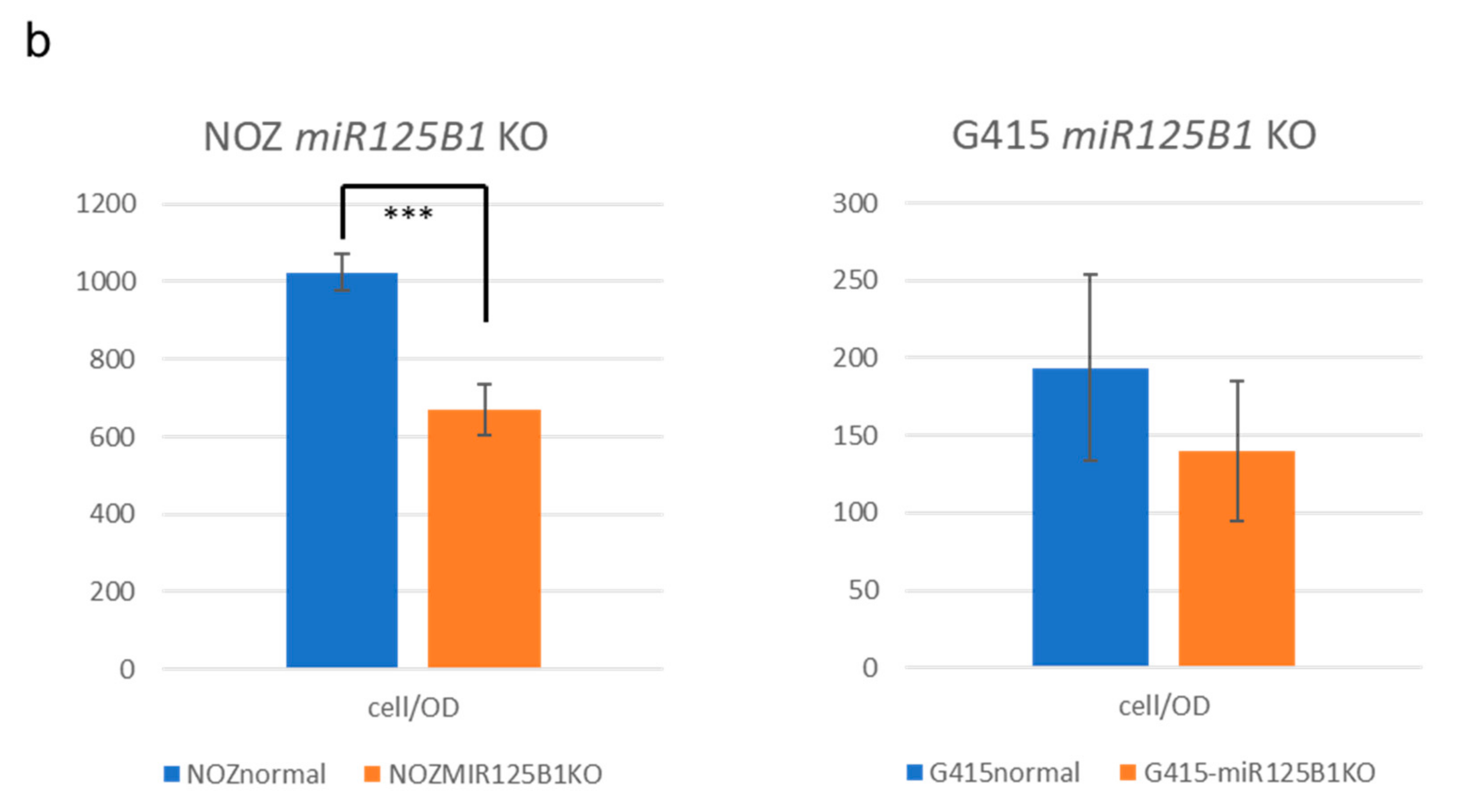
2.5. Non-Coding RNAs Are Involved in the Invasion and EMT of GBCs
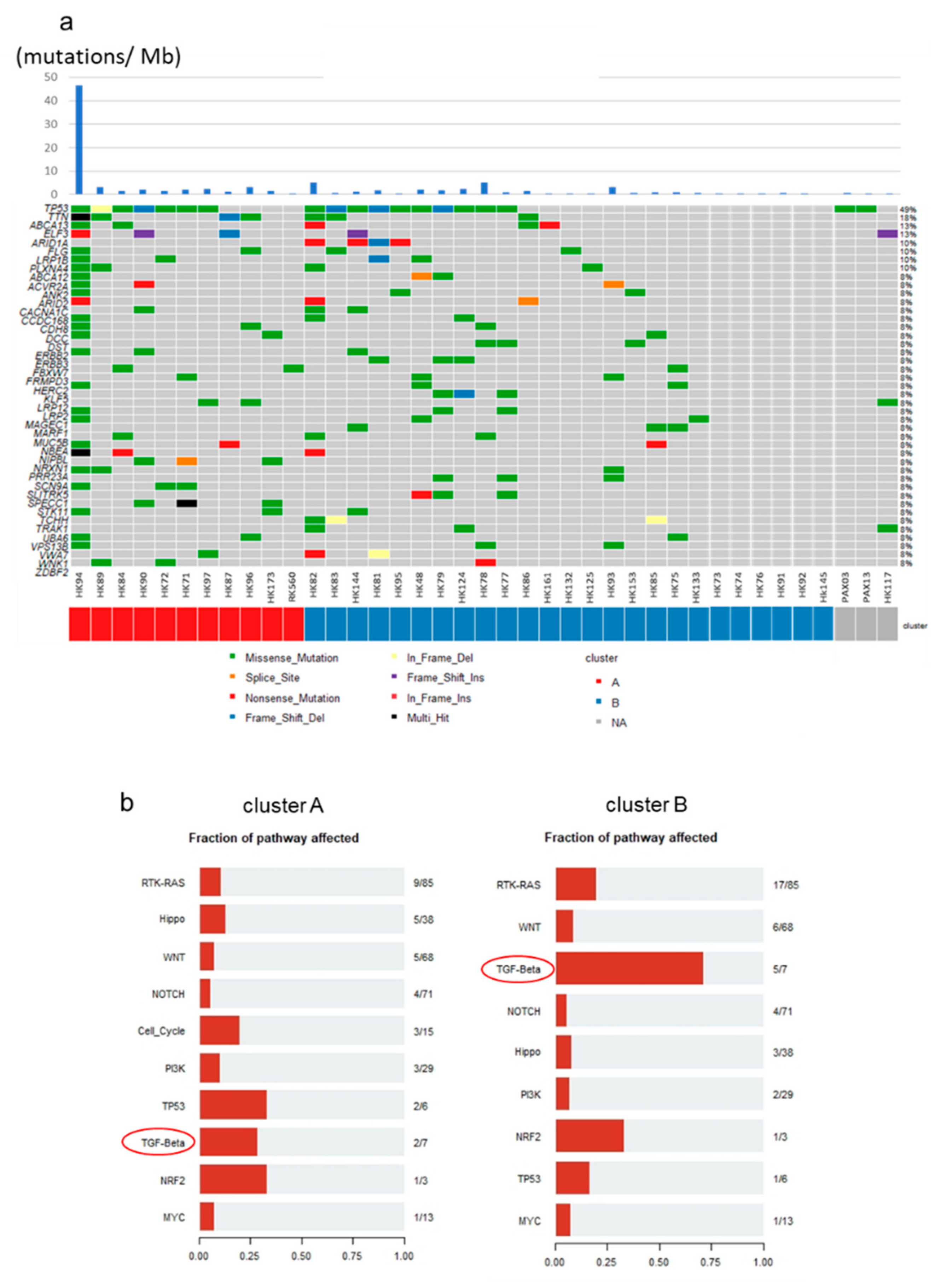
2.6. Mutational Profiles of GBC and Mutations of TGF-β Signaling Pathway Genes
2.7. Copy Number Variants (CNVs) and Fusion Events in GBCs
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Library Preparation and DNA or RNA Sequencing
4.3. Statistical Analysis
4.4. GSEA
4.5. SNV, INDELs, and Fusion Calling
4.6. DNA Copy Number Analysis
4.7. Immunohistochemistry
4.8. Cell Culture
4.9. Generation of CRISPR-Cas9-Mediated KO Cell Lines
4.10. Invasion and Cell Viability Assays
4.11. RNA-Seq Analysis of the KO Cells
4.12. Data Deposition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hundal, R.; Shaffer, E.A. Gallbladder cancer: Epidemiology and outcome. Clin. Epidemiol. 2014, 6, 99–109. [Google Scholar] [PubMed]
- Levy, A.D.; Rohrmann, C.A.; Murakata, L.A. Gallbladder Carcinoma: Radiologic-Pathologic Correlation. Radiographics 2001, 21, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T. Current Status and Future of Chemotherapy for Cholangiocarcinoma. Available online: https://doi.org/10.11210/tando.29.703 (accessed on 1 December 2020).
- Xu, S.; Zhan, M.; Jiang, C.; He, M.; Yang, L.; Shen, H.; Huang, S.; Huang, X.; Lin, R.; Shi, Y.; et al. Genome-wide CRISPR screen identifies ELP5 as a determinant of gemcitabine sensitivity in gallbladder cancer. Nat. Commun. 2019, 10, 5492. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Z.; Li, X.; Ye, J.; Wu, X.; Tan, Z.; Liu, C.; Shen, B.; Wang, X.A.; Wu, W.; et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014, 46, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Javle, M.; Pang, F.; Zhao, W.; Abdel-Wahab, R.; Chen, X.; Meric-Bernstam, F.; Chen, H.; Borad, M.J.; Liu, Y.; et al. Somatic genetic aberrations in gallbladder cancer: Comparison between Chinese and US patients. Hepatobiliary Surg. Nutr. 2019, 8, 604–614. [Google Scholar] [CrossRef]
- Pandey, A.; Stawiski, E.W.; Durinck, S.; Gowda, H.; Goldstein, L.D.; Barbhuiya, M.A.; Schroder, M.S.; Sreenivasamurthy, S.K.; Kim, S.W.; Phalke, S.; et al. Integrated genomic analysis reveals mutated ELF3 as a potential gallbladder cancer vaccine candidate. Nat. Commun. 2020, 11, 4225. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Jiang, J.; Yu, Y.; Tian, R.; Guo, X.; Li, X.; Shen, M.; Xu, M.; Zhu, F.; Shi, C.; et al. Direct targeting of SUZ12/ROCK2 by miR-200b/c inhibits cholangiocarcinoma tumourigenesis and metastasis. Br. J. Cancer 2013, 109, 3092–3104. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Terashima, M.; Ishimura, A.; Wanna-Udom, S.; Suzuki, T. MEG8 long noncoding RNA contributes to epigenetic progression of the epithelial-mesenchymal transition of lung and pancreatic cancer cells. J. Biol. Chem. 2018, 293, 18016–18030. [Google Scholar] [CrossRef]
- Jin, L.; Cai, Q.; Wang, S.; Wang, S.; Mondal, T.; Wang, J.; Quan, Z. Long noncoding RNA MEG3 regulates LATS2 by promoting the ubiquitination of EZH2 and inhibits proliferation and invasion in gallbladder cancer. Cell Death Dis. 2018, 9, 1017. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Tsuchikawa, T.; Okamura, K.; Nakamura, T.; Noji, T.; Asano, T.; Tanaka, K.; Shichinohe, T.; Mitsuhashi, T.; Hirano, S. Clinicopathological features and prognosis of advanced biliary carcinoma centered in the cystic duct. HPB 2018, 20, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Zissel, G.; Watz, H.; Dromann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human alveolar epithelial cells type II are capable of TGFbeta-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef]
- Sun, W.; Wei, F.Q.; Li, W.J.; Wei, J.W.; Zhong, H.; Wen, Y.H.; Lei, W.B.; Chen, L.; Li, H.; Lin, H.Q.; et al. A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br. J. Cancer 2017, 117, 1631–1643. [Google Scholar] [CrossRef]
- Takahashi, H.; Sakakura, K.; Toyoda, M.; Kaira, K.; Oyama, T.; Kudo, T.; Chikamatsu, K. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2017, 5, 8633–8647. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Plessers, J.; Dekimpe, E.; Van Woensel, M.; Roobrouck, V.D.; Bullens, D.M.; Pinxteren, J.; Verfaillie, C.M.; Van Gool, S.W. Clinical-Grade Human Multipotent Adult Progenitor Cells Block CD8+ Cytotoxic T Lymphocytes. Stem Cells Transl. Med. 2016, 5, 1607–1619. [Google Scholar] [CrossRef]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.F.; Joller, N.; Tan, D.J.; Teng, M.W.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef]
- Toor, S.M.; Sasidharan Nair, V.; Pfister, G.; Elkord, E. Effect of pembrolizumab on CD4(+) CD25(+), CD4(+) LAP(+) and CD4(+) TIM-3(+) T cell subsets. Clin. Exp. Immunol. 2019, 196, 345–352. [Google Scholar] [CrossRef]
- Wardell, C.P.; Fujita, M.; Yamada, T.; Simbolo, M.; Fassan, M.; Karlic, R.; Polak, P.; Kim, J.; Hatanaka, Y.; Maejima, K.; et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 2018, 68, 959–969. [Google Scholar] [CrossRef]
- Ng, A.Y.; Waring, P.; Ristevski, S.; Wang, C.; Wilson, T.; Pritchard, M.; Hertzog, P.; Kola, I. Inactivation of the transcription factor Elf3 in mice results in dysmorphogenesis and altered differentiation of intestinal epithelium. Gastroenterology 2002, 122, 1455–1466. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- McGrath, E.; Shin, H.; Zhang, L.; Phue, J.N.; Wu, W.W.; Shen, R.F.; Jang, Y.Y.; Revollo, J.; Ye, Z. Targeting specificity of APOBEC-based cytosine base editor in human iPSCs determined by whole genome sequencing. Nat. Commun. 2019, 10, 5353. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Ayhan, A.; Kuhn, E.; Wu, R.C.; Ogawa, H.; Bahadirli-Talbott, A.; Mao, T.L.; Sugimura, H.; Shih, I.M.; Wang, T.L. CCNE1 copy-number gain and overexpression identify ovarian clear cell carcinoma with a poor prognosis. Mod. Pathol. 2017, 30, 297–303. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.; Zhang, Y.; Yuan, M.; Yang, S.; Li, L.; Yang, H. Prognostic Values of CCNE1 Amplification and Overexpression in Cancer Patients: A Systematic Review and Meta-analysis. J. Cancer 2018, 9, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Ries, R.E.; Hylkema, T.; Alonzo, T.A.; Gerbing, R.B.; Santaguida, M.T.; Eidenschink Brodersen, L.; Pardo, L.; Cummings, C.L.; Loeb, K.R.; et al. Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin. Cancer Res. 2020, 26, 726–737. [Google Scholar] [CrossRef]
- Frattini, V.; Pagnotta, S.M.; Tala; Fan, J.J.; Russo, M.V.; Lee, S.B.; Garofano, L.; Zhang, J.; Shi, P.; Lewis, G.; et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 2018, 553, 222–227. [Google Scholar] [CrossRef]
- Zhu, C.; Wu, L.; Lv, Y.; Guan, J.; Bai, X.; Lin, J.; Liu, T.; Haohai, Z.; Yang, X.; Robson, S.C.; et al. The landscape of gene fusions in hepatocellular carcinoma. Mol. Oncol. 2019, 13, 1214–1222. [Google Scholar] [CrossRef]
- Gentilini, A.; Pastore, M.; Marra, F.; Raggi, C. The Role of Stroma in Cholangiocarcinoma: The Intriguing Interplay between Fibroblastic Component, Immune Cell Subsets and Tumor Epithelium. Int. J. Mol. Sci. 2018, 19, 2885. [Google Scholar] [CrossRef]
- Zhao, J.; Xiao, Z.; Li, T.; Chen, H.; Yuan, Y.; Wang, Y.A.; Hsiao, C.H.; Chow, D.S.; Overwijk, W.W.; Li, C. Stromal Modulation Reverses Primary Resistance to Immune Checkpoint Blockade in Pancreatic Cancer. ACS Nano 2018, 12, 9881–9893. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef]
- Xu, Z.; Pothula, S.P.; Wilson, J.S.; Apte, M.V. Pancreatic cancer and its stroma: A conspiracy theory. World J. Gastroenterol. 2014, 20, 11216–11229. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Ghidini, M.; Cascione, L.; Carotenuto, P.; Lampis, A.; Trevisani, F.; Previdi, M.C.; Hahne, J.C.; Said-Huntingford, I.; Raj, M.; Zerbi, A.; et al. Characterisation of the immune-related transcriptome in resected biliary tract cancers. Eur. J. Cancer 2017, 86, 158–165. [Google Scholar] [CrossRef]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef]
- Ottaviani, S.; Stebbing, J.; Frampton, A.E.; Zagorac, S.; Krell, J.; de Giorgio, A.; Trabulo, S.M.; Nguyen, V.T.M.; Magnani, L.; Feng, H.; et al. TGF-beta induces miR-100 and miR-125b but blocks let-7a through LIN28B controlling PDAC progression. Nat. Commun. 2018, 9, 1845. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef]
- Yang, L.; Wang, J.; Fan, Y.; Yu, K.; Jiao, B.; Su, X. Hsa_circ_0046264 up-regulated BRCA2 to suppress lung cancer through targeting hsa-miR-1245. Respir. Res. 2018, 19, 115. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Schmidt, M.A.; Marcano-Bonilla, L.; Roberts, L.R. Gallbladder cancer: Epidemiology and genetic risk associations. Chin. Clin. Oncol. 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Mayakonda, A.; Koeffler, H.P. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. BioRXiv 2016. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebata, N.; Fujita, M.; Sasagawa, S.; Maejima, K.; Okawa, Y.; Hatanaka, Y.; Mitsuhashi, T.; Oosawa-Tatsuguchi, A.; Tanaka, H.; Miyano, S.; et al. Molecular Classification and Tumor Microenvironment Characterization of Gallbladder Cancer by Comprehensive Genomic and Transcriptomic Analysis. Cancers 2021, 13, 733. https://doi.org/10.3390/cancers13040733
Ebata N, Fujita M, Sasagawa S, Maejima K, Okawa Y, Hatanaka Y, Mitsuhashi T, Oosawa-Tatsuguchi A, Tanaka H, Miyano S, et al. Molecular Classification and Tumor Microenvironment Characterization of Gallbladder Cancer by Comprehensive Genomic and Transcriptomic Analysis. Cancers. 2021; 13(4):733. https://doi.org/10.3390/cancers13040733
Chicago/Turabian StyleEbata, Nobutaka, Masashi Fujita, Shota Sasagawa, Kazuhiro Maejima, Yuki Okawa, Yutaka Hatanaka, Tomoko Mitsuhashi, Ayako Oosawa-Tatsuguchi, Hiroko Tanaka, Satoru Miyano, and et al. 2021. "Molecular Classification and Tumor Microenvironment Characterization of Gallbladder Cancer by Comprehensive Genomic and Transcriptomic Analysis" Cancers 13, no. 4: 733. https://doi.org/10.3390/cancers13040733
APA StyleEbata, N., Fujita, M., Sasagawa, S., Maejima, K., Okawa, Y., Hatanaka, Y., Mitsuhashi, T., Oosawa-Tatsuguchi, A., Tanaka, H., Miyano, S., Nakamura, T., Hirano, S., & Nakagawa, H. (2021). Molecular Classification and Tumor Microenvironment Characterization of Gallbladder Cancer by Comprehensive Genomic and Transcriptomic Analysis. Cancers, 13(4), 733. https://doi.org/10.3390/cancers13040733