
EphA2 and EGFR: Friends in Life, Partners in Crime. Can EphA2 Be a Predictive Biomarker of Response to Anti-EGFR Agents?



Abstract
Simple Summary
Abstract
1. Introduction
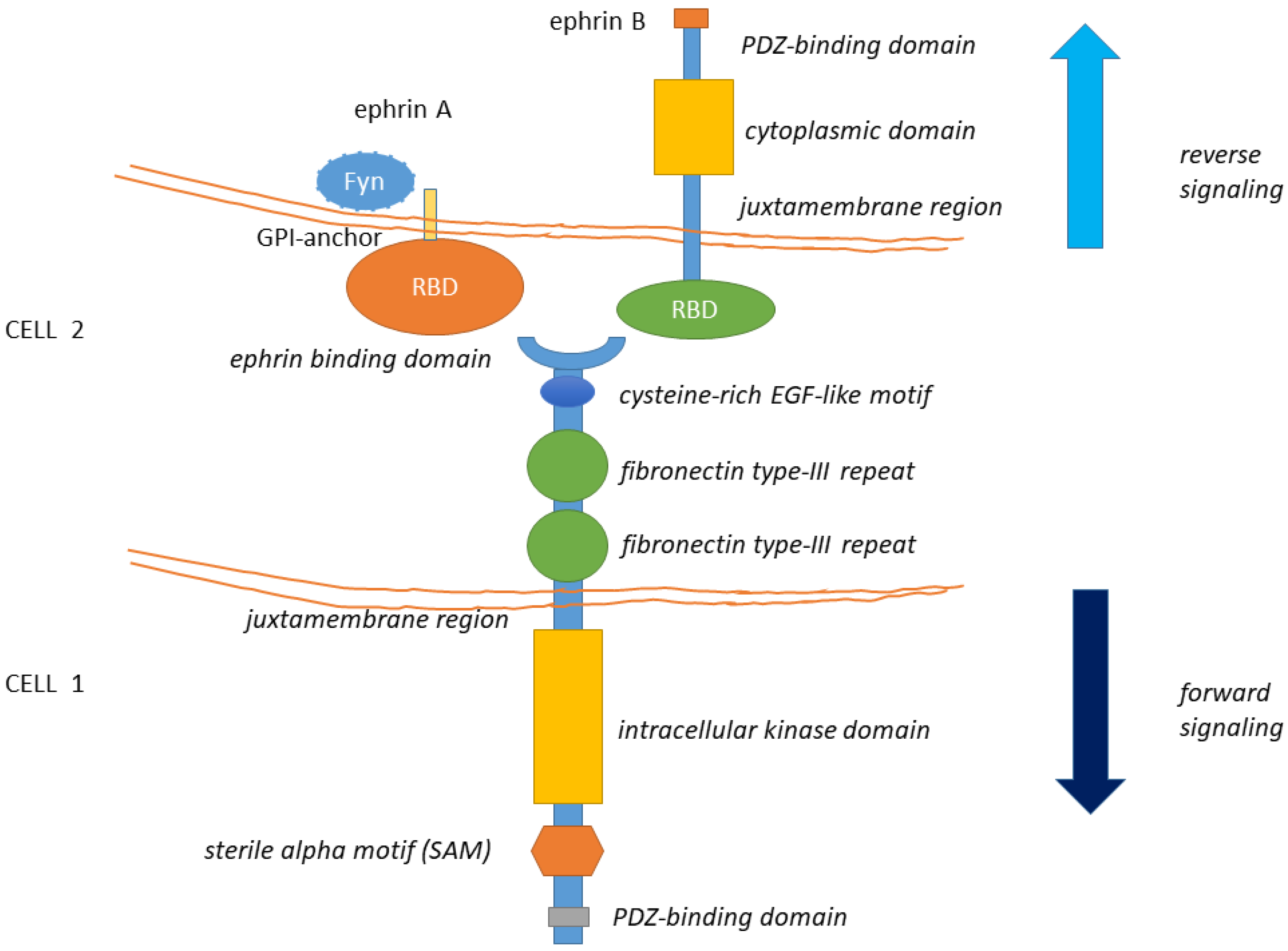
1.1. General Structure of Eph Receptors and Ephrin Ligands
1.2. General Features of Eph-Ephrin Signaling
2. EphA2 Signaling
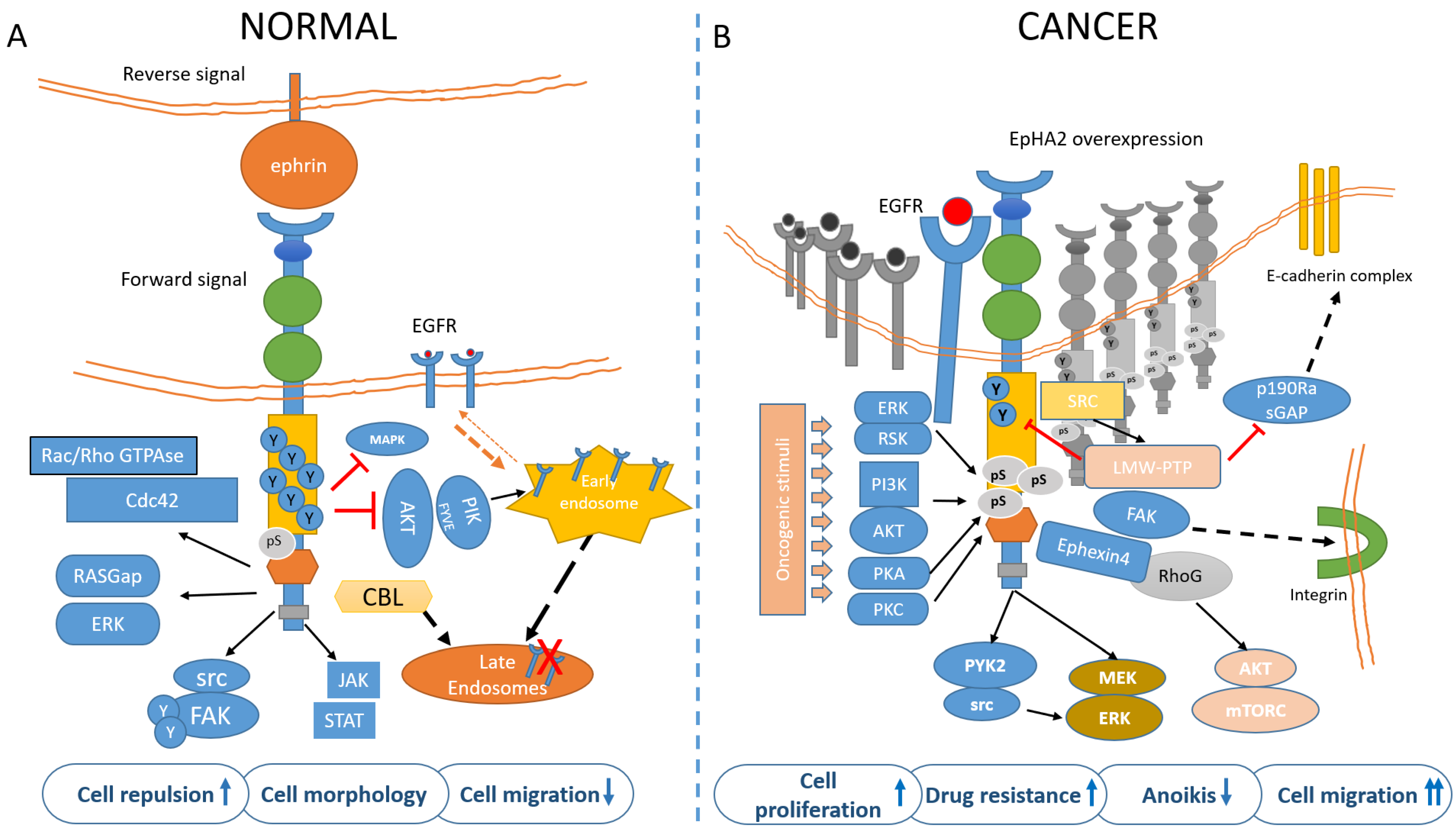
2.1. EphA2 Signaling in Normal Cells
2.2. EphA2 in Tissue Patterning
3. Molecular Determinants of EphA2 Signaling in Tumors
3.1. Intracellular Localization of EphA2
3.2. Expression Levels of EphA2
3.3. Ligand-Dependent EphA2 Signaling
3.4. Ligand-Independent Activation of EphA2
3.5. Tumor Context Modulates EphA2 Signaling
4. EphA2 Promotes Resistance to Therapy
EphA2 and CSCs
5. The EGFR-EphA2 Crosstalk in Cancer: Partners in Crime
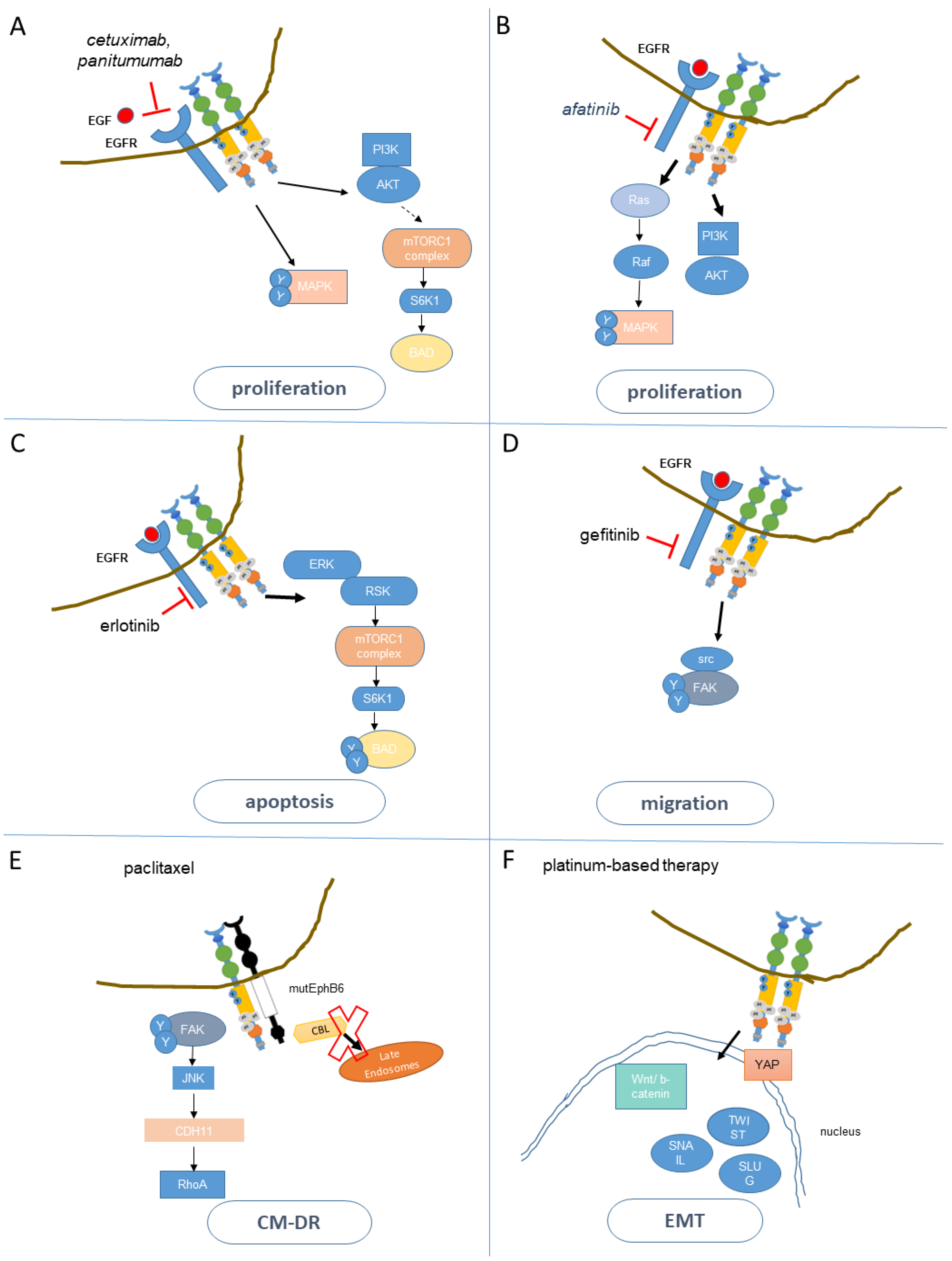
5.1. Resistance to Anti-EGFR Agents
5.2. EphA2 and the Resistance to Cetuximab
5.3. Mechanisms of Resistance to EGFR TKI
5.4. EphA2 and the Resistance to EGFR TKI
6. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Abl | abelson murine leukemia viral oncogene homolog 1 |
| AKT | protein kinase B |
| Ascl2 | Achaete-scute Complex Homologue 2 |
| BRAFV600E | B-Raf Proto-oncogene, Serine/Threonine Kinase |
| Cdc42 | cell division control protein 42 homolog |
| c-Myc | avian myelocytomatosis virus oncogene cellular homolog. |
| CRC | colorectal cancer |
| CSC | cancer stem cells |
| CTCs | circulating tumor cells |
| Cyclin-D1 | |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial to mesenchymal transition |
| ERK | extracellular-signal-regulated kinase |
| ERK1/2 | extracellular signal-regulated kinases 1/2 |
| FACS | fluorescence-activated cell sorting |
| FAK | focal adhesion kinase |
| FGF4 | fibroblast growth factor4 |
| FGFR | fibroblast growth factor receptor |
| FYN | oncogene related to SRC, FGR, YES |
| GEFs | guanidine nucleotide exchange factors |
| GPRC5A | G-protein coupled receptor 5A |
| Grb7 | growth factor receptor-bound protein 7 |
| HNSCC | squamous cell carcinoma of the head and neck |
| IGF-1 | Insulin-like-Growth-Factor-1 |
| JNK | c-Jun N-terminal kinase |
| K-Ras | related RAS viral oncogene homolog |
| Krt20 | cytokeratin 20 |
| Lgr5 | leucine-rich repeat-containing G-protein coupled receptor 5 |
| LMW-PTP | low molecular weight phosphotyrosine phosphatase |
| mAb | monoclonal antibody |
| MAPK | map kinase |
| MEK | mitogen-activated protein kinase |
| MET | mesenchymal to epithelial transition |
| mTOR | mechanistic target of rapamycin |
| N-Ras | related RAS Viral (R-Ras) oncogene homolog |
| PDGF | platelet-derived growth factor-beta |
| PDGFR | platelet-derived growth factor-beta receptor |
| PI3K | phosphoinositide 3-kinase |
| PIKFYVE | Phosphoinositide Kinase, FYVE-Type Zinc Finger Containing |
| PKA | protein Kinase A |
| PKC | protein Kinase C |
| PTEN | Phosphatase and Tensin Homolog |
| PYK | proline-rich protein tyrosine kinase 2 |
| Rac1 | ras-related C3 botulinum toxin substrate 1 |
| Ras-GAP | Ras-GTPase-activating protein |
| RET (RET/PTC) | REarranged during Transfection |
| Rho-GEFs | Rho Guanidine nucleotide Exchange Factors |
| Rho-GTP | Rho family of small GTP-binding proteins |
| RSK | ribosomal protein S6 kinase |
| siRNAs | antisense oligonucleotides |
| Src | proto-oncogene tyrosine-protein kinase |
| TCF | T-cell factor |
| TCF4 | T-cell factor 4 |
| TKI | tyrosine kinase inhibitors |
| TKR | tyrosine kinase receptor |
| VEGF | vascular endothelial growth factor |
| xCT | cystine/glutamate exchange transporter |
References
- Tuzi, N.L.; Gullick, W.J. Eph, the largest known family of putative growth factor receptors. Br. J. Cancer 1994, 69, 417–421. [Google Scholar] [CrossRef][Green Version]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph Receptor Signaling and Ephrins. Cold Spring Harb. Perspect. Biol. 2013, 5, a009159. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Journal club. A biologist is gratified to find reconciliation for a conflicted receptor. Nature 2009, 461, 149. [Google Scholar] [CrossRef] [PubMed]
- Himanen, J.P.; Goldgur, Y.; Miao, H.; Myshkin, E.; Guo, H.; Buck, M.; Nguyen, M.; Rajashankar, K.R.; Wang, B.; Nikolov, D.B. Ligand recognition by A-class Eph receptors: Crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex. EMBO Rep. 2009, 10, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Janes, P.W.; Griesshaber, B.; Atapattu, L.; Nievergall, E.; Hii, L.L.; Mensinga, A.; Chheang, C.; Day, B.W.; Boyd, A.W.; Bastiaens, P.I.; et al. Eph receptor function is modulated by heterooligomerization of A and B type Eph receptors. J. Cell Biol. 2011, 195, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Eph Nomenclature Committee. Unified Nomenclature for Eph Family Receptors and Their Ligands, the Ephrins. Cell 1997, 90, 403–404. [Google Scholar] [CrossRef]
- Wykosky, J.; Palma, E.; Gibo, D.M.; Ringler, S.L.; Turner, C.P.; Debinski, W. Soluble monomeric EphrinA1 is released from tumor cells and is a functional ligand for the EphA2 receptor. Oncogene 2008, 27, 7260–7273. [Google Scholar] [CrossRef]
- Egea, J.; Klein, R. Bidirectional Eph–ephrin signaling during axon guidance. Trends Cell Biol. 2007, 17, 230–238. [Google Scholar] [CrossRef]
- Binns, K.L.; Taylor, P.P.; Sicheri, F.; Pawson, T.; Holland, S.J. Phosphorylation of Tyrosine Residues in the Kinase Domain and Juxtamembrane Region Regulates the Biological and Catalytic Activities of Eph Receptors. Mol. Cell. Biol. 2000, 20, 4791–4805. [Google Scholar] [CrossRef]
- Wybenga-Groot, L.E.; Baskin, B.; Ong, S.H.; Tong, J.; Pawson, T.; Sicheri, F. Structural Basis for Autoinhibition of the EphB2 Receptor Tyrosine Kinase by the Unphosphorylated Juxtamembrane Region. Cell 2001, 106, 745–757. [Google Scholar] [CrossRef]
- Singla, N.; Erdjument-Bromage, H.; Himanen, J.P.; Muir, T.W.; Nikolov, D.B. A Semisynthetic Eph Receptor Tyrosine Kinase Provides Insight into Ligand-Induced Kinase Activation. Chem. Biol. 2011, 18, 361–371. [Google Scholar] [CrossRef]
- Murai, K.; Pasquale, E. ‘Eph’ective signaling: Forward, reverse and crosstalk. J. Cell Sci. 2003, 116, 2823–2832. [Google Scholar] [CrossRef]
- Atapattu, L.; Lackmann, M.; Janes, P.W. The role of proteases in regulating Eph/ephrin signaling. Cell Adhes. Migr. 2014, 8, 294–307. [Google Scholar] [CrossRef]
- Falivelli, G.; Lisabeth, E.M.; De La Torre, E.R.; Pérez-Tenorio, G.; Tosato, G.; Salvucci, O.; Pasquale, E.B. Attenuation of Eph Receptor Kinase Activation in Cancer Cells by Coexpressed Ephrin Ligands. PLoS ONE 2013, 8, e81445. [Google Scholar] [CrossRef]
- Takasugi, M.; Okada, R.; Takahashi, A.; Chen, D.V.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef]
- Liang, L.-Y.; Patel, O.; Janes, P.W.; Murphy, J.M.; Lucet, I.S. Eph receptor signalling: From catalytic to non-catalytic functions. Oncogene 2019, 38, 6567–6584. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Maru, Y.; Hagiwara, K.; Nishida, J.; Takaku, F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science 1987, 238, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Bartley, T.D.; Hunt, R.W.; Welcher, A.A.; Boyle, W.J.; Parker, V.P.; Lindberg, R.A.; Lu, H.S.; Colombero, A.M.; Elliott, R.L.; Guthrie, B.A.; et al. B61 is a ligand for the ECK receptor protein-tyrosine kinase. Nature 1994, 368, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Walker-Daniels, J.; Riese, D.J., 2nd; Kinch, M.S. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol. Cancer Res. 2002, 1, 79–87. [Google Scholar]
- Seiradake, E.; Schaupp, A.; Ruiz, D.D.T.; Kaufmann, R.; Mitakidis, N.; Harlos, K.; Aricescu, A.R.; Klein, R.; Jones, E.Y. Structurally encoded intraclass differences in EphA clusters drive distinct cell responses. Nat. Struct. Mol. Biol. 2013, 20, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Wang, B. EphA receptor signaling—Complexity and emerging themes. Semin. Cell Dev. Biol. 2012, 23, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Burnett, E.; Kinch, M.; Simon, E.; Wang, B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 1999, 2, 62–69. [Google Scholar] [CrossRef]
- Bin Fang, W.; Brantley-Sieders, D.M.; Hwang, Y.; Ham, A.-J.L.; Chen, J. Identification and Functional Analysis of Phosphorylated Tyrosine Residues within EphA2 Receptor Tyrosine Kinase. J. Biol. Chem. 2008, 283, 16017–16026. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.-J.; Henkemeyer, M. Ephrin reverse signaling in axon guidance and synaptogenesis. Semin. Cell Dev. Biol. 2012, 23, 58–64. [Google Scholar] [CrossRef]
- Wang, Y.; Shang, Y.; Li, J.; Chen, W.; Li, G.; Wan, J.; Liu, W.; Zhang, M. Specific Eph receptor-cytoplasmic effector signaling mediated by SAM–SAM domain interactions. eLife 2018, 7, e35677. [Google Scholar] [CrossRef] [PubMed]
- Noren, N.K.; Pasquale, E.B. Eph receptor–ephrin bidirectional signals that target Ras and Rho proteins. Cell. Signal. 2004, 16, 655–666. [Google Scholar] [CrossRef]
- Barquilla, A.; Pasquale, E.B. Eph Receptors and Ephrins: Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 465–487. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Miao, H.; Gerber, L.; Singh, J.; Denning, M.F.; Gilliam, A.C.; Wang, B. Disruption of EphA2 Receptor Tyrosine Kinase Leads to Increased Susceptibility to Carcinogenesis in Mouse Skin. Cancer Res. 2006, 66, 7050–7058. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Koyama, T.; Wakayama, Y.; Fukuhara, S.; Mochizuki, N. EphrinA/EphA signal facilitates insulin-like growth factor-I–induced myogenic differentiation through suppression of the Ras/extracellular signal–regulated kinase 1/2 cascade in myoblast cell lines. Mol. Biol. Cell 2011, 22, 3508–3519. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Anastasiadou, S.; Knöll, B. Ephrin-A5 Suppresses Neurotrophin Evoked Neuronal Motility, ERK Activation and Gene Expression. PLoS ONE 2011, 6, e26089. [Google Scholar] [CrossRef]
- Nie, D.-Y.; Di Nardo, A.; Han, J.M.; Baharanyi, H.; Kramvis, I.; Huynh, T.; Dabora, S.L.; Codeluppi, S.; Pandolfi, P.P.; Pasquale, E.B.; et al. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat. Neurosci. 2010, 13, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Niethamer, T.K.; Bush, J.O. Getting direction(s): The Eph/ephrin signaling system in cell positioning. Dev. Biol. 2019, 447, 42–57. [Google Scholar] [CrossRef]
- Cayuso, J.; Xu, Q.; Wilkinson, D.G. Mechanisms of boundary formation by Eph receptor and ephrin signaling. Dev. Biol. 2015, 401, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, C.; Sherman, A.; Chen, G.I.; Pasculescu, A.; Poliakov, A.; Hsiung, M.; Larsen, B.; Wilkinson, D.G.; Linding, R.; Pawson, T. Cell-Specific Information Processing in Segregating Populations of Eph Receptor Ephrin-Expressing Cells. Science 2009, 326, 1502–1509. [Google Scholar] [CrossRef]
- Wu, Z.; Ashlin, T.G.; Xu, Q.; Wilkinson, D.G. Role of forward and reverse signaling in Eph receptor and ephrin mediated cell segregation. Exp. Cell Res. 2019, 381, 57–65. [Google Scholar] [CrossRef]
- Holmberg, J.; Armulik, A.; Senti, K.-A.; Edoff, K.; Spalding, K.L.; Momma, S.; Cassidy, R.M.; Flanagan, J.G.; Frisén, J. Ephrin-A2 reverse signaling negatively regulates neural progenitor proliferation and neurogenesis. Genes Dev. 2005, 19, 462–471. [Google Scholar] [CrossRef]
- Jiao, J.-W.; Feldheim, D.A.; Chen, D.F. Ephrins as negative regulators of adult neurogenesis in diverse regions of the central nervous system. Proc. Natl. Acad. Sci. USA 2008, 105, 8778–8783. [Google Scholar] [CrossRef] [PubMed]
- Vaught, D.B.; Chen, J.; Brantley-Sieders, D.M. Regulation of Mammary Gland Branching Morphogenesis by EphA2 Receptor Tyrosine Kinase. Mol. Biol. Cell 2009, 20, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Orsulic, S.; Kemler, R. Expression of Eph receptors and ephrins is differentially regulated by E-cadherin. J. Cell Sci. 2000, 113, 1793–1802. [Google Scholar]
- Tanaka, M.; Kamata, R.; Sakai, R. EphA2 Phosphorylates the Cytoplasmic Tail of Claudin-4 and Mediates Paracellular Permeability. J. Biol. Chem. 2005, 280, 42375–42382. [Google Scholar] [CrossRef]
- De Robertis, M.; Mazza, T.; Fusilli, C.; LoIacono, L.; Poeta, M.L.; Sanchez, M.; Massi, E.; Lamorte, G.; Diodoro, M.G.; Pescarmona, E.; et al. EphB2 stem-related and EphA2 progression-related miRNA-based networks in progressive stages of CRC evolution: Clinical significance and potential miRNA drivers. Mol. Cancer 2018, 17, 169. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, I.M.; Göke, M.; Kanai, M.; Reinecker, H.-C.; Podolsky, D.K. Epithelial cell kinase-B61: An autocrine loop modulating intestinal epithelial migration and barrier function. Am. J. Physiol. 1997, 273, G824–G832. [Google Scholar] [CrossRef]
- Baldwin, C.; Chen, Z.W.; Bedirian, A.; Yokota, N.; Nasr, S.H.; Rabb, H.; Lemay, S. Upregulation of EphA2 during in vivo and in vitro renal ischemia-reperfusion injury: Role of Src kinases. Am. J. Physiol. Renal Physiol. 2006, 291, F960–F971. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Gooding, A.J.; Schiemann, W.P. Epithelial–Mesenchymal Transition Programs and Cancer Stem Cell Phenotypes: Mediators of Breast Cancer Therapy Resistance. Mol. Cancer Res. 2020, 18, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Zantek, N.D.; Azimi, M.; Fedor-Chaiken, M.; Wang, B.; Brackenbury, R.; Kinch, M.S. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1999, 10, 629–638. [Google Scholar]
- Shang, X.; Lin, X.; Howell, S.B. Claudin-4 controls the receptor tyrosine kinase EphA2 pro-oncogenic switch through beta-catenin. Cell Commun. Signal. 2014, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Faoro, L.; Singleton, P.A.; Cervantes, G.M.; Lennon, F.E.; Choong, N.W.; Kanteti, R.; Ferguson, B.D.; Husain, A.N.; Tretiakova, M.S.; Ramnath, N.; et al. EphA2 Mutation in Lung Squamous Cell Carcinoma Promotes Increased Cell Survival, Cell Invasion, Focal Adhesions, and Mammalian Target of Rapamycin Activation. J. Biol. Chem. 2010, 285, 18575–18585. [Google Scholar] [CrossRef] [PubMed]
- Mudali, S.V.; Fu, B.; Lakkur, S.S.; Luo, M.; Embuscado, E.E.; Iacobuzio-Donahue, C.A. Patterns of EphA2 protein expression in primary and metastatic pancreatic carcinoma and correlation with genetic status. Clin. Exp. Metastasis 2006, 23, 357–365. [Google Scholar] [CrossRef]
- Dohn, M.; Jiang, J.; Chen, X. Receptor tyrosine kinase EphA2 is regulated by p53-family proteins and induces apoptosis. Oncogene 2001, 20, 6503–6515. [Google Scholar] [CrossRef]
- Zelinski, D.P.; Zantek, N.D.; Walker-Daniels, J.; Peters, M.A.; Taparowsky, E.J.; Kinch, M.S. Estrogen and Myc negatively regulate expression of the EphA2 tyrosine kinase. J. Cell. Biochem. 2002, 85, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Kinch, M.S.; Carles-Kinch, K. Overexpression and functional alterations of the EphA2 tyrosine kinase in cancer. Clin. Exp. Metastasis 2003, 20, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Zelinski, D.P.; Zantek, N.D.; Stewart, J.C.; Irizarry, A.R.; Kinch, M.S. EphA2 overexpression causes tumorigenesis of mam-mary epithelial cells. Cancer Res. 2001, 61, 2301–2306. [Google Scholar] [PubMed]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. EphA2 as a Novel Molecular Marker and Target in Glioblastoma Multiforme. Mol. Cancer Res. 2005, 3, 541–551. [Google Scholar] [CrossRef]
- Miyazaki, T.; Kato, H.; Fukuchi, M.; Nakajima, M.; Kuwano, H. EphA2 overexpression correlates with poor prognosis in esophageal squamous cell carcinoma. Int. J. Cancer 2002, 103, 657–663. [Google Scholar] [CrossRef]
- Huang, C.; Yuan, W.; Lai, C.; Zhong, S.; Yang, C.; Wang, R.; Mao, L.; Chen, Z.; Chen, Z. EphA2-to-YAP pathway drives gastric cancer growth and therapy resistance. Int. J. Cancer 2020, 146, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Hu, Z.; Kinch, M.S.; Pan, C.-X.; Flockhart, D.A.; Kao, C.; Gardner, T.A.; Zhang, S.; Li, L.; Baldridge, L.A.; et al. High-Level Expression of EphA2 Receptor Tyrosine Kinase in Prostatic Intraepithelial Neoplasia. Am. J. Pathol. 2003, 163, 2271–2276. [Google Scholar] [CrossRef]
- Dunne, P.D.; Dasgupta, S.; Blayney, J.K.; McArt, D.G.; Redmond, K.L.; Weir, J.-A.; Bradley, C.A.; Sasazuki, T.; Shirasawa, S.; Wang, T.; et al. EphA2 Expression Is a Key Driver of Migration and Invasion and a Poor Prognostic Marker in Colorectal Cancer. Clin. Cancer Res. 2016, 22, 230–242. [Google Scholar] [CrossRef]
- Strimpakos, A.; Pentheroudakis, G.; Kotoula, V.; De Roock, W.; Kouvatseas, G.; Papakostas, P.; Makatsoris, T.; Papamichael, D.; Andreadou, A.; Sgouros, J.; et al. The Prognostic Role of Ephrin A2 and Endothelial Growth Factor Receptor Pathway Mediators in Patients with Advanced Colorectal Cancer Treated with Cetuximab. Clin. Colorectal Cancer 2013, 12, 267–274.e2. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, M.; LoIacono, L.; Fusilli, C.; Poeta, M.L.; Mazza, T.; Sanchez, M.; Marchionni, L.; Signori, E.; Lamorte, G.; Vescovi, A.L.; et al. Dysregulation of EGFR Pathway in EphA2 Cell Subpopulation Significantly Associates with Poor Prognosis in Colorectal Cancer. Clin. Cancer Res. 2016, 23, 159–170. [Google Scholar] [CrossRef]
- Thaker, P.H.; Deavers, M.; Celestino, J.; Thornton, A.; Fletcher, M.S.; Landen, C.N.; Kinch, M.S.; Kiener, P.A.; Sood, A.K. EphA2 Expression Is Associated with Aggressive Features in Ovarian Carcinoma. Clin. Cancer Res. 2004, 10, 5145–5150. [Google Scholar] [CrossRef]
- Han, L.; Dong, Z.; Qiao, Y.; Kristensen, G.B.; Holm, R.; Nesland, J.M.; Suo, Z. The clinical significance of EphA2 and Ephrin A-1 in epithelial ovarian carcinomas. Gynecol. Oncol. 2005, 99, 278–286. [Google Scholar] [CrossRef]
- Merritt, W.M.; Thaker, P.H.; Landen, C.N., Jr.; Deavers, M.; Fletcher, M.S.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Gershenson, D.; Kinch, M.S.; et al. Analysis of EphA2 expression and mutant p53 in ovarian carcinoma. Cancer Biol. Ther. 2006, 5, 1357–1360. [Google Scholar] [CrossRef][Green Version]
- Merritt, W.M.; Kamat, A.A.; Hwang, J.-Y.; Bottsford-Miller, J.; Lu, C.; Lin, Y.G.; Coffey, D.; Spannuth, W.A.; Nugent, E.; Han, L.Y.; et al. Clinical and biological impact of EphA2 overexpression and angiogenesis in endometrial cancer. Cancer Biol. Ther. 2010, 10, 1306–1314. [Google Scholar] [CrossRef]
- Wu, D.; Suo, Z.; Kristensen, G.B.; Li, S.; Troen, G.; Holm, R.; Nesland, J.M. Prognostic value of EphA2 and EphrinA-1 in squamous cell cervical carcinoma. Gynecol. Oncol. 2004, 94, 312–319. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Qiu, Y.; Huang, D.; Zhang, S.; Xie, L.; Qi, L.; Yu, C.; Zhou, X.; Hu, G.; et al. Clinical significance of EphA2 expression in squamous-cell carcinoma of the head and neck. J. Cancer Res. Clin. Oncol. 2010, 137, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Park, P.J.; Lai, W.; Maher, E.; Chakravarti, A.; Durso, L.; Jiang, X.; Yu, Y.; Brosius, A.; Thomas, M.; et al. A Genome-Wide Screen Reveals Functional Gene Clusters in the Cancer Genome and Identifies EphA2 as a Mitogen in Glioblastoma. Cancer Res. 2006, 66, 10815–10823. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Gu, J.-W.; Li, B.; Liu, W.; Wang, Y.-G.; Zhang, X.; Zhen, H.-N.; Fei, Z. Up-regulation of EphA2 and down-regulation of EphrinA1 are associated with the aggressive phenotype and poor prognosis of malignant glioma. Tumor Biol. 2010, 31, 477–488. [Google Scholar] [CrossRef]
- Wang, L.-F.; Fokas, E.; Bieker, M.; Rose, F.; Rexin, P.; Zhu, Y.; Pagenstecher, A.; Engenhart-Cabillic, R.; An, H.-X. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol. Rep. 2008, 19, 151–156. [Google Scholar] [CrossRef]
- Herrem, C.J.; Tatsumi, T.; Olson, K.S.; Shirai, K.; Finke, J.H.; Bukowski, R.M.; Zhou, M.; Richmond, A.L.; Derweesh, I.; Kinch, M.S.; et al. Expression of EphA2 is prognostic of disease-free interval and overall survival in surgically treated patients with renal cell carcinoma. Clin. Cancer Res. 2005, 11, 226–231. [Google Scholar]
- Xu, J.; Zhang, J.; Cui, L.; Zhang, H.; Zhang, S.; Bai, Y. High EphA2 protein expression in renal cell carcinoma is associated with a poor disease outcome. Oncol. Lett. 2014, 8, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Brannan, J.M.; Dong, W.; Prudkin, L.; Behrens, C.; Lotan, R.; Bekele, B.N.; Wistuba, I.; Johnson, F.M. Expression of the Receptor Tyrosine Kinase EphA2 Is Increased in Smokers and Predicts Poor Survival in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2009, 15, 4423–4430. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Shao, R.; Correa, A.M.; Behrens, C.; Johnson, F.M.; Raso, M.G.; Prudkin, L.; Solis, L.M.; Nunez, M.I.; Fang, B.; et al. Prognostic Significance of Combinations of RNA-Dependent Protein Kinase and EphA2 Biomarkers for NSCLC. J. Thorac. Oncol. 2013, 8, 301–308. [Google Scholar] [CrossRef]
- Kinch, M.S.; Moore, M.-B.; Harpole, D.H., Jr. Predictive value of the EphA2 receptor tyrosine kinase in lung cancer recurrence and survival. Clin. Cancer Res. 2003, 9, 613–618. [Google Scholar]
- Cui, X.-D.; Lee, M.-J.; Yu, G.-R.; Kim, I.-H.; Yu, H.-C.; Song, E.-Y.; Kim, D.-G. EFNA1 ligand and its receptor EphA2: Potential biomarkers for hepatocellular carcinoma. Int. J. Cancer 2009, 126, 940–949. [Google Scholar] [CrossRef]
- Yang, P.; Yuan, W.; He, J.; Wang, J.; Yu, L.; Jin, X.; Hu, Y.; Liao, M.; Chen, Z.; Zhang, Y. Overexpression of EphA2, MMP-9, and MVD-CD34 in hepatocellular carcinoma: Implications for tumor progression and prognosis. Hepatol. Res. 2009, 39, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Kataoka, H.; Sato, N.; Kanamori, M.; Ihara, M.; Igarashi, H.; Ravshanov, S.; Wang, Y.-J.; Li, Z.-Y.; Shimamura, T.; et al. EPHA2/EFNA1 expression in human gastric cancer. Cancer Sci. 2005, 96, 42–47. [Google Scholar] [CrossRef]
- Macrae, M.; Neve, R.M.; Rodriguez-Viciana, P.; Haqq, C.; Yeh, J.; Chen, C.; Gray, J.W.; McCormick, F. A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell 2005, 8, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.P.; Kandpal, R.P. Invasiveness of breast carcinoma cells and transcript profile: Eph receptors and ephrin ligands as molecular markers of potential diagnostic and prognostic application. Biochem. Biophys. Res. Commun. 2004, 318, 882–892. [Google Scholar] [CrossRef]
- Binda, E.; Visioli, A.; Giani, F.; Lamorte, G.; Copetti, M.; Pitter, K.L.; Huse, J.T.; Cajola, L.; Zanetti, N.; DiMeco, F.; et al. The EphA2 Receptor Drives Self-Renewal and Tumorigenicity in Stem-like Tumor-Propagating Cells from Human Glioblastomas. Cancer Cell 2012, 22, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Alonso, R.; Bustos, F.; Budzyk, M.; Kumar, P.; Helbig, A.O.; Hukelmann, J.; Lamond, A.I.; Lanner, F.; Zhou, H.; Petsalaki, E.; et al. Phosphoproteomics identifies a bimodal EPHA2 receptor switch that promotes embryonic stem cell differentiation. Nat. Commun. 2020, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Ota, S.; Kataoka, H.; Kanamori, M.; Li, Z.Y.; Band, H.; Tanaka, M.; Sugimura, H. Negative regulation of EphA2 receptor by Cbl. Biochem. Biophys. Res. Commun. 2002, 296, 214–220. [Google Scholar] [CrossRef]
- Miao, H.; Wei, B.-R.; Peehl, D.M.; Li, Q.; Alexandrou, T.; Schelling, J.R.; Rhim, J.S.; Sedor, J.R.; Burnett, E.; Wang, B. Activation of EphA receptor tyrosine kinase inhibits the Ras/MAPK pathway. Nat. Cell Biol. 2001, 3, 527–530. [Google Scholar] [CrossRef]
- Menges, C.W.; McCance, D.J. Constitutive activation of the Raf–MAPK pathway causes negative feedback inhibition of Ras–PI3K–AKT and cellular arrest through the EphA2 receptor. Oncogene 2007, 27, 2934–2940. [Google Scholar] [CrossRef]
- Miao, H.; Li, D.-Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 Mediates Ligand-Dependent Inhibition and Ligand-Independent Promotion of Cell Migration and Invasion via a Reciprocal Regulatory Loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef]
- Yang, N.-Y.; Fernandez, C.; Richter, M.; Xiao, Z.; Valencia, F.; Tice, D.A.; Pasquale, E.B. Crosstalk of the EphA2 receptor with a serine/threonine phosphatase suppresses the Akt-mTORC1 pathway in cancer cells. Cell. Signal. 2011, 23, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Neill, T.; Buraschi, S.; Goyal, A.; Sharpe, C.; Natkanski, E.; Schaefer, L.; Morrione, A.; Iozzo, R.V. EphA2 is a functional receptor for the growth factor progranulin. J. Cell Biol. 2016, 215, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Stallaert, W.; Bruggemann, Y.; Sabet, O.; Baak, L.; Gattiglio, M.; Bastiaens, P.I.H. Contact inhibitory Eph signaling suppresses EGF-promoted cell migration by decoupling EGFR activity from vesicular recycling. Sci. Signal. 2018, 11, eaat0114. [Google Scholar] [CrossRef]
- Parri, M.; Buricchi, F.; Giannoni, E.; Grimaldi, G.; Mello, T.; Raugei, G.; Ramponi, G.; Chiarugi, P. EphrinA1 Activates a Src/Focal Adhesion Kinase-mediated Motility Response Leading to Rho-dependent Actino/Myosin Contractility. J. Biol. Chem. 2007, 282, 19619–19628. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.; Hogan, C. Normal epithelial cells trigger EphA2-dependent RasV12 cell repulsion at the single cell level. Small GTPases 2019, 10, 305–310. [Google Scholar] [CrossRef]
- Zhou, Y.; Sakurai, H. Emerging and Diverse Functions of the EphA2 Noncanonical Pathway in Cancer Progression. Biol. Pharm. Bull. 2017, 40, 1616–1624. [Google Scholar] [CrossRef]
- Beauchamp, A.; Debinski, W. Ephs and ephrins in cancer: Ephrin-A1 signalling. Semin. Cell Dev. Biol. 2012, 23, 109–115. [Google Scholar] [CrossRef]
- Fang, W.B.; Ireton, R.C.; Zhuang, G.; Takahashi, T.; Reynolds, A.; Chen, J. Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J. Cell Sci. 2008, 121, 358–368. [Google Scholar] [CrossRef]
- Kikawa, K.D.; Vidale, D.R.; Van Etten, R.L.; Kinch, M.S. Regulation of the EphA2 Kinase by the Low Molecular Weight Tyrosine Phosphatase Induces Transformation. J. Biol. Chem. 2002, 277, 39274–39279. [Google Scholar] [CrossRef] [PubMed]
- Lori, G.; Gamberi, T.; Paoli, P.; Caselli, A.; Pranzini, E.; Marzocchini, R.; Modesti, A.; Raugei, G. LMW-PTP modulates glucose metabolism in cancer cells. Biochim. Biophys. Acta BBA Gen. Subj. 2018, 1862, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Raugei, G.; Ramponi, G.; Chiarugi, P. Low molecular weight protein tyrosine phosphatases: Small, but smart. Cell. Mol. Life Sci. 2002, 59, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Malentacchi, F.; Marzocchini, R.; Gelmini, S.; Orlando, C.; Serio, M.; Ramponi, G.; Raugei, G. Up-regulated expression of low molecular weight protein tyrosine phosphatases in different human cancers. Biochem. Biophys. Res. Commun. 2005, 334, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Kobayashi, M.; Hiramoto-Yamaki, N.; Harada, K.; Negishi, M.; Katoh, H. Ephexin4-mediated promotion of cell migration and anoikis resistance is regulated by serine 897 phosphorylation of EphA2. FEBS Open Bio 2013, 3, 78–82. [Google Scholar] [CrossRef]
- Murga, C.; Zohar, M.; Teramoto, H.; Gutkind, J.S. Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-κB. Oncogene 2002, 21, 207–216. [Google Scholar] [CrossRef]
- Fujimoto, S.; Negishi, M.; Katoh, H. RhoG Promotes Neural Progenitor Cell Proliferation in Mouse Cerebral Cortex. Mol. Biol. Cell 2009, 20, 4941–4950. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, N.; Negishi, M.; Katoh, H. RhoG regulates anoikis through a phosphatidylinositol 3-kinase-dependent mechanism. Exp. Cell Res. 2007, 313, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.D.; Anyiwe, K.; Schimmer, A.D. Anoikis resistance and tumor metastasis. Cancer Lett. 2008, 272, 177–185. [Google Scholar] [CrossRef]
- Barquilla, A.; Lamberto, I.; Noberini, R.; Heynen-Genel, S.; Brill, L.M.; Pasquale, E.B. Protein kinase A can block EphA2 receptor–mediated cell repulsion by increasing EphA2 S897 phosphorylation. Mol. Biol. Cell 2016, 27, 2757–2770. [Google Scholar] [CrossRef]
- Gehring, M.P.; Pasquale, E.B. Protein kinase C phosphorylates the EphA2 receptor on serine 892 in the regulatory linker connecting the kinase and SAM domains. Cell. Signal. 2020, 73, 109668. [Google Scholar] [CrossRef]
- Harada, K.; Hiramoto-Yamaki, N.; Negishi, M.; Katoh, H. Ephexin4 and EphA2 mediate resistance to anoikis through RhoG and phosphatidylinositol 3-kinase. Exp. Cell Res. 2011, 317, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.R.; Kanvinde, P.; King, C.; Pasquale, E.B.; Hristova, K. The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures. Commun. Biol. 2018, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Chavent, M.; Karia, D.; Kalli, A.C.; Domański, J.; Duncan, A.L.; Hedger, G.; Stansfeld, P.J.; Seiradake, E.; Jones, E.Y.; Sansom, M.S.P. Interactions of the EphA2 Kinase Domain with PIPs in Membranes: Implications for Receptor Function. Structure 2018, 26, 1025–1034.e2. [Google Scholar] [CrossRef]
- Wen, Q.; Chen, Z.; Chen, Z.; Chen, J.; Wang, R.; Huang, C.; Yuan, W. EphA2 affects the sensitivity of oxaliplatin by inducing EMT in oxaliplatin-resistant gastric cancer cells. Oncotarget 2017, 8, 47998–48011. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Chen, L.; Wu, W.; Wang, J.; Zheng, X.; Chen, Z.; Jiang, Q.; Han, J.; Wei, L.; Wang, L.; et al. EPH receptor A2 governs a feedback loop that activates Wnt/β-catenin signaling in gastric cancer. Cell Death Dis. 2018, 9, 1146. [Google Scholar] [CrossRef]
- Hamaoka, Y.; Negishi, M.; Katoh, H. Tyrosine kinase activity of EphA2 promotes its S897 phosphorylation and glioblastoma cell proliferation. Biochem. Biophys. Res. Commun. 2018, 499, 920–926. [Google Scholar] [CrossRef]
- Allocca, C.; Cirafici, A.M.; Laukkanen, M.O.; Castellone, M.D. Serine 897 Phosphorylation of EPHA2 Is Involved in Signaling of Oncogenic ERK1/2 Drivers in Thyroid Cancer Cells. Thyroid 2020. [Google Scholar] [CrossRef]
- Graves, P.R.; Din, S.U.; Ashamalla, M.; Ashamalla, H.; Gilbert, T.S.K.; Graves, L.M. Ionizing radiation induces EphA2 S897 phosphorylation in a MEK/ERK/RSK-dependent manner. Int. J. Radiat. Biol. 2017, 93, 929–936. [Google Scholar] [CrossRef]
- Cui, X.-D.; Lee, M.-J.; Kim, J.-H.; Hao, P.-P.; Liu, L.; Yu, G.-R.; Kim, D.-G. Activation of mammalian target of rapamycin complex 1 (mTORC1) and Raf/Pyk2 by growth factor-mediated Eph receptor 2 (EphA2) is required for cholangiocarcinoma growth and metastasis. Hepatology 2013, 57, 2248–2260. [Google Scholar] [CrossRef]
- Taddei, M.L.; Parri, M.; Angelucci, A.; Bianchini, F.; Marconi, C.; Giannoni, E.; Raugei, G.; Bologna, M.; Calorini, L.; Chiarugi, P. EphA2 Induces Metastatic Growth Regulating Amoeboid Motility and Clonogenic Potential in Prostate Carcinoma Cells. Mol. Cancer Res. 2011, 9, 149–160. [Google Scholar] [CrossRef]
- Taddei, M.L.; Parri, M.; Angelucci, A.; Onnis, B.; Bianchini, F.; Giannoni, E.; Raugei, G.; Calorini, L.; Rucci, N.; Teti, A.; et al. Kinase-Dependent and -Independent Roles of EphA2 in the Regulation of Prostate Cancer Invasion and Metastasis. Am. J. Pathol. 2009, 174, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, K.; Katoh, H. The cystine/glutamate antiporter xCT is a key regulator of EphA2 S897 phosphorylation under glucose-limited conditions. Cell. Signal. 2019, 62, 109329. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.-P.; Xiao, T.; Li, Q.-G.; Lu, S.-S.; Zhu, W.; Liu, Y.-Y.; Qiu, J.-Y.; Song, Z.-H.; Huang, W.; Yi, H.; et al. Y772 phosphorylation of EphA2 is responsible for EphA2-dependent NPC nasopharyngeal carcinoma growth by Shp2/Erk-1/2 signaling pathway. Cell Death Dis. 2020, 11, 709. [Google Scholar] [CrossRef] [PubMed]
- Bin Fang, W.; Brantley-Sieders, D.M.; Parker, M.A.; Reith, A.D.; Chen, J. A kinase-dependent role for EphA2 receptor in promoting tumor growth and metastasis. Oncogene 2005, 24, 7859–7868. [Google Scholar] [CrossRef]
- Sheng, Y.; Wei, J.; Zhang, Y.; Gao, X.; Wang, Z.; Yang, J.; Yan, S.; Zhu, Y.; Zhang, Z.; Xu, D.; et al. Mutated EPHA2 is a target for combating lymphatic metastasis in intrahepatic cholangiocarcinoma. Int. J. Cancer 2019, 144, 2440–2452. [Google Scholar] [CrossRef]
- Tan, Y.-H.C.; Srivastava, S.; Won, B.M.; Kanteti, R.; Arif, Q.; Husain, A.N.; Li, H.; Vigneswaran, W.T.; Pang, K.-M.; Kulkarni, P.; et al. EPHA2 mutations with oncogenic characteristics in squamous cell lung cancer and malignant pleural mesothelioma. Oncogenesis 2019, 8, 49. [Google Scholar] [CrossRef]
- Sáinz-Jaspeado, M.; Huertas-Martinez, J.; Lagares-Tena, L.; Liberal, J.M.; Mateo-Lozano, S.; De Álava, E.; De Torres, C.; Mora, J.; Del Muro, X.G.; Tirado, O.M. EphA2-Induced Angiogenesis in Ewing Sarcoma Cells Works through bFGF Production and Is Dependent on Caveolin-1. PLoS ONE 2013, 8, e71449. [Google Scholar] [CrossRef]
- Garcia-Monclús, S.; López-Alemany, R.; Almacellas-Rabaiget, O.; Herrero-Martín, D.; Huertas-Martinez, J.; Lagares-Tena, L.; Alba-Pavón, P.; Hontecillas-Prieto, L.; Mora, J.; De Álava, E.; et al. EphA2 receptor is a key player in the metastatic onset of Ewing sarcoma. Int. J. Cancer 2018, 143, 1188–1201. [Google Scholar] [CrossRef]
- Weng, G.; Zhao, W.; Yin, Y.; Wang, S.; Du, L.; Liu, N.; Mu, D.; Yu, Q.; Yuan, S. Genomic alterations of whole exome sequencing in esophageal squamous cell carcinoma before and after radiotherapy. J. Thorac. Dis. 2020, 12, 5945–5957. [Google Scholar] [CrossRef] [PubMed]
- Parri, M.; Taddei, M.L.; Bianchini, F.; Calorini, L.; Chiarugi, P. EphA2 Reexpression Prompts Invasion of Melanoma Cells Shifting from Mesenchymal to Amoeboid-like Motility Style. Cancer Res. 2009, 69, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Smalley, I.; Emmons, M.F.; Sharma, R.; Izumi, V.; Messina, J.; Koomen, J.M.; Pasquale, E.B.; Forsyth, P.A.; Smalley, K.S.M. Noncanonical EphA2 Signaling Is a Driver of Tumor-Endothelial Cell Interactions and Metastatic Dissemi-nation in BRAF Inhibitor Resistant Melanoma. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Miao, B.; Ji, Z.; Tan, L.; Taylor, M.; Zhang, J.; Choi, H.G.; Frederick, D.T.; Kumar, R.; Wargo, J.A.; Flaherty, K.T.; et al. EPHA2 Is a Mediator of Vemurafenib Resistance and a Novel Therapeutic Target in Melanoma. Cancer Discov. 2015, 5, 274–287. [Google Scholar] [CrossRef]
- Paraiso, K.H.T.; Das Thakur, M.; Fang, B.; Koomen, J.M.; Fedorenko, I.V.; John, J.K.; Tsao, H.; Flaherty, K.T.; Sondak, V.K.; Messina, J.L.; et al. Ligand-Independent EPHA2 Signaling Drives the Adoption of a Targeted Therapy–Mediated Metastatic Melanoma Phenotype. Cancer Discov. 2015, 5, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.-M.; Journe, F. Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers 2020, 12, 1323. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Ruan, H.; Li, S.; Bao, L.; Zhang, X.-P. Enhanced YB1/EphA2 axis signaling promotes acquired resistance to sunitinib and metastatic potential in renal cell carcinoma. Oncogene 2020, 39, 6113–6128. [Google Scholar] [CrossRef]
- Chen, C.-T.; Liao, L.-Z.; Lu, C.-H.; Huang, Y.-H.; Lin, Y.-K.; Lin, J.-H.; Chow, L.-P. Quantitative phosphoproteomic analysis identifies the potential therapeutic target EphA2 for overcoming sorafenib resistance in hepatocellular carcinoma cells. Exp. Mol. Med. 2020, 52, 497–513. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Xu, S.-Q.; Palladino, C.; Belfiore, A.; Iozzo, R.V.; Morrione, A. Progranulin/EphA2 axis: A novel oncogenic mechanism in bladder cancer. Matrix Biol. 2020, 93, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Lv, H.; Shen, N.; Wang, X.-M.; Tang, S.; Xiong, B.; Ding, J.; Geng, M.-Y.; Huang, M. EPHA2 feedback activation limits the response to PDEδ inhibition in KRAS-dependent cancer cells. Acta Pharmacol. Sin. 2019, 41, 270–277. [Google Scholar] [CrossRef]
- Akada, M.; Harada, K.; Negishi, M.; Katoh, H. EphB6 promotes anoikis by modulating EphA2 signaling. Cell. Signal. 2014, 26, 2879–2884. [Google Scholar] [CrossRef] [PubMed]
- Bulk, E.; Yu, J.; Hascher, A.; Koschmieder, S.; Wiewrodt, R.; Krug, U.; Timmermann, B.; Marra, A.; Hillejan, L.; Wiebe, K.; et al. Mutations of the EPHB6 Receptor Tyrosine Kinase Induce a Pro-Metastatic Phenotype in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e44591. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Bataille, F.; Meyer, S.; Becker, B.; Roesch, A.; Landthaler, M.; Vogt, T. Loss of EphB6 expression in metastatic melanoma. Int. J. Oncol. 2003, 23, 1553–1559. [Google Scholar] [CrossRef]
- Yoon, S.; Choi, J.-H.; Kim, S.J.; Lee, E.-J.; Shah, M.; Choi, S.; Woo, H.G. EPHB6 mutation induces cell adhesion-mediated paclitaxel resistance via EPHA2 and CDH11 expression. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Moyano-Galceran, L.; Pietilä, E.A.; Turunen, S.P.; Corvigno, S.; Hjerpe, E.; Bulanova, D.; Joneborg, U.; Alkasalias, T.; Miki, Y.; Yashiro, M.; et al. Adaptive RSK-EphA2-GPRC5A signaling switch triggers chemotherapy resistance in ovarian cancer. EMBO Mol. Med. 2020, 12, e11177. [Google Scholar] [CrossRef] [PubMed]
- Brantley-Sieders, D.M.; Zhuang, G.; Hicks, D.; Bin Fang, W.; Hwang, Y.; Cates, J.M.; Coffman, K.; Jackson, D.; Bruckheimer, E.; Muraoka-Cook, R.S.; et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J. Clin. Investig. 2008, 118, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Brantley-Sieders, D.M.; Vaught, D.; Yu, J.; Xie, L.; Wells, S.; Jackson, D.; Muraoka-Cook, R.; Arteaga, C.; Chen, J. Elevation of Receptor Tyrosine Kinase EphA2 Mediates Resistance to Trastuzumab Therapy. Cancer Res. 2010, 70, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Markosyan, N.; Li, J.; Sun, Y.H.; Richman, L.P.; Lin, J.H.; Yan, F.; Quinones, L.; Sela, Y.; Yamazoe, T.; Gordon, N.; et al. Tumor cell–intrinsic EPHA2 suppresses antitumor immunity by regulating PTGS2 (COX-2). J. Clin. Investig. 2019, 129, 3594–3609. [Google Scholar] [CrossRef]
- Tanabe, A.; Sahara, H. The Metabolic Heterogeneity and Flexibility of Cancer Stem Cells. Cancers 2020, 12, 2780. [Google Scholar] [CrossRef]
- Varun, B.R.; Jayanthi, P.; Ramani, P. Cancer stem cells: A comprehensive review on identification and therapeutic implications. J. Oral Maxillofac. Pathol. 2020, 24, 190. [Google Scholar] [CrossRef]
- Walsh, H.R.; Cruickshank, B.M.; Brown, J.M.; Marcato, P. The Flick of a Switch: Conferring Survival Advantage to Breast Cancer Stem Cells through Metabolic Plasticity. Front. Oncol. 2019, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic Adaptations in Cancer Stem Cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Huang, J.; Xiao, D.; Li, G.; Ma, J.; Chen, P.; Yuan, W.; Hou, F.; Ge, J.; Zhong, M.; Tang, Y.; et al. EphA2 promotes epithelial–mesenchymal transition through the Wnt/β-catenin pathway in gastric cancer cells. Oncogene 2013, 33, 2737–2747. [Google Scholar] [CrossRef]
- Codony-Servat, J.; Codony-Servat, C.; Cardona, A.F.; Giménez-Capitán, A.; Drozdowskyj, A.; Berenguer, J.; Bracht, J.W.P.; Ito, M.; Karachaliou, N.; Rosell, R. Cancer Stem Cell Biomarkers in EGFR-Mutation–Positive Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2019, 20, 167–177. [Google Scholar] [CrossRef]
- Song, W.; Ma, Y.; Wang, J.; Brantley-Sieders, D.; Chen, J. JNK Signaling Mediates EPHA2-Dependent Tumor Cell Proliferation, Motility, and Cancer Stem Cell–like Properties in Non–Small Cell Lung Cancer. Cancer Res. 2014, 74, 2444–2454. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.B.; Pedersen, M.W.; Stockhausen, M.-T.; Grandal, M.V.; Van Deurs, B.; Poulsen, H.S. Activation of the EGFR Gene Target EphA2 Inhibits Epidermal Growth Factor–Induced Cancer Cell Motility. Mol. Cancer Res. 2007, 5, 283–293. [Google Scholar] [CrossRef]
- Larsen, A.B.; Stockhausen, M.-T.; Poulsen, H.S. Cell adhesion and EGFR activation regulate EphA2 expression in cancer. Cell. Signal. 2010, 22, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yamada, N.; Tanaka, T.; Hori, T.; Yokoyama, S.; Hayakawa, Y.; Yano, S.; Fukuoka, J.; Koizumi, K.; Saiki, I.; et al. Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA. Nat. Commun. 2015, 6, 7679. [Google Scholar] [CrossRef]
- Porazinski, S.; De Navascués, J.; Yako, Y.; Hill, W.; Jones, M.R.; Maddison, R.; Fujita, Y.; Hogan, C. EphA2 Drives the Segregation of Ras-Transformed Epithelial Cells from Normal Neighbors. Curr. Biol. 2016, 26, 3220–3229. [Google Scholar] [CrossRef]
- Troiani, T.; Napolitano, S.; Della Corte, C.M.; Martini, G.; Martinelli, E.; Morgillo, F.; Ciardiello, F. Therapeutic value of EGFR inhibition in CRC and NSCLC: 15 years of clinical evidence. ESMO Open 2016, 1, e000088. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Prewett, M.; Rockwell, P.; Goldstein, N.I. CCR 20th Anniversary Commentary: A Chimeric Antibody, C225, Inhibits EGFR Activation and Tumor Growth. Clin. Cancer Res. 2015, 21, 227–229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef]
- Martinelli, E.; Ciardiello, D.; Martini, G.; Troiani, T.; Cardone, C.; Vitiello, P.; Normanno, N.; Rachiglio, A.; Maiello, E.; Latiano, T.; et al. Implementing anti-epidermal growth factor receptor (EGFR) therapy in metastatic colorectal cancer: Challenges and future perspectives. Ann. Oncol. 2020, 31, 30–40. [Google Scholar] [CrossRef]
- Arrington, A.K.; Heinrich, E.L.; Lee, W.; Duldulao, M.; Patel, S.; Sanchez, J.; Garcia-Aguilar, J.; Kim, J. Prognostic and Predictive Roles of KRAS Mutation in Colorectal Cancer. Int. J. Mol. Sci. 2012, 13, 12153–12168. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.-B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.-F.; Côté, J.-F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS Mutation Status Is Predictive of Response to Cetuximab Therapy in Colorectal Cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef]
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol. Cancer 2018, 17, 53. [Google Scholar] [CrossRef]
- Cuyàs, E.; Queralt, B.; Martin-Castillo, B.; Bosch-Barrera, J.; Menendez, J.A. EphA2 receptor activation with ephrin-A1 ligand restores cetuximab efficacy in NRAS-mutant colorectal cancer cells. Oncol. Rep. 2017, 38, 263–270. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.R.R.; Picone, A.; Ferraro, G.; Zanghì, M.; Toscano, G.; Giordano, A.; Adamo, V. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget 2015, 6, 26814–26825. [Google Scholar] [CrossRef] [PubMed]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic Analysis of Epidermal Growth Factor Receptor Somatic Mutant Proteins Shows Increased Sensitivity to the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, Erlotinib. Cancer Res. 2006, 66, 8163–8171. [Google Scholar] [CrossRef] [PubMed]
- Saldaña-Rivera, L.; Bello, M.; Méndez-Luna, D. Structural insight into the binding mechanism of ATP to EGFR and L858R, and T790M and L858R/T790 mutants. J. Biomol. Struct. Dyn. 2019, 37, 4671–4684. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef]
- Rosell, R.; Chaib, I.; Santarpia, M. Targeting MET amplification in EGFR-mutant non-small-cell lung cancer. Lancet Respir. Med. 2020, 8, 1068–1070. [Google Scholar] [CrossRef]
- Okimoto, R.A.; Bivona, T.G. AXL receptor tyrosine kinase as a therapeutic target in NSCLC. Lung Cancer 2015, 6, 27–34. [Google Scholar] [CrossRef]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Bivona, T.G.; Hieronymus, H.; Parker, J.S.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.B.; Miller, V.A.; Costa, C.; et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Ebi, H.; Sano, T.; Nanjo, S.; Ishikawa, D.; Sato, M.; Hasegawa, Y.; Sekido, Y.; et al. EGFR-TKI Resistance Due to BIM Polymorphism Can Be Circumvented in Combination with HDAC Inhibition. Cancer Res. 2013, 73, 2428–2434. [Google Scholar] [CrossRef]
- Weng, C.-H.; Chen, L.-Y.; Lin, Y.-C.; Shih, J.-Y.; Lin, Y.-C.; Tseng, R.-Y.; Chiu, A.-C.; Yeh, Y.-H.; Liu, C.; Lin, Y.-T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Wang, S.; Tan, L.; Hastings, A.K.; Song, W.; Lovly, C.M.; Meador, C.B.; Ye, F.; Lu, P.; Balko, J.M.; et al. EPHA2 Blockade Overcomes Acquired Resistance to EGFR Kinase Inhibitors in Lung Cancer. Cancer Res. 2016, 76, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.; Busto, M.E.D.C.; Kramer, K.; Médard, G.; Kuster, B. Chemical Proteomics Uncovers EPHA2 as a Mechanism of Acquired Resistance to Small Molecule EGFR Kinase Inhibition. J. Proteome Res. 2015, 14, 2617–2625. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Z.; Zhang, M.; Huang, W.; Li, Z.; Wang, S.; Zhang, C.; Dong, B.; Gao, J.; Shen, L. EPHA2 blockade reverses acquired resistance to afatinib induced by EPHA2-mediated MAPK pathway activation in gastric cancer cells and avatar mice. Int. J. Cancer 2019, 145, 2440–2449. [Google Scholar] [CrossRef]
- Martini, G.; Cardone, C.; Vitiello, P.P.; Belli, V.; Napolitano, S.; Troiani, T.; Ciardiello, D.; Della Corte, C.M.; Morgillo, F.; Matrone, N.; et al. EPHA2 Is a Predictive Biomarker of Resistance and a Potential Therapeutic Target for Improving Antiepidermal Growth Factor Receptor Therapy in Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Syeda, F.; Walker, J.R.; Finerty, P.J., Jr.; Cuerrier, D.; Wojciechowski, A.; Liu, Q.; Dhe-Paganon, S.; Gray, N.S. Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4467–4470. [Google Scholar] [CrossRef]
- Kuo, M.T.; Long, Y.; Tsai, W.-B.; Li, Y.-Y.; Chen, H.H.; Feun, L.G.; Savaraj, N. Collaboration Between RSK-EphA2 and Gas6-Axl RTK Signaling in Arginine Starvation Response That Confers Resistance to EGFR Inhibitors. Transl. Oncol. 2020, 13, 355–364. [Google Scholar] [CrossRef]
- Shaurova, T.; Zhang, L.; Goodrich, D.W.; Hershberger, P.A. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front. Genet. 2020, 11, 281. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, P.; Sun, B.; Zhang, S.; Cai, W.; Han, C.; Li, L.; Lu, H.; Zhao, X. Epithelial-Mesenchymal Transition Regulated by EphA2 Contributes to Vasculogenic Mimicry Formation of Head and Neck Squamous Cell Carcinoma. BioMed Res. Int. 2014, 2014, 803914. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Monache, S.D.; Sferra, R.; Vitale, F.; Cristiano, L.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The Small Molecule Ephrin Receptor Inhibitor, GLPG1790, Reduces Renewal Capabilities of Cancer Stem Cells, Showing Anti-Tumour Efficacy on Preclinical Glioblastoma Models. Cancers 2019, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, M.; Poeta, M.L.; Signori, E.; Fazio, V.M. Current understanding and clinical utility of miRNAs regulation of colon cancer stem cells. Semin. Cancer Biol. 2018, 53, 232–247. [Google Scholar] [CrossRef]
- Khan, A.Q.; Ahmed, E.I.; Elareer, N.R.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of miRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells 2019, 8, 840. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | mRNA Protein | Linked to | Cases (n) | Ref |
|---|---|---|---|---|
| Esophageal Squamous Cell Carcinoma | Protein | loco regional metastases; pathological grade; reduced OS | 80 | Miyazaki et al., 2002 [57] |
| Gastric Cancer | Protein | cancer recurrence (in association with YAP) | 47 | Huang et al., 2020 [58] |
| Prostate cancer | Protein | pathological grading | 93 | Zeng et al., 2003 [59] |
| Colorectal cancer | mRNA protein | CSC markers (CD44 and Lgr5); reduced OS | 338 | Dunne et al., 2016 [60] |
| Colorectal cancer | mRNA | poor prognosis and response to cetuximab | 226 | Strimpakos et al., 2013 [61] |
| Colorectal cancer | mRNA | tumor progression and poor OS (EphA2 with miR-423-5p, CREB1, ADAMTS14) | 1663 (TGCA) | De Robertis et al., 2018 [43] |
| Colorectal cancer | mRNA | worse PFS despite EGFRhigh (cetuximab-treated patients) | 80 (TGCA) | De Robertis et al., 2017 [62] |
| Ovarian carcinoma | Protein | aggressive features and median survival | 79 | Thaker et al., 2004 [63] |
| Ovarian cancer | mRNA protein | poor survival | 118 | Han et al., 2005 [64] |
| Epithelial Ovarian Cancer | poor survival (stronger when combined with p53null status) | 79 | Merritt et al., 2006 [65] | |
| Endometrial cancer | Protein | higher pathological grade and clinical stage; shorter disease-specific survival (DSS) | 139 | Merritt et al., 2011 [66] |
| Cervical carcinoma | mRNA | decreased overall survival (OS) | 206 | Wu et al., 2004 [67] |
| Head and neck squamous cell carcinoma | mRNA protein | higher clinical stage, recurrence, and lymph node metastasis; reduced disease-free survival (DFS) and OS | 98 | Liu et al., 2011 [68] |
| Glioblastoma | mRNA protein | increased pathological grade; reduced OS | 21 | Liu et al., 2006 [69] |
| Malignant glioma | protein | decreased DFS and OS (oppositely to EphrinA1) | 78 | Li et al., 2010 [70] |
| Glioblastoma multiforme | protein | Reduced OS | 40 | Wang et al., 2008 [71] |
| Renal Cell Carcinoma | protein | increased pathological grade, reduced DFS and OS | 34 | Herrem et al., 2005 [72] |
| Renal Cell Carcinoma | protein | reduced OS | 62 | Xu et al., 2014 [73] |
| Non-Small-Cell-Lung-Cancer | protein | smoking history; reduced PFS and OS | 279 | Brannan et al., 2009 [74] |
| Non-Small-Cell-Lung-Cancer | protein | reduced overall survival (Stronger when associated with PKR) | 218 | Guo et al., 2013 [75] |
| Non-Small-Cell-Lung-Cancer | protein | brain metastases; reduced OS | 270 | Kinch et al., 2003 [76] |
| Hepatocellular carcinoma | mRNA protein | higher pathological grade; and reduced OS | 40 | Cui et al., 2010 [77] |
| Hepatocellular carcinoma | protein | decreased OS | 129 | Yang et al., 2009 [78] |
| Gastric cancer | protein | higher in high-risk macroscopic grade 3 and 4 tumors | 49 | Nakamura et al., 2005 [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cioce, M.; Fazio, V.M. EphA2 and EGFR: Friends in Life, Partners in Crime. Can EphA2 Be a Predictive Biomarker of Response to Anti-EGFR Agents? Cancers 2021, 13, 700. https://doi.org/10.3390/cancers13040700
Cioce M, Fazio VM. EphA2 and EGFR: Friends in Life, Partners in Crime. Can EphA2 Be a Predictive Biomarker of Response to Anti-EGFR Agents? Cancers. 2021; 13(4):700. https://doi.org/10.3390/cancers13040700
Chicago/Turabian StyleCioce, Mario, and Vito Michele Fazio. 2021. "EphA2 and EGFR: Friends in Life, Partners in Crime. Can EphA2 Be a Predictive Biomarker of Response to Anti-EGFR Agents?" Cancers 13, no. 4: 700. https://doi.org/10.3390/cancers13040700
APA StyleCioce, M., & Fazio, V. M. (2021). EphA2 and EGFR: Friends in Life, Partners in Crime. Can EphA2 Be a Predictive Biomarker of Response to Anti-EGFR Agents? Cancers, 13(4), 700. https://doi.org/10.3390/cancers13040700