
Pathological Changes in Pancreatic Carcinogenesis: A Review


Oncology Pathology, Department of Pathology and Host-Defense, Faculty of Medicine, Kagawa University, 1750-1 Ikenobe, Miki-cho, Kita-gun, Kagawa 761-0793, Japan
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(4), 686; https://doi.org/10.3390/cancers13040686
Submission received: 24 January 2021
/
Revised: 4 February 2021
/
Accepted: 5 February 2021
/
Published: 8 February 2021
(This article belongs to the Special Issue Carcinogenesis)
Abstract
:Simple Summary
Pancreatic cancer has an extremely poor prognosis. Pathological characteristics of pancreatic carcinogenesis, including precancerous lesions and cancers, might provide valuable information for the development of early diagnosis and effective treatments. Autopsy studies have revealed pathological characteristics of precancerous lesions. Animal studies using hamsters and mice have revealed the mechanisms of carcinogenesis. We have summarized pathological changes in the pancreas of humans and experimental animals.
Abstract
Despite advances in diagnostics and therapeutics, the prognosis of pancreatic cancer remains dismal. Because of a lack of early diagnostic methods, aggressive local progression, and high incidence of distant metastasis, most pancreatic cancers are inoperable; therefore, the characteristics of early pancreatic cancer have not been well understood. Autopsy studies revealed the characteristics of prediagnostic pancreatic malignancies, including precancerous lesions, early stage pancreatic cancer, and pancreatic cancer without clinical symptoms (occult cancers). Animal models using hamsters and genetically engineered mice have focused on mechanisms of carcinogenesis, thereby providing insights into risk factors and prevention and serving as a preclinical test for the development of novel diagnostic and treatment modalities. In this review, we have summarized pathological changes in the pancreas of humans and experimental animals during carcinogenesis.
1. Introduction
The annual incidence of pancreatic ductal adenocarcinoma (PDAC) has been increasing, and it is the leading cause of cancer-related deaths worldwide [1,2]. Tobacco use, heavy alcohol consumption, diabetes, obesity, pancreatitis, vitamin D deficiency, solar radiation, aging, and family history are major risk factors for the incidence of PDAC [3]. Despite advances in diagnostics and therapeutics, the prognosis of PDAC remains poor, with an overall 5-year survival rate of below 10% [4,5]. Because of the lack of early diagnostic methods, aggressive local progression, and high incidence of distant metastasis, approximately 70% of PDACs are diagnosed at advanced stages when patients present with symptoms. Most PDACs are inoperable because the characteristics of early stage PDACs are not well understood. Prediagnostic pancreatic malignancies, including precancerous lesions, early stage PDAC, and PDAC without clinical symptoms (occult cancers), can be detected only on autopsy [6].
Recent molecular studies have shown that the development of PDAC occurs at least in part through the intraepithelial proliferation/dysplasia–cancer sequence. Approximately 90% of PDACs can arise from pancreatic intraepithelial neoplasia (PanIN), with a small proportion arising from intraductal papillary mucinous neoplasm (IPMN) or mucinous cystic neoplasm (MCN). Recently, it has been reported that the centro–acinar region might be the origin of PDAC, and PanINs or atypical epithelium is considered to arise in the acinar–ductular transformation [7,8]. Among them, high-grade PanINs might be an immediate precursor to PDAC. The potentially curative treatment is radical surgical resection. However, improvements in overall survival after resection have been observed only in cases in which the tumor is small and confined to the pancreas or, ideally, pre-invasive [7,8]. Thus, early diagnosis of PDAC, especially in its pre-invasive stage, that is, high-grade PanIN (carcinoma in situ), is most important [9].
Many studies that have used genetically engineered mice have been published since the 2000s, and studies have used the Syrian hamster model since its development in the mid-1970s. These studies focus on the mechanisms of carcinogenesis, thereby providing insights into risk factors and prevention and serving as a preclinical test for the development of novel diagnostic and treatment modalities [10]. In this review, we have summarized pathological changes in the pancreas of humans and experimental animals during carcinogenesis.
2. Pathology of Pancreatic Tumors
2.1. Primary Pancreatic Tumors
Autopsy studies have shown that the incidence of macroscopic pancreatic lesions is 6% [11]. Primary pancreatic tumors are observed in 3% of cases, and most primary tumors are PDACs (2%), followed by neuroendocrine tumors (0.3%) and intraductal papillary mucinous neoplasms (0.1%). High-grade PanINs are detected in 0.04% of the cases. Occult PDAC is discovered incidentally in 8% of PDAC cases [11].
PDAC is characterized by a robust fibro-inflammatory response, namely, desmoplastic reaction [12]. The desmoplastic reaction is the result of a complex interplay of cancer cells, vascular cells, pancreatic stellate cells, and inflammatory cells, and it promotes proliferation, invasion, and chemoresistance in PDAC cells [13]. Distant metastasis is an important prognostic factor for PDAC. Twenty-seven percent of occult cancer cases involve distant metastasis, and most of them are localized in the pancreatic tail. Asymptomatic cancers of the pancreatic tail have a greater ability to metastasize to distant sites than those of the pancreatic head and body. These results indicate the aggressive features of PDACs.
2.2. Secondary Pancreatic Tumors
Secondary pancreatic tumors resulting from the invasion and metastasis of malignant tumors were detected in 175 cases (2%). The primary sites of metastatic tumors were the stomach (17%), lung (17%), colorectum (6%), esophagus (5%), gall bladder (3%), and kidney (2%). The primary sites of direct invasion were the stomach (19%), common bile duct (3%), gall bladder (1%), and liver (1%) [11]. The infiltration rate of hematologic tumors was 16%. This indicates that we should pay attention to various primary and secondary tumors in the pancreas.
3. Pathology of Precancerous Lesions of the Pancreas
3.1. Cancer-Related Lesions of the Pancreas
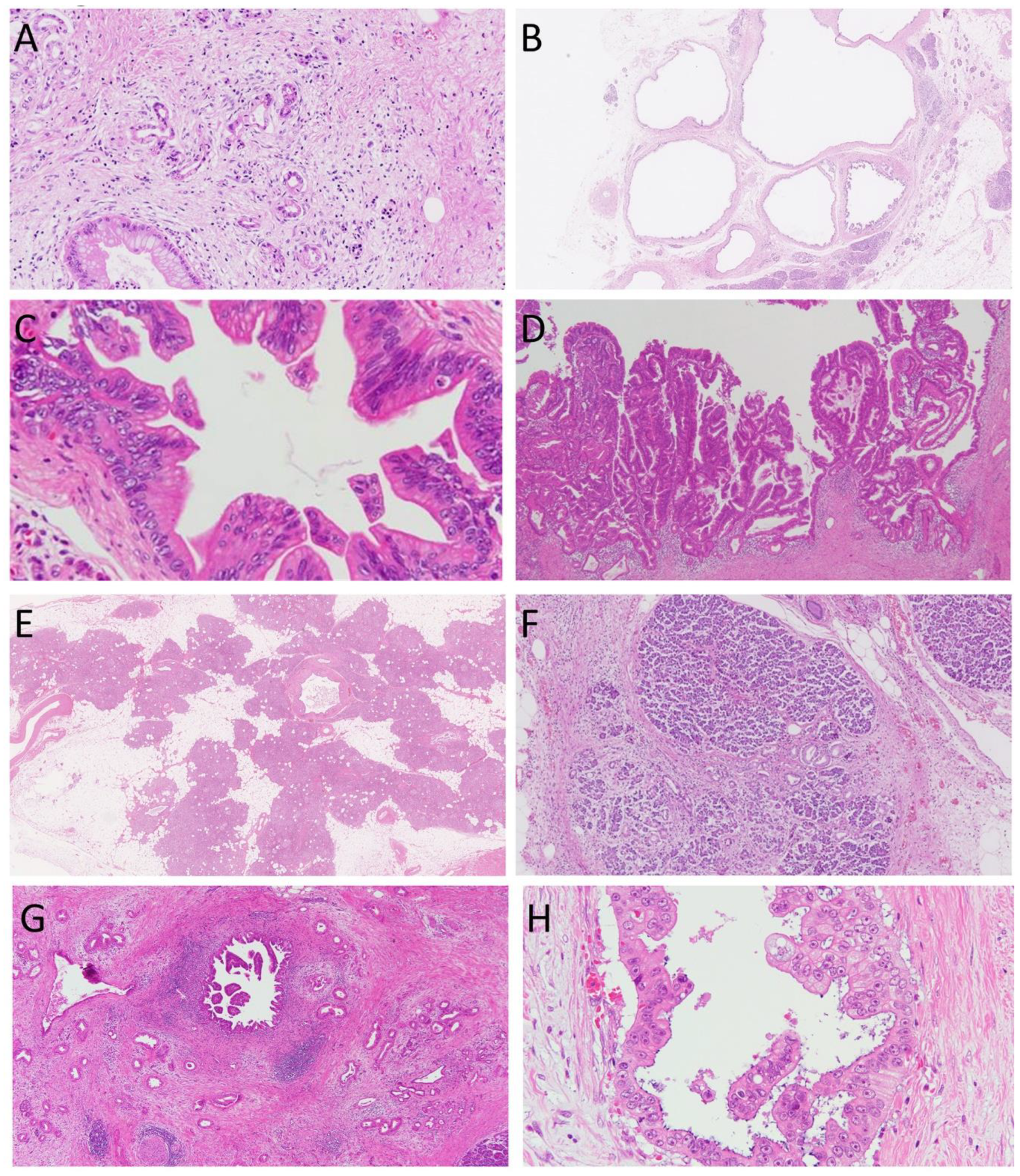
Cancer-related lesions are classified into precancerous lesions and surrogate markers of cancers (Table 1) [14]. ADM, cyst, PanIN, and IPMN (Figure 1A–D) are precancerous lesions of PDAC, whereas fatty replacement and lobulocentric atrophy (Figure 1E,F) are surrogate markers of PDAC. It is difficult to detect PanIN lesions using currently available imaging modalities, including endoscopic ultrasound (EUS), computed tomography, and magnetic resonance imaging; therefore, we need to establish imaging indicators of higher grade PanIN for the identification of PDAC at an early stage (Figure 1G,H).
3.2. Pancreatic Intraepithelial Neoplasia (PanIN)
Previous autopsy studies have reported that the incidence of high-grade dysplasia/carcinoma in situ in the pancreas was 2.9–4.0% [15,16,17,18]. In contrast, high-grade PanIN in patients with PDAC was 40% [19]. We evaluated PanIN lesions in 173 consecutive autopsy cases without PDAC or IPMN by subjecting the entire pancreas for microscopic examination [15]. High-grade PanIN was found in 4% of examined cases associated with a high incidence of diabetes mellitus and/or older age. High-grade PanINs were always multifocal, and the number of high-grade PanIN foci was positively associated with that of low-grade PanIN. High-grade PanIN was located more frequently in the pancreatic body and tail than in the pancreatic head and predominantly involved small interlobular/intralobular ducts rather than the main duct.
Microscopically, aged pancreas shows a decrease in pancreatic parenchymal cells, including endocrine (islet) and exocrine (acinar and duct) cells, resulting in the induction of oxidative stress. They are replaced by fat cells and fibroblasts. The decreased number of pancreatic parenchymal cells is closely associated with pancreatic dysfunction. A lot of studies indicate that PanINs are usually accompanied by fibrosis [18]. Both dysplasia and hyperplasia of the pancreatic ducts are often associated with fibrosis, especially in the pancreatic body and tail [20]. Multicentric PanINs are associated with lobular atrophy of the pancreatic parenchyma and chronic pancreatitis [21]. A study involving a familial PDAC cohort indicated that pancreatic parenchymal atrophy might be secondary to the obstruction of small pancreatic ducts by the proliferative epithelium of PanIN [21]. Fibrosis plays a key role in carcinogenesis steps of the pancreas, via the production of cytokines and extracellular matrices by fibroblasts [22]. Furthermore, an increasing PanIN grade was associated with fibrosis and chronic pancreatitis on EUS [9]. PanINs were associated with fat infiltration of the pancreas, intralobular fibrosis, a high BMI, and subcutaneous and intravisceral fat [23].
3.3. Pancreatic Cystic Lesions
Pancreatic cyst is one of the precursors of PDAC. IPMNs are the most common type of pancreatic cystic neoplasms, accounting for at least 20% of all resected pancreatic cystic neoplasms. Other than IPMN, there are various cystic lesions, such as MCN, serous cystic neoplasm, duct ectasia, and macrocysts with a mucinous lining [14,24].
We analyzed the pancreas in 280 consecutive autopsy cases without PDAC or IPMN by subjecting the entire pancreas for microscopic examination [15]. We found 236 cysts in 93 patients (33%), and 9 cysts (4%) had high-grade dysplasia, similar to the result of a previous report (3%) [16]. In contrast, there were 15 non-cystic lesions with high-grade dysplasia. In total, 24 high-grade dysplastic lesions were noted in 15 patients (5%). Of the 15 patients with high-grade dysplastic lesions, in 10 patients, the condition was accompanied by pancreatic cysts. Patients with cysts showed a higher incidence of high-grade dysplasia (11%; p = 0.005) than patients without cysts (3%). All cysts with high-grade dysplasia were located in the branch duct of the pancreatic head/body, while 20% of non-cystic lesions with high-grade dysplasia were located in the main pancreatic duct. This suggests that cystic lesions with high-grade dysplasia may have characteristics different from those of non-cystic high-grade dysplasia.
The incidence of cystic and non-cystic high-grade dysplasia is low; therefore, cysts themselves are less likely to be a precursor of PDAC. However, rare cysts have malignant potential. Patients with multiple and large cysts may have an increased risk of developing into PDAC. Furthermore, cystic high-grade dysplasia might include incipient IPMNs of the gastric type with high-grade dysplasia.
Various carcinogens, environmental factors, and aging-related telomere dysfunction contribute to accumulation of genetic abnormalities [25]. These genetic events, oxidative stress, and parenchymal atrophy may accelerate carcinogenesis, and obstruction of the pancreatic ducts due to proliferative epithelial lesions and/or inflammation may induce retention cyst formation. Secretion of abundant mucin by hyperplastic or metaplastic duct epithelium may also induce pancreatic cyst formation.
In a recent study via radiological imaging and pancreatic juice cytology, a high-grade PanIN presented with a localized stricture of the main pancreatic duct [26]. However, high-grade PanINs that underwent autopsy are often detected in the branch pancreatic duct [15]. There might be differences in pathogenesis and risk factors between high-grade dysplasia involving branch ducts, resulting in cystic changes vs. high-grade dysplasia involving the main pancreatic duct, leading to stenosis of the main duct. Small branch duct lesions are usually asymptomatic. It is important to develop new laboratory tests to detect these lesions.
4. Animal Models of Pancreatic Carcinogenesis
4.1. Hamster
Animal models are necessary to develop early diagnostic tools and explore new therapeutic approaches. Syrian golden hamster has been used as an animal PDAC model, as it reflects the human physiology. Multiple chemicals have been used to model tumorigenesis [27] (Table 2). Subcutaneous injection of N-nitrosobis(2-oxopropyl)amine (BOP) had induced PDACs in up to 90% of animals within 3–12 months, the histology of which is similar to that of human tumors [28]. An initial subcutaneous injection of BOP 70 mg/kg body weight, followed by three cycles of ethionine-methionine rescue-induced pancreatic regeneration 12 days later produced pancreatic carcinoma after 10 weeks [29]. Ethanol and NNK induced PanIN and PDAC at over 50% incidence [30]. Furthermore, bile reflux into the pancreatic ducts produced intraductal papillary carcinoma in hamsters [31]. K-ras activation by point mutations and p16 inactivation by aberrant methylation of 5′ CpG islands or by homologous deletions have been observed in both hamster and human PDACs [32]. The hamster model has been used to validate and dissect numerous conditions thought to modulate human cancer risk [32].
4.2. Mouse
The first genetically engineered PDAC mouse model was Pdx1-Cre; LSL-KrasG12D mice [27,33] (Table 2). It expressed an oncogenic KrasG12D allele from its endogenous promoter in the developing pancreas through Cre-mediated recombination driven by Pdx1 regulatory elements. Pdx1 is a homeodomain protein critical for early pancreatic development, and both differentiated exocrine and endocrine cell types in the mature pancreas arise from a Pdx1-expressing progenitor population. It developed PanIN lesions, and a minor proportion of them (<10%) developed invasive metastatic adenocarcinomas after several months.
Complex Kras-based genetically engineered mouse models showed a higher frequency of metastasis than basic Kras-based genetically engineered mouse models [27]. Engineering mice that express oncogenic Kras in the pancreas in combination with a conditional Ink4a/Arf of the p53-null allele [34,35] or a conditional knock-in mutant p53R172H [36] developed lethal metastatic PDACs with complete penetrance and shorter latency. Engineering mice that express oncogenic Kras in the pancreas in combination with a conditional SMAD4 [37,38], Notch1 [39], or Tgfbr2-null allele [40] or a conditional knock-in mutant Brca2 [41] also developed metastatic PDACs.
Engineering mouse models provide insights into the origin of PDAC. Activating KrasLSL in adult acinar cells produces PanIN lesions [42]. Acinar-derived PanINs progressed to invasive PDAC in the context of Tp53 depletion [43]. In contrast, activating KrasLSL in adult duct cells produced almost no PanINs [44]. These reports indicate that PDAC originates from differentiated acinar cells and non-ductal cells. Furthermore, acinar-derived PanINs in the KrasLSL model do not show acinar characteristics, suggesting that lesion formation requires extensive phenotype reprogramming, named acinar–ductal reprogramming [10,45]. Inflammation might enhance ductal reprogramming of Kras mutant acinar cells. Acinar–ductal reprogramming occurs in mutant acinar cells, and it is different from acinar–ductal metaplasia, which is the transient upregulation of duct markers in wild-type acinar cells in response to injury. However, the presence or absence of acinar–ductal reprogramming in the human pancreas is unclear, warranting further studies.
4.3. Other Models
Genetically engineered animal models accurately mimic the pathophysiological features of human PDAC including initiation from precancerous lesions and containing cancer cells, stromal components, and immune cells. However, it usually requires a long tumor latency period. It is often difficult to obtain appropriate genetically engineered animal models. Therefore, several alternative methods have been developed.
Xenograft is the transplantation of living cells, tissues, or organs from one species to another. Usually, human-derived cancer cells or cancer tissue are implanted into immunodeficient rodents. Xenograft models using pancreatic satellite cells can show desmoplasia, which is the main characteristic feature of PDAC [46]. It can provide short tumor latency and uniformity in the initial tumor burden, but it lacks a functional immune system [46]. Furthermore, cancer cells and stromal components belong to different species. More recently, the development of patient-derived xenografts has provided great benefit for the development of personalized therapies since drugs could be directly tested on each individual patient’s tumor. The PDX model has facilitated translational studies on PDAC.
Allograft is the transplantation of living cells, tissues, or organs between the same species. It can provide cancer tissues with heterogeneous stromal components and a functional immune system, but all the components are of non-human origin [46].
There are various transplanted tumor models, such as orthotopic or ectopic implantation; metastasis model of the liver, lung, or lymph node; and perineural invasion model [40]. However, the establishment of transplanted tumor models usually requires effort and time as well as materials. Therefore, organoid models of PDAC have been developed to mimic human PDAC [47,48]. It can control and reproduce the three-dimensional structure of PDAC in the short term. It can be used to study tumorigenesis, tumor development, and treatment effects for each patient and provide great benefit to develop individualized treatments.
5. Conclusions
Various experimental models have been used to study the features of PDAC. Pathological changes in the carcinogenesis of the pancreas are important, and they aid in understanding the mechanisms of PDAC progression and developing novel strategies for the early detection and treatment of PDAC. We need to develop a clinical test for early PDAC to improve the prognosis of PDAC.
Author Contributions
Writing—Original Draft Preparation, K.Y. and Y.M.; Writing—Review & Editing, J.Y. and Y.N.-N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant-in-aid from Gender Information Site of Kagawa University to K.Y.
Acknowledgments
We thank Maiko Tada for preparing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- GBD 2017 Pancreatic Cancer Collaborators. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Pandol, S.; Gukovskaya, A.; Edderkaoui, M.; Dawson, D.; Eibl, G.; Lugea, A. Epidemiology, risk factors, and the promotion of pancreatic cancer: Role of the stellate cell. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. S2), 127–134. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [Green Version]
- Ahrendt, S.A.; Pitt, H.A. Surgical management of pancreatic cancer. Oncology 2002, 16, 725–734; discussion 734, 736–738, 740, 743. [Google Scholar]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Aichler, M.; Seiler, C.; Tost, M.; Siveke, J.; Mazur, P.K.; Da Silva-Buttkus, P.; Bartsch, D.K.; Langer, P.; Chiblak, S.; Durr, A.; et al. Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: A comparative study in transgenic mice and human tissues. J. Pathol. 2012, 226, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Esposito, I.; Seiler, C.; Bergmann, F.; Kleeff, J.; Friess, H.; Schirmacher, P. Hypothetical progression model of pancreatic cancer with origin in the centroacinar-acinar compartment. Pancreas 2007, 35, 212–217. [Google Scholar] [CrossRef]
- LeBlanc, J.K.; Chen, J.H.; Al-Haddad, M.; Luz, L.; McHenry, L.; Sherman, S.; Juan, M.; Dewitt, J. Can endoscopic ultrasound predict pancreatic intraepithelial neoplasia lesions in chronic pancreatitis?: A retrospective study of pathologic correlation. Pancreas 2014, 43, 849–854. [Google Scholar] [CrossRef]
- Murtaugh, L.C. Pathogenesis of pancreatic cancer: Lessons from animal models. Toxicol. Pathol. 2014, 42, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Ishiwata, T.; Yachida, S.; Suzuki, A.; Hamashima, Y.; Hamayasu, H.; Yoshimura, H.; Honma, N.; Aida, J.; Takubo, K.; et al. Clinicopathological Features of 15 Occult and 178 Clinical Pancreatic Ductal Adenocarcinomas in 8339 Autopsied Elderly Patients. Pancreas 2016, 45, 234–240. [Google Scholar] [CrossRef]
- Longo, V.; Brunetti, O.; Gnoni, A.; Cascinu, S.; Gasparini, G.; Lorusso, V.; Ribatti, D.; Silvestris, N. Angiogenesis in pancreatic ductal adenocarcinoma: A controversial issue. Oncotarget 2016, 7, 58649–58658. [Google Scholar] [CrossRef] [Green Version]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [CrossRef]
- Matsuda, Y. Age-related morphological changes in the pancreas and their association with pancreatic carcinogenesis. Pathol. Int. 2019, 69, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Furukawa, T.; Yachida, S.; Nishimura, M.; Seki, A.; Nonaka, K.; Aida, J.; Takubo, K.; Ishiwata, T.; Kimura, W.; et al. The Prevalence and Clinicopathological Characteristics of High-Grade Pancreatic Intraepithelial Neoplasia: Autopsy Study Evaluating the Entire Pancreatic Parenchyma. Pancreas 2017, 46, 658–664. [Google Scholar] [CrossRef]
- Kimura, W.; Nagai, H.; Kuroda, A.; Muto, T.; Esaki, Y. Analysis of small cystic lesions of the pancreas. Int. J. Pancreatol. 1995, 18, 197–206. [Google Scholar]
- Mukada, T.; Yamada, S. Dysplasia and carcinoma in situ of the exocrine pancreas. Tohoku J. Exp. Med. 1982, 137, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Luttges, J.; Reinecke-Luthge, A.; Mollmann, B.; Menke, M.A.; Clemens, A.; Klimpfinger, M.; Sipos, B.; Kloppel, G. Duct changes and K-ras mutations in the disease-free pancreas: Analysis of type, age relation and spatial distribution. Virchows Arch. 1999, 435, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Andea, A.; Sarkar, F.; Adsay, V.N. Clinicopathological correlates of pancreatic intraepithelial neoplasia: A comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod. Pathol. 2003, 16, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Detlefsen, S.; Sipos, B.; Feyerabend, B.; Kloppel, G. Pancreatic fibrosis associated with age and ductal papillary hyperplasia. Virchows Arch. 2005, 447, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Brune, K.; Abe, T.; Canto, M.; O’Malley, L.; Klein, A.P.; Maitra, A.; Volkan Adsay, N.; Fishman, E.K.; Cameron, J.L.; Yeo, C.J.; et al. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am. J. Surg. Pathol. 2006, 30, 1067–1076. [Google Scholar] [PubMed]
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebours, V.; Gaujoux, S.; d’Assignies, G.; Sauvanet, A.; Ruszniewski, P.; Levy, P.; Paradis, V.; Bedossa, P.; Couvelard, A. Obesity and Fatty Pancreatic Infiltration Are Risk Factors for Pancreatic Precancerous Lesions (PanIN). Clin. Cancer Res. 2015, 21, 3522–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basturk, O.; Hong, S.M.; Wood, L.D.; Adsay, N.V.; Albores-Saavedra, J.; Biankin, A.V.; Brosens, L.A.; Fukushima, N.; Goggins, M.; Hruban, R.H.; et al. A Revised Classification System and Recommendations from the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am. J. Surg. Pathol. 2015, 39, 1730–1741. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ishiwata, T.; Izumiyama-Shimomura, N.; Hamayasu, H.; Fujiwara, M.; Tomita, K.; Hiraishi, N.; Nakamura, K.; Ishikawa, N.; Aida, J.; et al. Gradual telomere shortening and increasing chromosomal instability among PanIN grades and normal ductal epithelia with and without cancer in the pancreas. PLoS ONE 2015, 10, e0117575. [Google Scholar] [CrossRef]
- Yokode, M.; Akita, M.; Fujikura, K.; Kim, M.J.; Morinaga, Y.; Yoshikawa, S.; Terada, T.; Matsukiyo, H.; Tajiri, T.; Abe-Suzuki, S.; et al. High-grade PanIN presenting with localised stricture of the main pancreatic duct: A clinicopathological and molecular study of 10 cases suggests a clue for the early detection of pancreatic cancer. Histopathology 2018, 73, 247–258. [Google Scholar] [CrossRef]
- Saloman, J.L.; Albers, K.M.; Cruz-Monserrate, Z.; Davis, B.M.; Edderkaoui, M.; Eibl, G.; Epouhe, A.Y.; Gedeon, J.Y.; Gorelick, F.S.; Grippo, P.J.; et al. Animal Models: Challenges and Opportunities to Determine Optimal Experimental Models of Pancreatitis and Pancreatic Cancer. Pancreas 2019, 48, 759–779. [Google Scholar] [CrossRef]
- Takahashi, M.; Hori, M.; Ishigamori, R.; Mutoh, M.; Imai, T.; Nakagama, H. Fatty pancreas: A possible risk factor for pancreatic cancer in animals and humans. Cancer Sci. 2018, 109, 3013–3023. [Google Scholar] [CrossRef]
- Mizumoto, K.; Kitazawa, S.; Ito, S.; Takashima, Y.; Tsutsumi, M.; Denda, A.; Konishi, Y. Cycles of repeated augmentation pressure in rapid production of pancreatic and cholangiocellular carcinomas in hamsters initiated with N-nitrosobis(2-oxopropyl)amine. Carcinogenesis 1989, 10, 1457–1459. [Google Scholar] [CrossRef]
- Banerjee, J.; Papu John, A.M.; Al-Wadei, M.H.; Schuller, H.M. Prevention of pancreatic cancer in a hamster model by cAMP decrease. Oncotarget 2016, 7, 44430–44441. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Tajima, Y.; Kuroki, T.; Mishima, T.; Kitasato, A.; Fukuda, K.; Tsutsumi, R.; Kanematsu, T. Bile-reflux into the pancreatic ducts is associated with the development of intraductal papillary carcinoma in hamsters. J. Surg. Res. 2006, 136, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Hori, M.; Mutoh, M.; Wakabayashi, K.; Nakagama, H. Experimental animal models of pancreatic carcinogenesis for prevention studies and their relevance to human disease. Cancers 2011, 3, 582–602. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; Vickers, S.M.; Adsay, N.V.; Jhala, N.C.; Kim, H.G.; Schoeb, T.R.; Grizzle, W.E.; Klug, C.A. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res 2007, 67, 8121–8130. [Google Scholar] [CrossRef] [Green Version]
- Hanlon, L.; Avila, J.L.; Demarest, R.M.; Troutman, S.; Allen, M.; Ratti, F.; Rustgi, A.K.; Stanger, B.Z.; Radtke, F.; Adsay, V.; et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res 2010, 70, 4280–4286. [Google Scholar] [CrossRef] [Green Version]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Cassidy, L.D.; Pisupati, V.; Jonasson, J.G.; Bjarnason, H.; Eyfjord, J.E.; Karreth, F.A.; Lim, M.; Barber, L.M.; Clatworthy, S.A.; et al. Germline Brca2 heterozygosity promotes Kras(G12D)-driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 2010, 18, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.; Tsou, L.; Wang, H.; Gaiser, S.; Chang, D.Z.; Daniluk, J.; Bi, Y.; Grote, T.; Longnecker, D.S.; Logsdon, C.D. Ras activity levels control the development of pancreatic diseases. Gastroenterology 2009, 137, 1072–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Strobel, O.; Dor, Y.; Alsina, J.; Stirman, A.; Lauwers, G.; Trainor, A.; Castillo, C.F.; Warshaw, A.L.; Thayer, S.P. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 2007, 133, 1999–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suklabaidya, S.; Dash, P.; Das, B.; Suresh, V.; Sasmal, P.K.; Senapati, S. Experimental models of pancreatic cancer desmoplasia. Lab. Investig. 2018, 98, 27–40. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, K.; Guo, M.; Liu, Y.; Zheng, J. Progress in Animal Models of Pancreatic Ductal Adenocarcinoma. J. Cancer 2020, 11, 1555–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Cancer-related lesions of the pancreas. (A), acinar-to-ductal metaplasia, (B), cyst, (C), pancreatic intraepithelial neoplasia, (D), intraductal papillary mucinous neoplasm, (E), fatty replacement, (F), lobulocentric atrophy, (G), and (H), pancreatic ductal carcinoma. Hematoxylin and eosin staining. Original magnification, (A,C,H), ×400; (B,E), ×40; (D,F,G), ×100.
Figure 1.
Cancer-related lesions of the pancreas. (A), acinar-to-ductal metaplasia, (B), cyst, (C), pancreatic intraepithelial neoplasia, (D), intraductal papillary mucinous neoplasm, (E), fatty replacement, (F), lobulocentric atrophy, (G), and (H), pancreatic ductal carcinoma. Hematoxylin and eosin staining. Original magnification, (A,C,H), ×400; (B,E), ×40; (D,F,G), ×100.

{kind=link}
Table 1.
Cancer-related pathological changes in the human pancreas.
| Precursor lesions of pancreatic cancer |
| Acinar-to-ductal metaplasia (ADM) |
| Cyst |
| Pancreatic intraepithelial neoplasia (PanIN) |
| Intraductal papillary mucinous neoplasm (IPMN) |
| Surrogate markers of pancreatic cancer |
| Focal fatty replacement |
| Lobulocentric atrophy |
| Duct dilatation/Cyst/IPMN |
Table 2.
Cancer-related pathological changes in the pancreas of various animals.
| Hamster | Precursor Lesions | |
|---|---|---|
| BOP | PanIN | Takahashi, 2018 |
| BOP, ethionine-methionine | PanIN | Mizumoto, 1989 |
| Bile reflux into the pancreatic duct | IPMN | Adachi, 2006 |
| Mouse | ||
| LSL-KrasG12D; Pdx1-Cre | PanIN | Hingorani, 2003 |
| LSL-KrasG12D; Ptf/p48-Cre | PanIN | Hingorani, 2003 |
| KIC model, Pdx-Cre; LSL-KrasG12D; Ink4a/Arflox/lox | PanIN | Aguirre, 2003 |
| Pdx-Cre; LSL-KrasG12D; Trp53lox/lox; Ink4a/Arflox/lox | PanIN | Bardeesy, 2006 |
| KPC model, Pdx1-Cre; LSL-KrasG12D; LSL-Trp53R172H/+ | PanIN | Hingorani, 2005 |
| KD model, Pdx-Cre; LSL-KrasG12D; SMAD4lox/lox | PanIN, IPMN | Kojima, 2007 |
| Ptf1a-Cre; LSL-KrasG12D; SMAD4lox/lox | PanIN, MCN | Izeradjene, 2007 |
| Ptf1a-Cre; LSL-KrasG12D; Notch1lox/lox | PanIN | Hanlon, 2010 |
| Ptf1a-Cre; LSL-KrasG12D; Tgfr2lox/lox | PanIN | Ijichi, 2006 |
| Pdx1-Cre; LSL-KrasG12D; Brca2tr/Δ11 | PanIN | Skoulidis, 2010 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yamakawa, K.; Ye, J.; Nakano-Narusawa, Y.; Matsuda, Y. Pathological Changes in Pancreatic Carcinogenesis: A Review. Cancers 2021, 13, 686. https://doi.org/10.3390/cancers13040686
AMA Style
Yamakawa K, Ye J, Nakano-Narusawa Y, Matsuda Y. Pathological Changes in Pancreatic Carcinogenesis: A Review. Cancers. 2021; 13(4):686. https://doi.org/10.3390/cancers13040686
Chicago/Turabian StyleYamakawa, Keiko, Juanjuan Ye, Yuko Nakano-Narusawa, and Yoko Matsuda. 2021. "Pathological Changes in Pancreatic Carcinogenesis: A Review" Cancers 13, no. 4: 686. https://doi.org/10.3390/cancers13040686
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.