Spatial Heterogeneity in Large Resected Diffuse Large B-Cell Lymphoma Bulks Analysed by Massively Parallel Sequencing of Multiple Synchronous Biopsies

 ,
, 
 , , , , ,
, , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
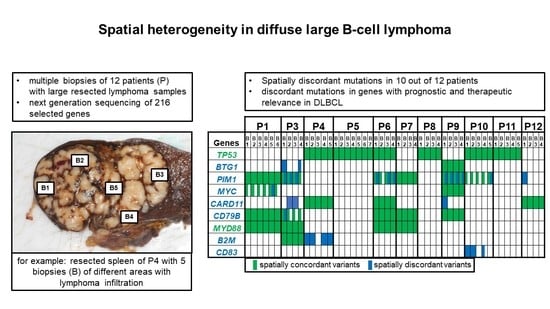
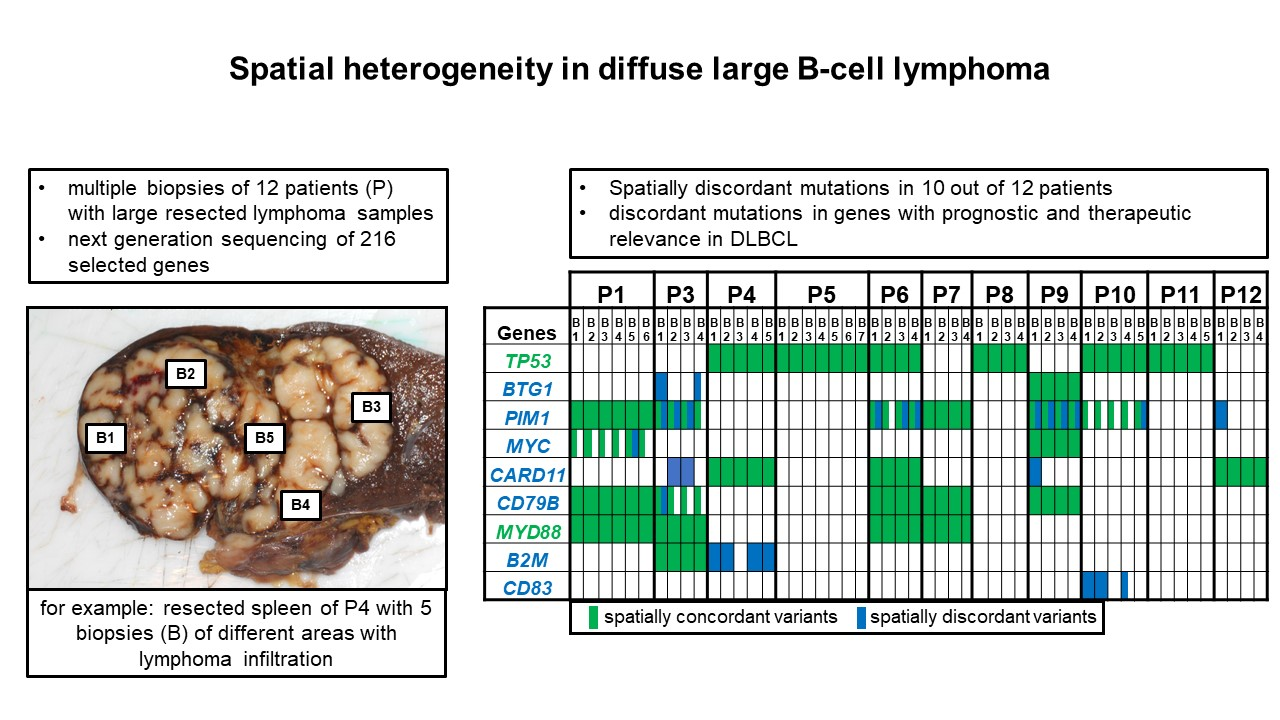
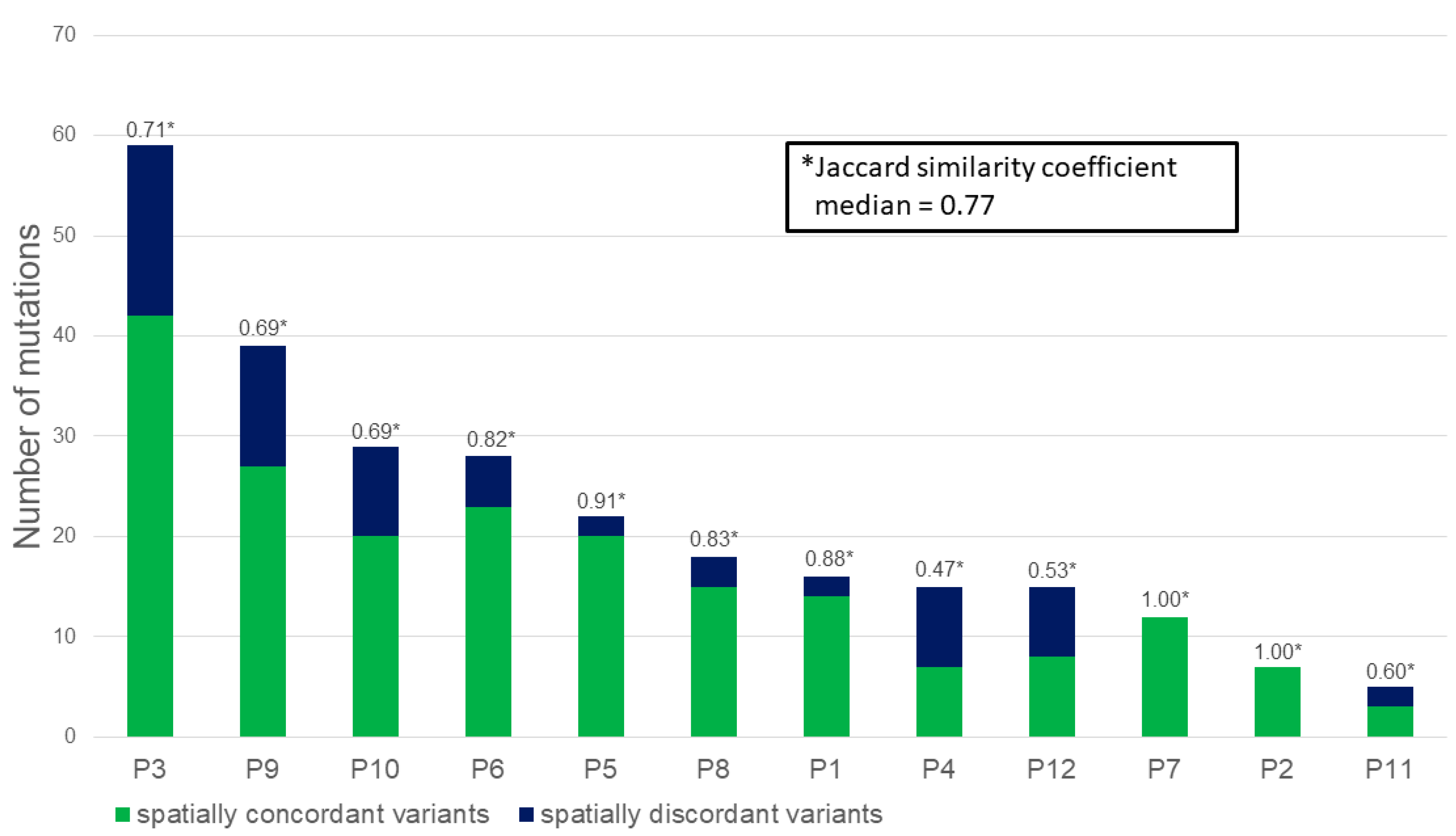
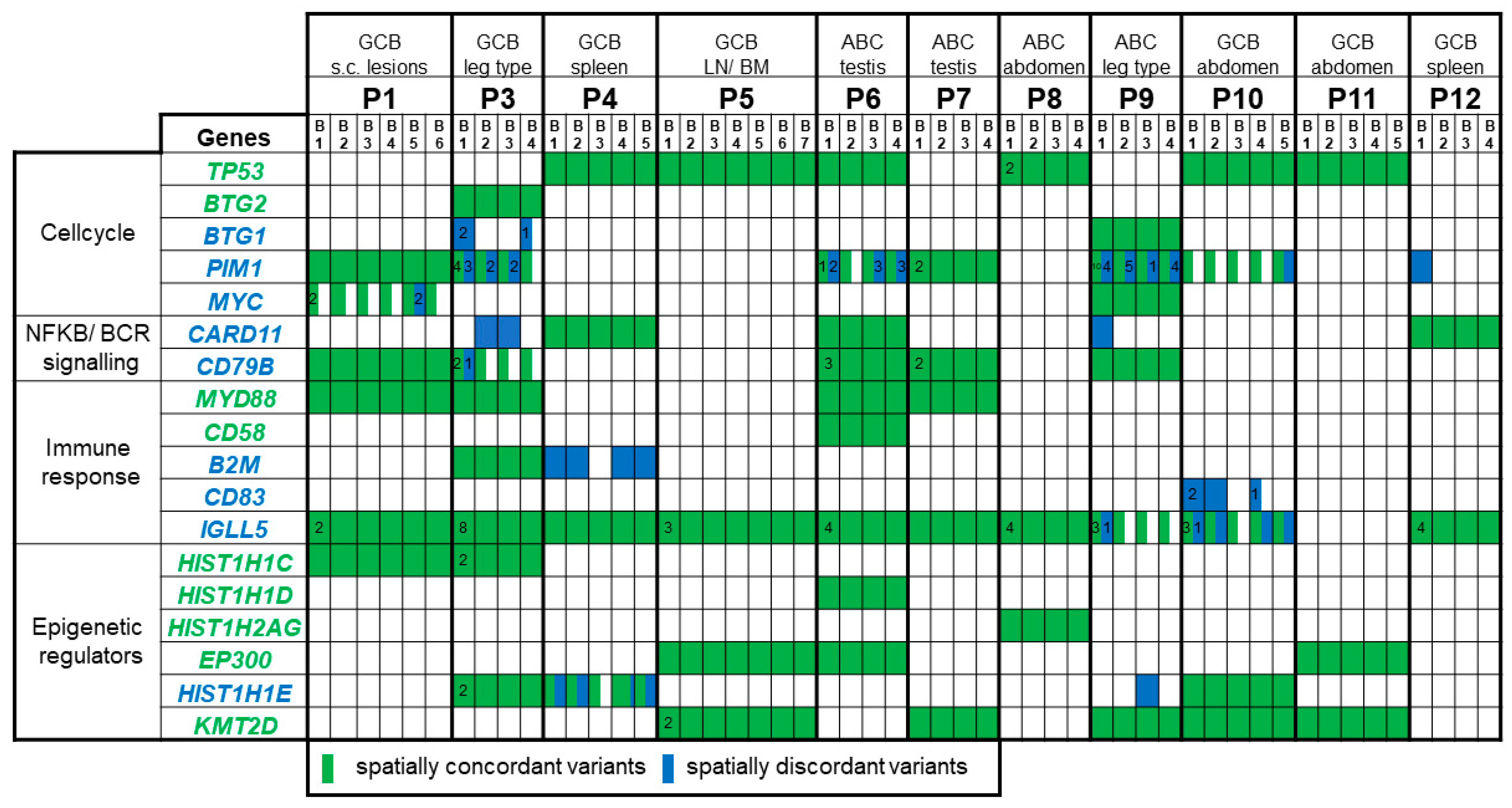
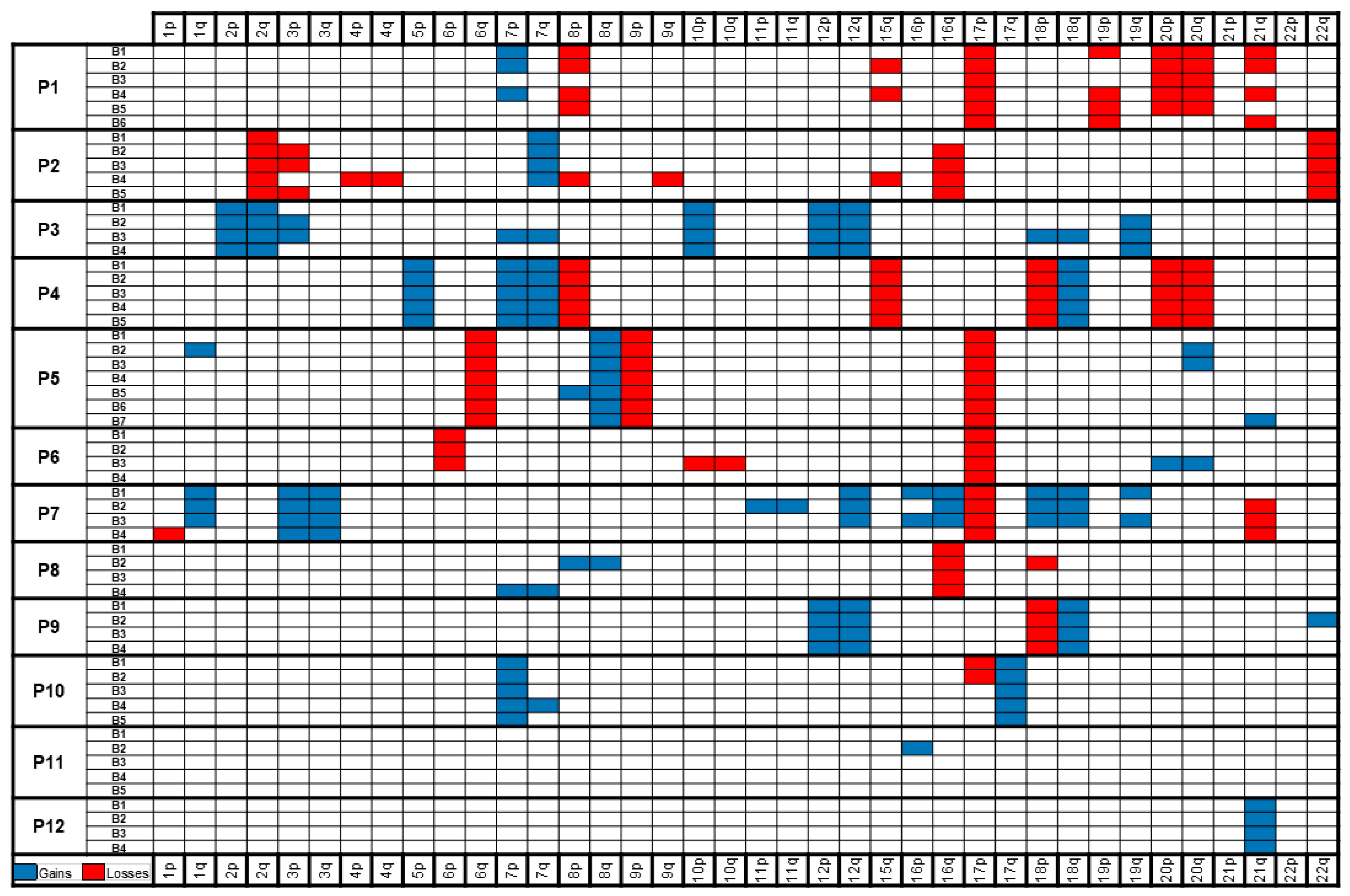
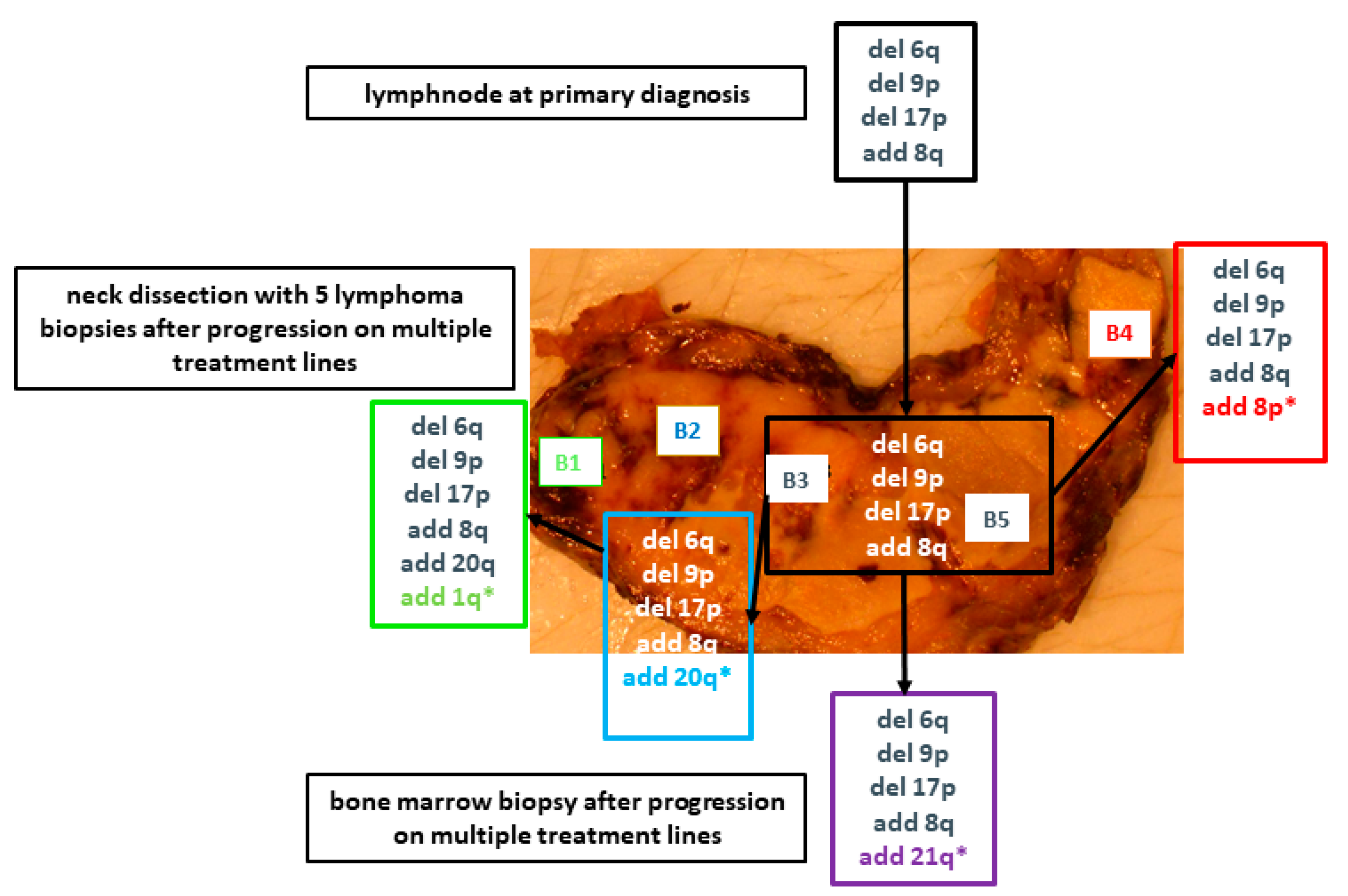
2. Results
2.1. Patient Characteristics
2.2. Massively Parallel Sequencing
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Targeted Massively Parallel Sequencing
4.3. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morton, L.M.; Wang, S.S.; Devesa, S.S.; Hartge, P.; Weisenburger, D.D.; Linet, M.S. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood 2006, 107, 265–276. [Google Scholar] [CrossRef]
- Smith, A.; Crouch, S.; Lax, S.; Li, J.; Painter, D.; Howell, D.; Patmore, R.; Jack, A.; Roman, E. Lymphoma incidence, survival and prevalence 2004–2014: Sub-type analyses from the UK’s Haematological Malignancy Research Network. Br. J. Cancer 2015, 112, 1575–1584. [Google Scholar] [CrossRef]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadie, M.; Simonetti, A.; Lutz, J.M.; et al. Incidence of hematologic malignancies in Europe by morphologic subtype: Results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef]
- Moller, M.B.; Pedersen, N.T.; Christensen, B.E. Diffuse large B-cell lymphoma: Clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br. J. Haematol. 2004, 124, 151–159. [Google Scholar] [CrossRef]
- Gisselbrecht, C.; Glass, B.; Mounier, N.; Singh Gill, D.; Linch, D.C.; Trneny, M.; Bosly, A.; Ketterer, N.; Shpilberg, O.; Hagberg, H.; et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J. Clin. Oncol. 2010, 28, 4184–4190. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494. [Google Scholar] [CrossRef]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshall, J.; Ramakrishna, M.; et al. The life history of 21 breast cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef]
- Melchardt, T.; Hufnagl, C.; Weinstock, D.M.; Kopp, N.; Neureiter, D.; Trankenschuh, W.; Hackl, H.; Weiss, L.; Rinnerthaler, G.; Hartmann, T.N.; et al. Clonal evolution in relapsed and refractory diffuse large B-cell lymphoma is characterized by high dynamics of subclones. Oncotarget 2016, 7, 51494–51502. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef]
- Zenz, T.; Kreuz, M.; Fuge, M.; Klapper, W.; Horn, H.; Staiger, A.M.; Winter, D.; Helfrich, H.; Huellein, J.; Hansmann, M.L.; et al. TP53 mutation and survival in aggressive B cell lymphoma. Int. J. Cancer 2017, 141, 1381–1388. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996. [Google Scholar] [CrossRef]
- Jo, J.; Yoon, D.H.; Lee, S.W.; Park, C.S.; Huh, J.; Lee, K.; Kang, E.H.; Kim, S.; Suh, C. Abbreviated chemotherapy for limited-stage diffuse large B-cell lymphoma after complete resection. Blood Res. 2014, 49, 115–119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoon, D.H.; Sohn, B.S.; Oh, S.Y.; Lee, W.S.; Lee, S.M.; Yang, D.H.; Huh, J.; Suh, C. Feasibility of abbreviated cycles of immunochemotherapy for completely resected limited-stage CD20+ diffuse large B-cell lymphoma (CISL 12-09). Oncotarget 2017, 8, 13367–13374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, S.J.; Kang, H.J.; Kim, J.S.; Oh, S.Y.; Choi, C.W.; Lee, S.I.; Won, J.H.; Kim, M.K.; Kwon, J.H.; Mun, Y.C.; et al. Comparison of treatment strategies for patients with intestinal diffuse large B-cell lymphoma: Surgical resection followed by chemotherapy versus chemotherapy alone. Blood 2011, 117, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Araf, S.; Wang, J.; Korfi, K.; Pangault, C.; Kotsiou, E.; Rio-Machin, A.; Rahim, T.; Heward, J.; Clear, A.; Iqbal, S.; et al. Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia 2018, 32, 1261–1265. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.; Kuppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346. [Google Scholar] [CrossRef]
- Miao, Y.; Medeiros, L.J.; Li, Y.; Li, J.; Young, K.H. Genetic alterations and their clinical implications in DLBCL. Nat. Rev. Clin. Oncol. 2019, 16, 634–652. [Google Scholar] [CrossRef]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef]
- Grommes, C.; Pastore, A.; Palaskas, N.; Tang, S.S.; Campos, C.; Schartz, D.; Codega, P.; Nichol, D.; Clark, O.; Hsieh, W.Y.; et al. Ibrutinib Unmasks Critical Role of Bruton Tyrosine Kinase in Primary CNS Lymphoma. Cancer Discov. 2017, 7, 1018–1029. [Google Scholar] [CrossRef]
- Sebastian, E.; Alcoceba, M.; Martin-Garcia, D.; Blanco, O.; Sanchez-Barba, M.; Balanzategui, A.; Marin, L.; Montes-Moreno, S.; Gonzalez-Barca, E.; Pardal, E.; et al. High-resolution copy number analysis of paired normal-tumor samples from diffuse large B cell lymphoma. Ann. Hematol. 2016, 95, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Dharanipragada, P.; Parekh, N. Genome-wide characterization of copy number variations in diffuse large B-cell lymphoma with implications in targeted therapy. Precis. Clin. Med. 2019, 2, 246–258. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Melchardt, T.; Troppan, K.; Weiss, L.; Hufnagl, C.; Neureiter, D.; Trankenschuh, W.; Hopfinger, G.; Magnes, T.; Deutsch, A.; Neumeister, P.; et al. A modified scoring of the NCCN-IPI is more accurate in the elderly and is improved by albumin and beta2 -microglobulin. Br. J. Haematol. 2015, 168, 239–245. [Google Scholar] [CrossRef]
- Melchardt, T.; Troppan, K.; Weiss, L.; Hufnagl, C.; Neureiter, D.; Trankenschuh, W.; Schlick, K.; Huemer, F.; Deutsch, A.; Neumeister, P.; et al. Independent Prognostic Value of Serum Markers in Diffuse Large B-Cell Lymphoma in the Era of the NCCN-IPI. J. Natl. Compr. Cancer Netw. 2015, 13, 1501–1508. [Google Scholar] [CrossRef][Green Version]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Muller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Berger, M.F.; Davis, M.J.; Blumenstiel, B.; Defelice, M.; Pochanard, P.; Ducar, M.; Van Hummelen, P.; Macconaill, L.E.; Hahn, W.C.; et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012, 2, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Jurinovic, V.; Kridel, R.; Hoster, E.; Staiger, A.M.; Szczepanowski, M.; Pott, C.; Kopp, N.; Murakami, M.; Horn, H.; et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: A retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015, 16, 1111–1122. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | COO | Age (y) | Gender | NCCN-IPI | Sample Type | Biopsies (Number) | Treatment | PFS (mo) | OS (mo) | Status |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GCB | 41 | f | low-int. | subcut. lesions | 6 | R-CHOP, R-EPOCH, auto-STC | 47 | 47 | alive |
| 2 | Non- GCB | 80 | f | high | adrenal gland, spleen, pancreas | 5 | 1x dexa | 1 | 1 | dead |
| 3 | GCB | 77 | f | high-int. | lower leg | 4 | 1x lip.doxo., rituximab | 2 | 7 | dead |
| 4 | GCB | 51 | m | high | spleen | 5 | R-CHOP | 44 | 44 | alive |
| 5 | GCB | 54 | m | low | LN, ND, BM | 7 | multiple incl. R-CHOP, DHAP, auto-SCT | 1 | 10 | dead |
| 6 | Non- GCB | 82 | m | high-int. | testis | 4 | R-DHAP | 42 | 42 | alive |
| 7 | Non- GCB | 78 | m | low-int. | testis | 4 | R-COMP | 44 | 44 | alive |
| 8 | Non- GCB | 62 | f | high-int. | pancreas, spleen, adrenal gland | 4 | R-CHOP | 63 | 63 | alive |
| 9 | Non- GCB | 79 | f | high-int. | subcut. lesions | 4 | R-COMP | 23 | 23 | alive |
| 10 | GCB | 54 | f | high-int. | lung, spleen | 5 | R-CHOP | 39 | 39 | alive |
| 11 | GCB | 39 | f | low | small intestine | 5 | R-CHOP | 31 | 31 | alive |
| 12 | GCB | 60 | f | low-int. | spleen | 4 | R-COMP | 35 | 35 | alive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magnes, T.; Wagner, S.; Thorner, A.R.; Neureiter, D.; Klieser, E.; Rinnerthaler, G.; Weiss, L.; Huemer, F.; Schlick, K.; Zaborsky, N.; et al. Spatial Heterogeneity in Large Resected Diffuse Large B-Cell Lymphoma Bulks Analysed by Massively Parallel Sequencing of Multiple Synchronous Biopsies. Cancers 2021, 13, 650. https://doi.org/10.3390/cancers13040650
Magnes T, Wagner S, Thorner AR, Neureiter D, Klieser E, Rinnerthaler G, Weiss L, Huemer F, Schlick K, Zaborsky N, et al. Spatial Heterogeneity in Large Resected Diffuse Large B-Cell Lymphoma Bulks Analysed by Massively Parallel Sequencing of Multiple Synchronous Biopsies. Cancers. 2021; 13(4):650. https://doi.org/10.3390/cancers13040650
Chicago/Turabian StyleMagnes, Teresa, Sandro Wagner, Aaron R. Thorner, Daniel Neureiter, Eckhard Klieser, Gabriel Rinnerthaler, Lukas Weiss, Florian Huemer, Konstantin Schlick, Nadja Zaborsky, and et al. 2021. "Spatial Heterogeneity in Large Resected Diffuse Large B-Cell Lymphoma Bulks Analysed by Massively Parallel Sequencing of Multiple Synchronous Biopsies" Cancers 13, no. 4: 650. https://doi.org/10.3390/cancers13040650
APA StyleMagnes, T., Wagner, S., Thorner, A. R., Neureiter, D., Klieser, E., Rinnerthaler, G., Weiss, L., Huemer, F., Schlick, K., Zaborsky, N., Steiner, M., Greil, R., Egle, A., & Melchardt, T. (2021). Spatial Heterogeneity in Large Resected Diffuse Large B-Cell Lymphoma Bulks Analysed by Massively Parallel Sequencing of Multiple Synchronous Biopsies. Cancers, 13(4), 650. https://doi.org/10.3390/cancers13040650