
Molecular Mechanisms of Senescence and Implications for the Treatment of Myeloid Malignancies



Abstract
Simple Summary
Abstract
1. Introduction on Cellular Senescence
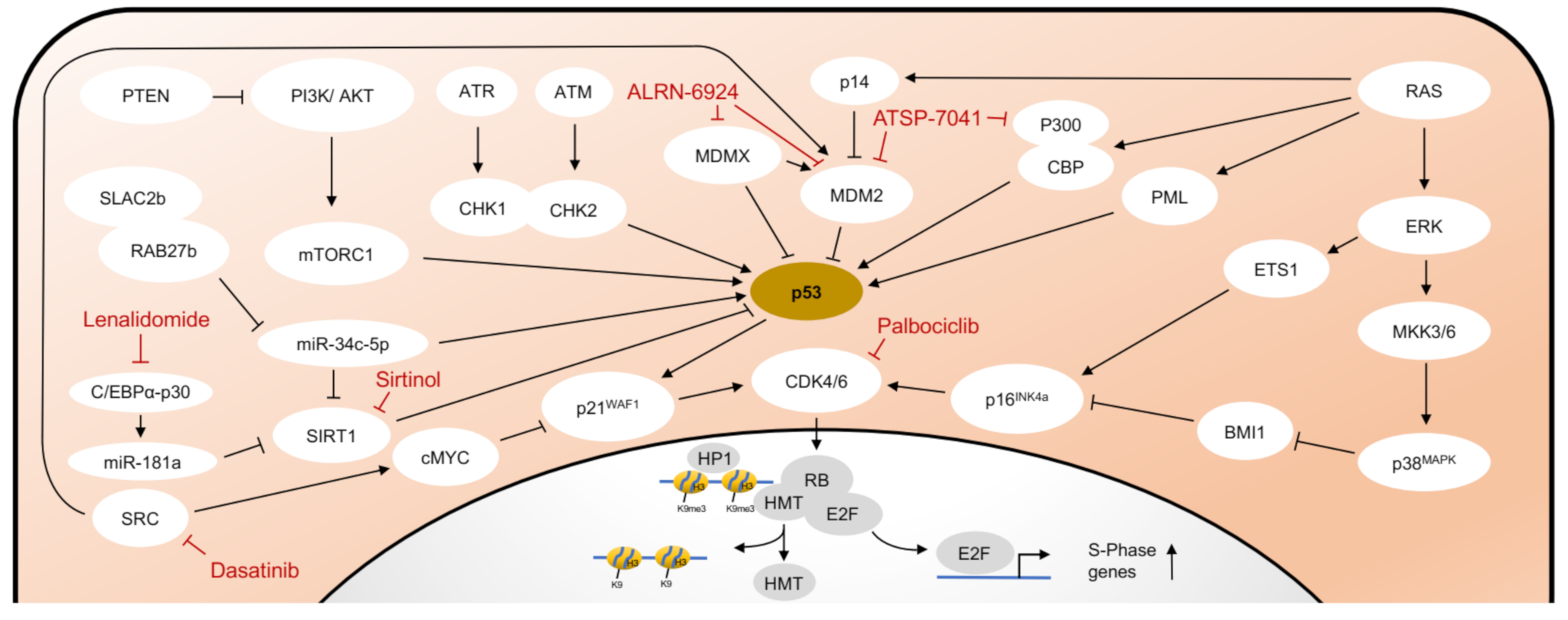
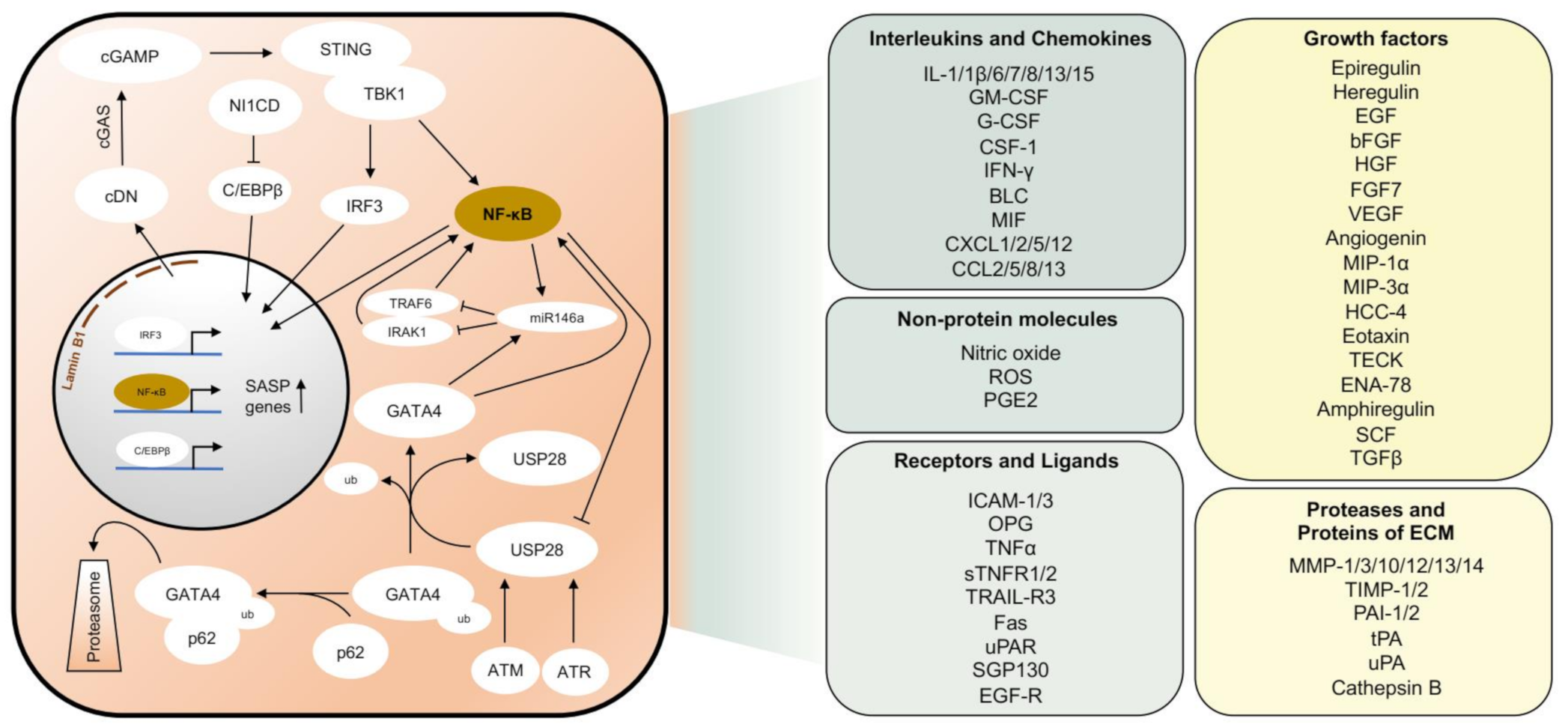
2. Molecular Mechanisms of Senescence
2.1. DNA Damage Response (DDR)
2.2. Senescence Associated Molecular Pathways Are Interconnected
2.3. The Senescence Associated Secretory Phenotype (SASP)
3. Treatment Strategies for Myeloid Malignancies that Involve Cellular Senescence
3.1. CDK and PARP Inhibition
3.2. Inhibition of MDM2/MDMX
3.3. Inhibition of SIRT1
3.4. Inhibition of Telomerase Activity
3.5. Epigenetic Modifiers
3.6. Influencing the Bone Marrow Microenvironment
3.7. Senolytics
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Chiu, C.P.; Dragowska, W.; Kim, N.W.; Vaziri, H.; Yui, J.; Thomas, T.E.; Harley, C.B.; Lansdorp, P.M. Differential expression of telomerase activity in hematopoietic progenitors from adult human bone marrow. Stem. Cells 1996, 14, 239–248. [Google Scholar] [CrossRef]
- Afshari, C.A.; Vojta, P.J.; Annab, L.A.; Futreal, P.A.; Willard, T.B.; Barrett, J.C. Investigation of the role of G1/S cell cycle mediators in cellular senescence. Exp. Cell Res. 1993, 209, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, S.W.; Rush, D.; Ellsworth, J.L.; Schimke, R.T. Defining cellular senescence in IMR-90 cells: A flow cytometric analysis. Proc. Natl. Acad. Sci. USA 1988, 85, 9086–9090. [Google Scholar] [CrossRef] [PubMed]
- Hensler, P.J.; Pereira-Smith, O.M. Human replicative senescence. A molecular study. Am. J. Pathol. 1995, 147, 1–8. [Google Scholar]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef]
- Tu, Z.; Aird, K.M.; Bitler, B.G.; Nicodemus, J.P.; Beeharry, N.; Xia, B.; Yen, T.J.; Zhang, R. Oncogenic RAS regulates BRIP1 expression to induce dissociation of BRCA1 from chromatin, inhibit DNA repair, and promote senescence. Dev. Cell 2011, 21, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, M.; Yan, J.; Liu, T.; Pan, G.; Yang, H.; Pei, M.; He, F. Alcohol Induces Cellular Senescence and Impairs Osteogenic Potential in Bone Marrow-Derived Mesenchymal Stem Cells. Alcohol. Alcohol. 2017, 52, 289–297. [Google Scholar] [CrossRef]
- Kim, C.O.; Huh, A.J.; Han, S.H.; Kim, J.M. Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch. Gerontol. Geriatr. 2012, 54, e35–e41. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Noda, A.; Ning, Y.; Venable, S.F.; Pereira-Smith, O.M.; Smith, J.R. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp. Cell Res. 1994, 211, 90–98. [Google Scholar] [CrossRef]
- Muller, M. Cellular senescence: Molecular mechanisms, in vivo significance, and redox considerations. Antioxid. Redox. Signal 2009, 11, 59–98. [Google Scholar] [CrossRef]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar]
- Ohshima, S. Apoptosis in stress-induced and spontaneously senescent human fibroblasts. Biochem. Biophys. Res. Commun. 2004, 324, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; Van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Krishna, D.R.; Sperker, B.; Fritz, P.; Klotz, U. Does pH 6 beta-galactosidase activity indicate cell senescence? Mech. Ageing Dev. 1999, 109, 113–123. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Evans, M.K. Techniques to Induce and Quantify Cellular Senescence. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Stewart, S.A.; Ben-Porath, I.; Carey, V.J.; O’Connor, B.F.; Hahn, W.C.; Weinberg, R.A. Erosion of the telomeric single-strand overhang at replicative senescence. Nat. Genet. 2003, 33, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012, 31, 2839–2851. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Dobbelstein, M.; Freedman, D.A.; Shenk, T.; Levine, A.J. Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 1998, 17, 554–564. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; Van Laar, T.; Dekker, P.; Bazuine, M.; Van Ham, R.C.; Van der Houven van Oordt, W.; Hateboer, G.; Van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: Implications for targeting mTOR during malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef]
- Liu, P.; Lu, Z.; Wu, Y.; Shang, D.; Zhao, Z.; Shen, Y.; Zhang, Y.; Zhu, F.; Liu, H.; Tu, Z. Cellular Senescence-Inducing Small Molecules for Cancer Treatment. Curr. Cancer Drug Targets 2019, 19, 109–119. [Google Scholar] [CrossRef]
- Lavin, M.F.; Kozlov, S. ATM activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.C.; Tarnawski, A.S. PTEN regulatory functions in tumor suppression and cell biology. Med. Sci. Monit. 2004, 10, RA235–RA241. [Google Scholar] [PubMed]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef]
- Peng, D.; Wang, H.; Li, L.; Ma, X.; Chen, Y.; Zhou, H.; Luo, Y.; Xiao, Y.; Liu, L. miR-34c-5p promotes eradication of acute myeloid leukemia stem cells by inducing senescence through selective RAB27B targeting to inhibit exosome shedding. Leukemia 2018, 32, 1180–1188. [Google Scholar] [CrossRef]
- Zhou, B.; Li, C.; Qi, W.; Zhang, Y.; Zhang, F.; Wu, J.X.; Hu, Y.N.; Wu, D.M.; Liu, Y.; Yan, T.T.; et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 2012, 55, 2032–2043. [Google Scholar] [CrossRef]
- Zhou, D.; Xu, P.; Zhou, X.; Diao, Z.; Ouyang, J.; Yan, G.; Chen, B. MiR-181a enhances drug sensitivity of mixed lineage leukemia-rearranged acute myeloid leukemia by increasing poly(ADP-ribose) polymerase1 acetylation. Leuk. Lymphoma 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwangbo, J.; Wong, P.K. p38 MAPK-Mediated Bmi-1 down-regulation and defective proliferation in ATM-deficient neural stem cells can be restored by Akt activation. PLoS ONE 2011, 6, e16615. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; De Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e2654. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Gallagher, T.R.; Cavey, J.R.; Sheppard, P.W.; Ralston, S.H.; Layfield, R.; Searle, M.S. Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J. Biol. Chem. 2008, 283, 5427–5440. [Google Scholar] [CrossRef]
- Mazzucco, A.E.; Smogorzewska, A.; Kang, C.; Luo, J.; Schlabach, M.R.; Xu, Q.; Patel, R.; Elledge, S.J. Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program. Genes Dev. 2017, 31, 1933–1938. [Google Scholar] [CrossRef]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and Temporal Control of Senescence. Trends Cell Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, N.; Ghione, S.; Laurens, V.; Plenchette, S.; Bettaieb, A.; Paul, C. Senescence and Cancer: Role of Nitric Oxide (NO) in SASP. Cancers 2020, 12, 1145. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Leite de Oliveira, R.; Bernards, R. Anti-cancer therapy: Senescence is the new black. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Miettinen, T.P.; Peltier, J.; Hartlova, A.; Gierlinski, M.; Jansen, V.M.; Trost, M.; Bjorklund, M. Thermal proteome profiling of breast cancer cells reveals proteasomal activation by CDK4/6 inhibitor palbociclib. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Hu, W.; Sung, T.; Jessen, B.A.; Thibault, S.; Finkelstein, M.B.; Khan, N.K.; Sacaan, A.I. Mechanistic Investigation of Bone Marrow Suppression Associated with Palbociclib and its Differentiation from Cytotoxic Chemotherapies. Clin. Cancer Res. 2016, 22, 2000–2008. [Google Scholar] [CrossRef]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef]
- Uras, I.Z.; Maurer, B.; Nebenfuehr, S.; Zojer, M.; Valent, P.; Sexl, V. Therapeutic Vulnerabilities in FLT3-Mutant AML Unmasked by Palbociclib. Int. J. Mol. Sci. 2018, 19, 3987. [Google Scholar] [CrossRef]
- Scholl, S.; Fleischmann, M.; Schnetzke, U.; Heidel, F.H. Molecular Mechanisms of Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia: Ongoing Challenges and Future Treatments. Cells 2020, 9, 2493. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; De Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Giansanti, M.; Voso, M.T.; Lo-Coco, F.; Graziani, G. Targeting ADP-ribosylation by PARP inhibitors in acute myeloid leukaemia and related disorders. Biochem. Pharmacol. 2019, 167, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Consalvo, M.I.; Aloisio, F.; Fabiani, E.; Giansanti, M.; Di Cristino, F.; Falconi, G.; Tentori, L.; Di Veroli, A.; Curzi, P.; et al. Cytotoxicity and Differentiating Effect of the Poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Myelodysplastic Syndromes. Cancers 2019, 11, 1373. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci. Rep. 2018, 8, 2410. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; Gonzalez-Gualda, E.; Doherty, G.J.; Munoz-Espin, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Kantarjian, H.M.; Estey, E.; Manshouri, T.; Chan, C.Y.; Rahman Elsaied, A.; Kornblau, S.M.; Cortes, J.; Thomas, D.A.; Pierce, S.; et al. The prognostic significance of p16(INK4a)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982. [Google Scholar] [CrossRef]
- Han, X.; Medeiros, L.J.; Zhang, Y.H.; You, M.J.; Andreeff, M.; Konopleva, M.; Bueso-Ramos, C.E. High Expression of Human Homologue of Murine Double Minute 4 and the Short Splicing Variant, HDM4-S, in Bone Marrow in Patients with Acute Myeloid Leukemia or Myelodysplastic Syndrome. Clin. Lymphoma Myeloma Leuk. 2016, 16, S30–S38. [Google Scholar] [CrossRef] [PubMed]
- Peller, S.; Rotter, V. TP53 in hematological cancer: Low incidence of mutations with significant clinical relevance. Hum. Mutat. 2003, 21, 277–284. [Google Scholar] [CrossRef]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Saxena, K.; Konopleva, M. An expert overview of emerging therapies for acute myeloid leukemia: Novel small molecules targeting apoptosis, p53, transcriptional regulation and metabolism. Expert Opin. Investig. Drugs 2020, 29, 973–988. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov. 2020. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02909972 (accessed on 10 January 2021).
- Kannan, S.; Partridge, A.W.; Lane, D.P.; Verma, C.S. The Dual Interactions of p53 with MDM2 and p300: Implications for the Design of MDM2 Inhibitors. Int. J. Mol. Sci. 2019, 20, 5996. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Send in the senolytics. Nat. Biotechnol. 2020, 38, 1371–1377. [Google Scholar] [CrossRef]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Lee, S.H.; Um, S.J.; Kim, E.J. CBX8 antagonizes the effect of Sirtinol on premature senescence through the AKT-RB-E2F1 pathway in K562 leukemia cells. Biochem. Biophys. Res. Commun. 2016, 469, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell. Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Q.; Hu, Y.; Xu, L.; Jiang, Y.; Zhang, C.; Ding, L.; Jiang, R.; Sun, J.; Sun, H.; et al. miR-181a increases FoxO1 acetylation and promotes granulosa cell apoptosis via SIRT1 downregulation. Cell Death Dis. 2017, 8, e3088. [Google Scholar] [CrossRef] [PubMed]
- Schwind, S.; Maharry, K.; Radmacher, M.D.; Mrozek, K.; Holland, K.B.; Margeson, D.; Whitman, S.P.; Hickey, C.; Becker, H.; Metzeler, K.H.; et al. Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2010, 28, 5257–5264. [Google Scholar] [CrossRef]
- Townsley, D.M.; Dumitriu, B.; Young, N.S. Bone marrow failure and the telomeropathies. Blood 2014, 124, 2775–2783. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, M.; Sun, X.; Sun, J. Telomerase activity and telomere length in acute leukemia: Correlations with disease progression, subtypes and overall survival. Int. J. Lab. Hematol. 2010, 32, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, D.; Onozawa, M.; Miyashita, N.; Yokoyama, S.; Nakagawa, M.; Hashimoto, D.; Teshima, T. Short-term treatment with imetelstat sensitizes hematopoietic malignant cells to a genotoxic agent via suppression of the telomerase-mediated DNA repair process. Leuk. Lymphoma 2020, 61, 2722–2732. [Google Scholar] [CrossRef]
- Baerlocher, G.M.; Oppliger Leibundgut, E.; Ottmann, O.G.; Spitzer, G.; Odenike, O.; McDevitt, M.A.; Roth, A.; Daskalakis, M.; Burington, B.; Stuart, M.; et al. Telomerase Inhibitor Imetelstat in Patients with Essential Thrombocythemia. N. Engl. J. Med. 2015, 373, 920–928. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Kishtagari, A.; Watts, J. Biological and clinical implications of telomere dysfunction in myeloid malignancies. Ther. Adv. Hematol. 2017, 8, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Fenaux, P.; Van Eygen, K.; Raza, A.; Santini, V.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; et al. Imetelstat Achieves Meaningful and Durable Transfusion Independence in High Transfusion-Burden Patients with Lower-Risk Myelodysplastic Syndromes in a Phase II Study. J. Clin. Oncol. 2021, 39, 48–56. [Google Scholar] [CrossRef]
- Sawa, M.; Yamamoto, K.; Yokozawa, T.; Kiyoi, H.; Hishida, A.; Kajiguchi, T.; Seto, M.; Kohno, A.; Kitamura, K.; Itoh, Y.; et al. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int. J. Hematol. 2005, 82, 42–47. [Google Scholar] [CrossRef]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef]
- Bommi, P.V.; Dimri, M.; Sahasrabuddhe, A.A.; Khandekar, J.; Dimri, G.P. The polycomb group protein BMI1 is a transcriptional target of HDAC inhibitors. Cell Cycle 2010, 9, 2663–2673. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, K.; Zhou, Y.; Xiao, Y.; Deng, M.; Jiang, Z.; Ye, W.; Wang, X.; Wei, X.; Li, J.; et al. A New Strategy to Target Acute Myeloid Leukemia Stem and Progenitor Cells Using Chidamide, a Histone Deacetylase Inhibitor. Curr. Cancer Drug Targets 2015, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.C.; Zhu, C.Y.; Yan, F.; Wang, M.Z.; Chen, X.S.; Wang, X.K.; Pang, B.X.; Li, Y.H.; Liu, D.H.; et al. Chidamide increases the sensitivity of refractory or relapsed acute myeloid leukemia cells to anthracyclines via regulation of the HDAC3 -AKT-P21-CDK2 signaling pathway. J. Exp. Clin. Cancer Res. 2020, 39, 278. [Google Scholar] [CrossRef]
- Li, Q.; Huang, J.C.; Liao, D.Y.; Wu, Y. Chidamide plus decitabine synergistically induces apoptosis of acute myeloid leukemia cells by upregulating PERP. Am. J. Transl. Res. 2020, 12, 3461–3475. [Google Scholar]
- He, B.; Wang, Q.; Liu, X.; Lu, Z.; Han, J.; Pan, C.; Carter, B.Z.; Liu, Q.; Xu, N.; Zhou, H. A novel HDAC inhibitor chidamide combined with imatinib synergistically targets tyrosine kinase inhibitor resistant chronic myeloid leukemia cells. Biomed. Pharmacother. 2020, 129, 110390. [Google Scholar] [CrossRef] [PubMed]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; Van Rooijen, E.; Werner-Klein, M.; et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell 2018, 33, 785. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Miyagi, S.; Pedersen, M.T.; Kooistra, S.M.; Johansen, J.V.; Helin, K. Jmjd2/Kdm4 demethylases are required for expression of Il3ra and survival of acute myeloid leukemia cells. Genes Dev. 2016, 30, 1278–1288. [Google Scholar] [CrossRef]
- Agger, K.; Nishimura, K.; Miyagi, S.; Messling, J.E.; Rasmussen, K.D.; Helin, K. The KDM4/JMJD2 histone demethylases are required for hematopoietic stem cell maintenance. Blood 2019, 134, 1154–1158. [Google Scholar] [CrossRef]
- Jin, C.; Yang, L.; Xie, M.; Lin, C.; Merkurjev, D.; Yang, J.C.; Tanasa, B.; Oh, S.; Zhang, J.; Ohgi, K.A.; et al. Chem-seq permits identification of genomic targets of drugs against androgen receptor regulation selected by functional phenotypic screens. Proc. Natl. Acad. Sci. USA 2014, 111, 9235–9240. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Fung, T.K.; Zeisig, B.B.; Holmes, K.; Rane, J.K.; Mowen, K.A.; Finn, M.G.; Lenhard, B.; Chan, L.C.; So, C.W. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016, 29, 32–48. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Gottgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef]
- Rui, L.; Emre, N.C.; Kruhlak, M.J.; Chung, H.J.; Steidl, C.; Slack, G.; Wright, G.W.; Lenz, G.; Ngo, V.N.; Shaffer, A.L.; et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 2010, 18, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, A.M.; Sun, Y.; Hellmich, C.; Marlein, C.R.; Mistry, J.; Forde, E.; Piddock, R.E.; Shafat, M.S.; Morfakis, A.; Mehta, T.; et al. Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 2019, 133, 446–456. [Google Scholar] [CrossRef]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Serrano, M. Senescence and Cancer: In the Name of Immunosuppression. Cancer Cell 2016, 30, 507–508. [Google Scholar] [CrossRef][Green Version]
- Jacamo, R.; Mu, H.; Zhang, Q.; Chachad, D.; Zhiqiang, W.; Ma, W.; Zhang, M.; Mak, P.; Mak, D.; Ruvolo, P.; et al. Effects of CCL2/CCR2 Blockade in Acute Myeloid Leukemia. Blood 2015, 126, 1348. [Google Scholar] [CrossRef]
- Macanas-Pirard, P.; Quezada, T.; Navarrete, L.; Broekhuizen, R.; Leisewitz, A.; Nervi, B.; Ramirez, P.A. The CCL2/CCR2 Axis Affects Transmigration and Proliferation but Not Resistance to Chemotherapy of Acute Myeloid Leukemia Cells. PLoS ONE 2017, 12, e0168888. [Google Scholar] [CrossRef] [PubMed]
- Lompardia, S.L.; Papademetrio, D.L.; Mascaro, M.; Alvarez, E.M.; Hajos, S.E. Human leukemic cell lines synthesize hyaluronan to avoid senescence and resist chemotherapy. Glycobiology 2013, 23, 1463–1476. [Google Scholar] [CrossRef]
- Lompardia, S.; Diaz, M.; Pibuel, M.; Papademetrio, D.; Poodts, D.; Mihalez, C.; Alvarez, E.; Hajos, S. Hyaluronan abrogates imatinib-induced senescence in chronic myeloid leukemia cell lines. Sci. Rep. 2019, 9, 10930. [Google Scholar] [CrossRef]
- Kultti, A.; Pasonen-Seppanen, S.; Jauhiainen, M.; Rilla, K.J.; Karna, R.; Pyoria, E.; Tammi, R.H.; Tammi, M.I. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp. Cell Res. 2009, 315, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Lompardia, S.L.; Diaz, M.; Papademetrio, D.L.; Pibuel, M.; Alvarez, E.; Hajos, S.E. 4-methylumbelliferone and imatinib combination enhances senescence induction in chronic myeloid leukemia cell lines. Investig. New Drugs 2017, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Katlinskaya, Y.V.; Carbone, C.J.; Yu, Q.; Fuchs, S.Y. Type 1 interferons contribute to the clearance of senescent cell. Cancer Biol. Ther. 2015, 16, 1214–1219. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Schoenwaelder, S.M.; Jarman, K.E.; Gardiner, E.E.; Hua, M.; Qiao, J.; White, M.J.; Josefsson, E.C.; Alwis, I.; Ono, A.; Willcox, A.; et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011, 118, 1663–1674. [Google Scholar] [CrossRef]
- Gandhi, L.; Camidge, D.R.; Ribeiro de Oliveira, M.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef]
- Roberts, A.W.; Advani, R.H.; Kahl, B.S.; Persky, D.; Sweetenham, J.W.; Carney, D.A.; Yang, J.; Busman, T.B.; Enschede, S.H.; Humerickhouse, R.A.; et al. Phase 1 study of the safety, pharmacokinetics, and antitumour activity of the BCL2 inhibitor navitoclax in combination with rituximab in patients with relapsed or refractory CD20+ lymphoid malignancies. Br. J. Haematol. 2015, 170, 669–678. [Google Scholar] [CrossRef]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.N.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.; et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 2020, 11, 1996. [Google Scholar] [CrossRef]
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 2826–2833. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.J.; Cho, K.A.; Oh, Y.S.; Park, S.C. Role of Src-specific phosphorylation site on focal adhesion kinase for senescence-associated apoptosis resistance. Apoptosis 2006, 11, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, A.; Roninson, I.B.; Gamelin, E.; Coqueret, O. Src inhibits adriamycin-induced senescence and G2 checkpoint arrest by blocking the induction of p21waf1. Cancer Res. 2005, 65, 8927–8935. [Google Scholar] [CrossRef] [PubMed]
- Batuello, C.N.; Hauck, P.M.; Gendron, J.M.; Lehman, J.A.; Mayo, L.D. Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proc. Natl. Acad. Sci. USA 2015, 112, 1749–1754. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Corrigendum to ‘Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease’ EBioMedicine 47 (2019) 446–456. EBioMedicine 2020, 52, 102595. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. JAMA 2018, 320, 1319–1320. [Google Scholar] [CrossRef]
- Kivioja, J.L.; Thanasopoulou, A.; Kumar, A.; Kontro, M.; Yadav, B.; Majumder, M.M.; Javarappa, K.K.; Eldfors, S.; Schwaller, J.; Porkka, K.; et al. Dasatinib and navitoclax act synergistically to target NUP98-NSD1(+)/FLT3-ITD(+) acute myeloid leukemia. Leukemia 2019, 33, 1360–1372. [Google Scholar] [CrossRef]
- Weisberg, E.; Liu, Q.; Nelson, E.; Kung, A.L.; Christie, A.L.; Bronson, R.; Sattler, M.; Sanda, T.; Zhao, Z.; Hur, W.; et al. Using combination therapy to override stromal-mediated chemoresistance in mutant FLT3-positive AML: Synergism between FLT3 inhibitors, dasatinib/multi-targeted inhibitors and JAK inhibitors. Leukemia 2012, 26, 2233–2244. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Mikula-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Ksiazek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell Mol. Life Sci. 2020, 77, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; Van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e116. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.S.L.; Narita, M. Short-term gain, long-term pain: The senescence life cycle and cancer. Genes Dev. 2019, 33, 127–143. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov. 2020. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00255346 (accessed on 12 January 2021).
- US National Library of Medicine. ClinicalTrials.gov. 2020. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02942290 (accessed on 12 January 2021).
- US National Library of Medicine. ClinicalTrials.gov. 2020. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04041050 (accessed on 12 January 2021).
- Identification and Elimination of Damaged and/or Senescent Cells. U.S. Patent Application 20200123607, 4 April 2020.



{kind=link}
{kind=link}
| Drug | ClinicalTrials.gov ID | Phase | Target | Combined Drug | Diseases | Status | Estimated Study Completion Date |
|---|---|---|---|---|---|---|---|
| Palbociclib | NCT03132454 | I | CDK4/6 | Sorafenib or Decitabine or Dexamethasone or alone | Relapsed/refractory acute leukemia | Recruiting | June 2021 |
| NCT03844997 | I/II | CDK4/6 | CPX-351 | Primary AML | Recruiting | January 2021 | |
| Talazoparib | NCT02878785 | I/II | PARP | Decitabine | Primary AML, Relapsed/refractory AML | Active, not recruiting | December 2022 |
| Veliparib | NCT03289910 | II | PARP | Carboplatin or Topotecan | Primary AML, Relapsed/refractory AML, sAML arising from MDS, aCML, CMML, ET, PV, Myelofibrosis | Recruiting | June 2021 |
| Imetelstat | NCT04576156 | III | Telomerase | none | Intermediate-2 or high-risk Myelofibrosis | Recruiting | May 2024 |
| Chidamide | NCT03031262 | I/II | HDAC | Cytarabine | Postremission therapy of CBF-AML | Recruiting | December 2022 |
| NCT03453255 | I/II | HDAC | Decitabine, Homoharringtonine, and Cytarabine | Postremission therapy of AML with t(8;21) | Unknown | December 2020 | |
| Navitoclax | NCT04472598 | III | BCL-2, BCL-XL, BCL-W | Ruxolitinib | Myelofibrosis | Recruiting | July 2028 |
| NCT04041050 | I | BCL-2, BCL-XL, BCL-W | Ruxolitinib or Celecoxib or alone | MPN | Recruiting | May 2023 | |
| Venetoclax | NCT03826992 | I | BCL-2 | CPX-351 | Relapsed/refractory acute leukemia | Recruiting | January 2023 |
| NCT03573024 | II | BCL-2 | Azacitidine | Primary AML | Recruiting | June 2023 | |
| Dasatinib | NCT02013648 | III | SRC | Cytarabine and Daunorubicin or Idarubicin | Primary CBF-AML | Recruiting | February 2024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ernst, P.; Heidel, F.H. Molecular Mechanisms of Senescence and Implications for the Treatment of Myeloid Malignancies. Cancers 2021, 13, 612. https://doi.org/10.3390/cancers13040612
Ernst P, Heidel FH. Molecular Mechanisms of Senescence and Implications for the Treatment of Myeloid Malignancies. Cancers. 2021; 13(4):612. https://doi.org/10.3390/cancers13040612
Chicago/Turabian StyleErnst, Philipp, and Florian H. Heidel. 2021. "Molecular Mechanisms of Senescence and Implications for the Treatment of Myeloid Malignancies" Cancers 13, no. 4: 612. https://doi.org/10.3390/cancers13040612
APA StyleErnst, P., & Heidel, F. H. (2021). Molecular Mechanisms of Senescence and Implications for the Treatment of Myeloid Malignancies. Cancers, 13(4), 612. https://doi.org/10.3390/cancers13040612