Cutaneous Findings in Neurofibromatosis Type 1

 ,
, 
 , and
, and
Abstract
Simple Summary
Abstract
1. Background
2. Genetic
3. Mosaicism
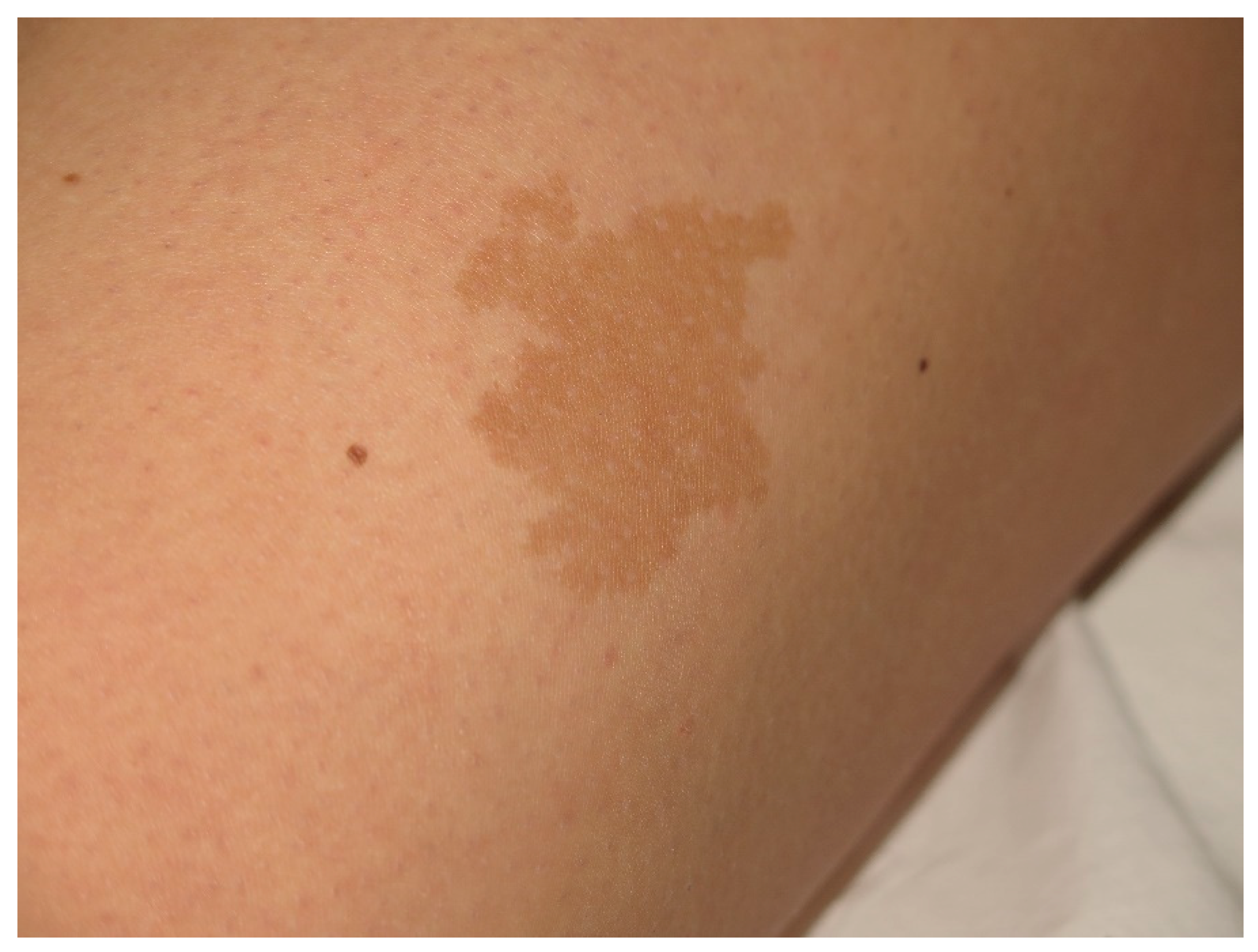
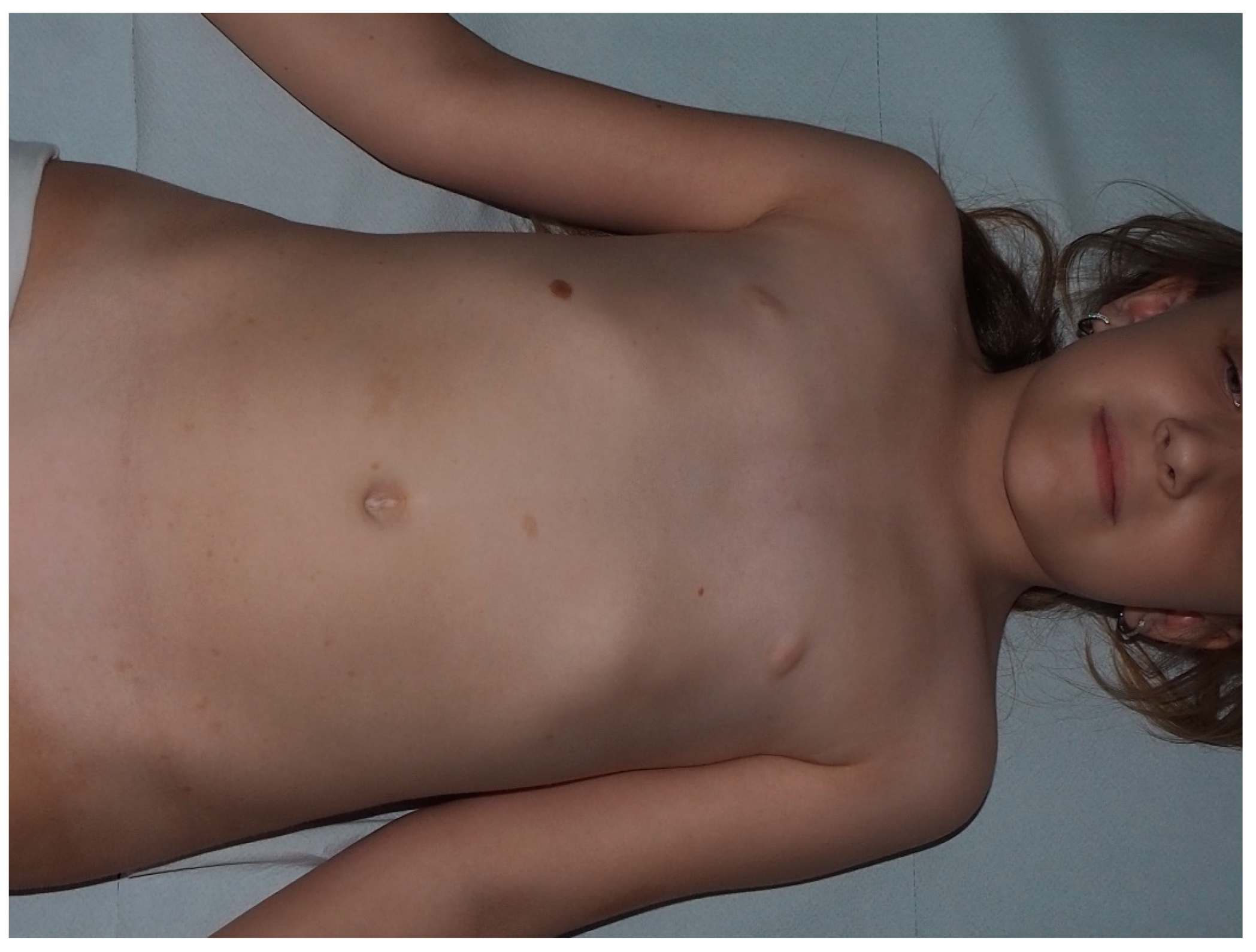
4. Cafe-Au-Lait Macules
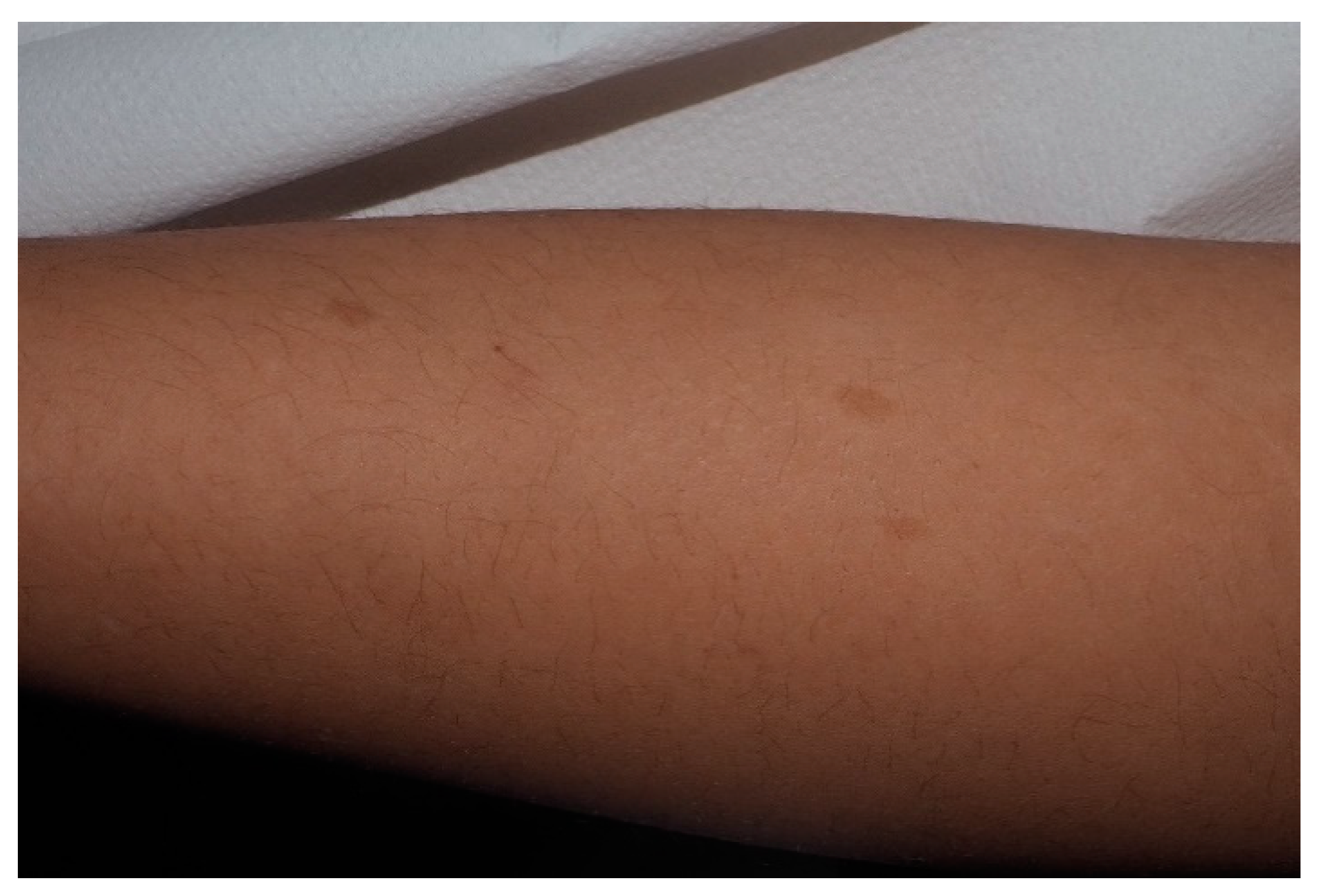
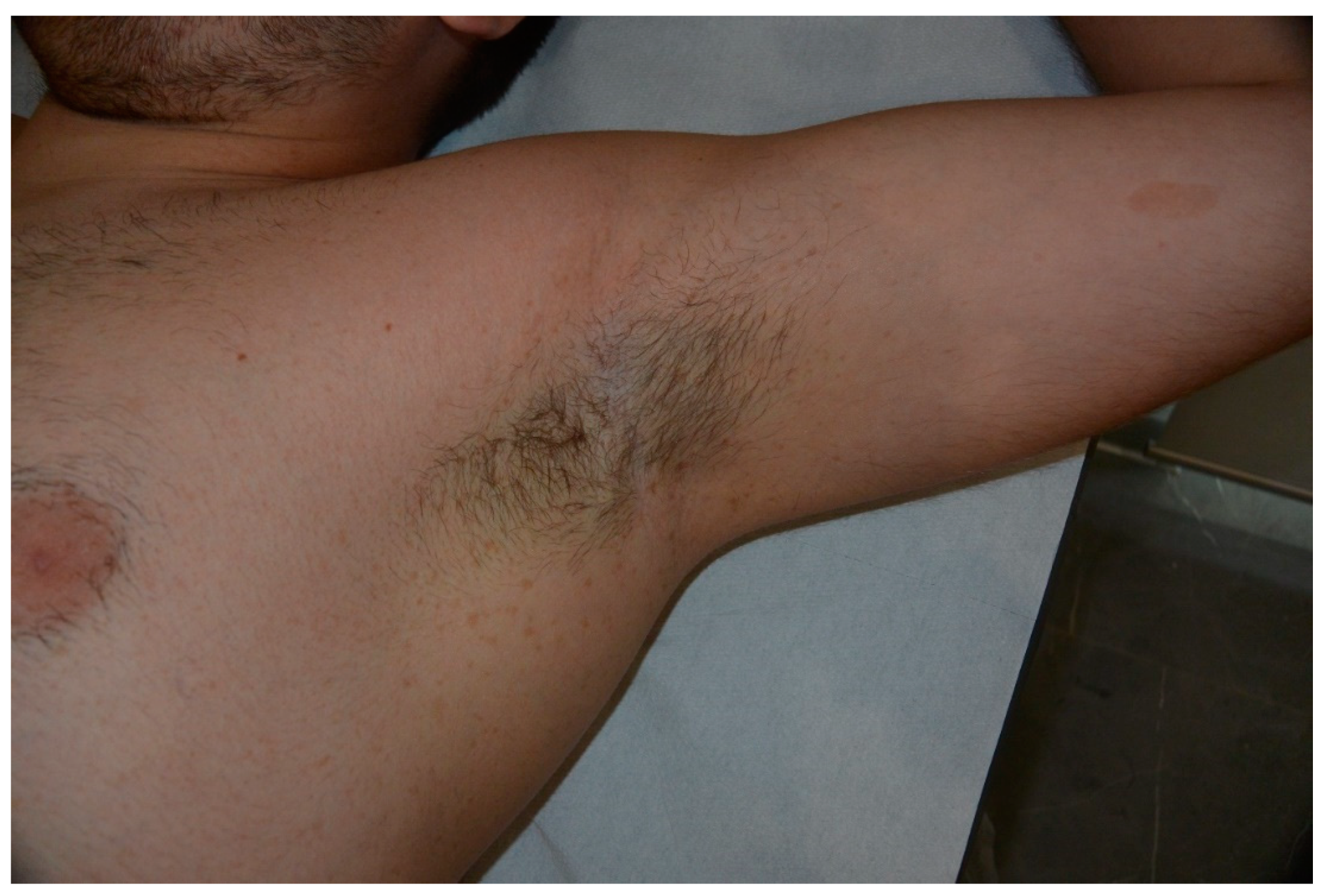
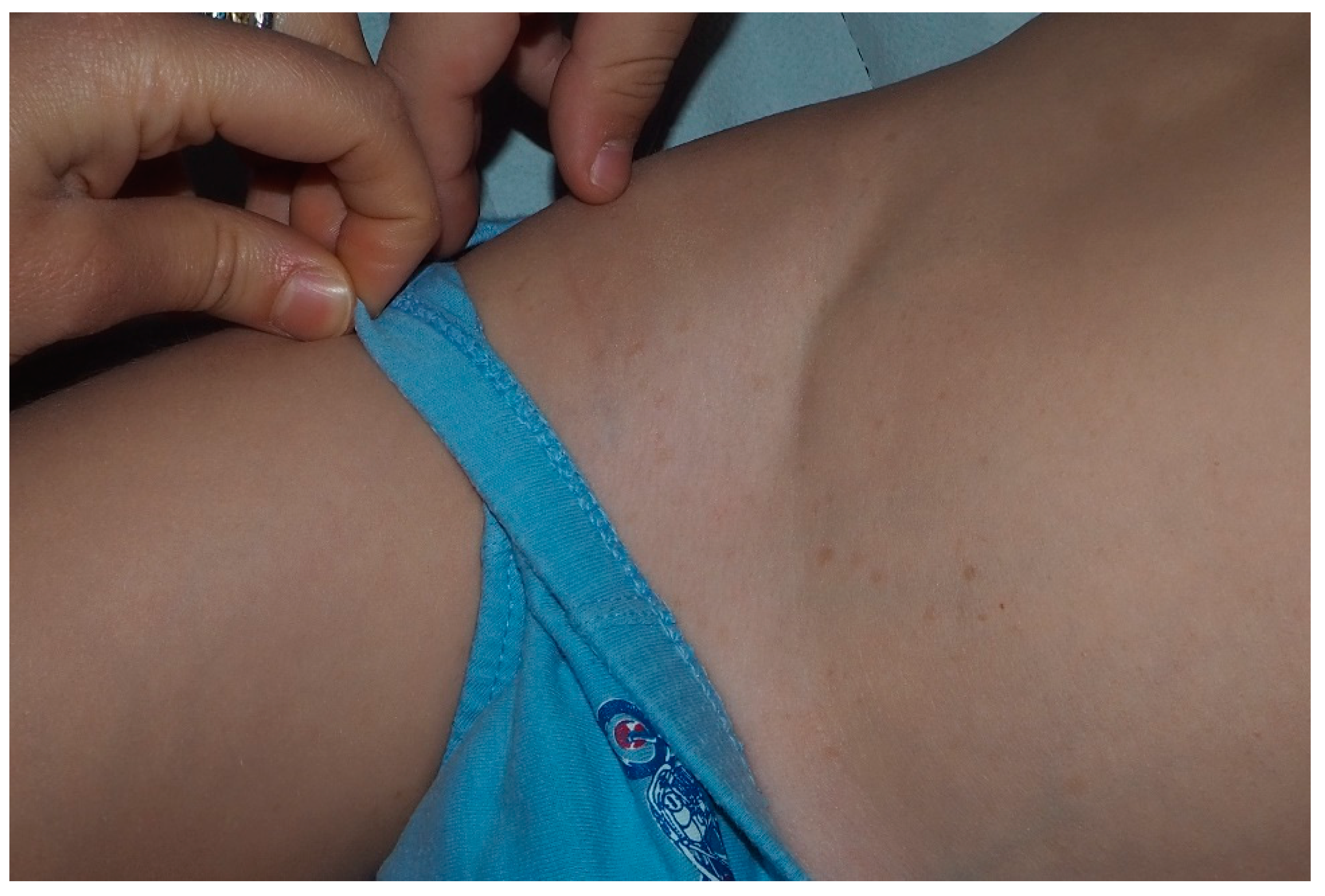
5. Freckling
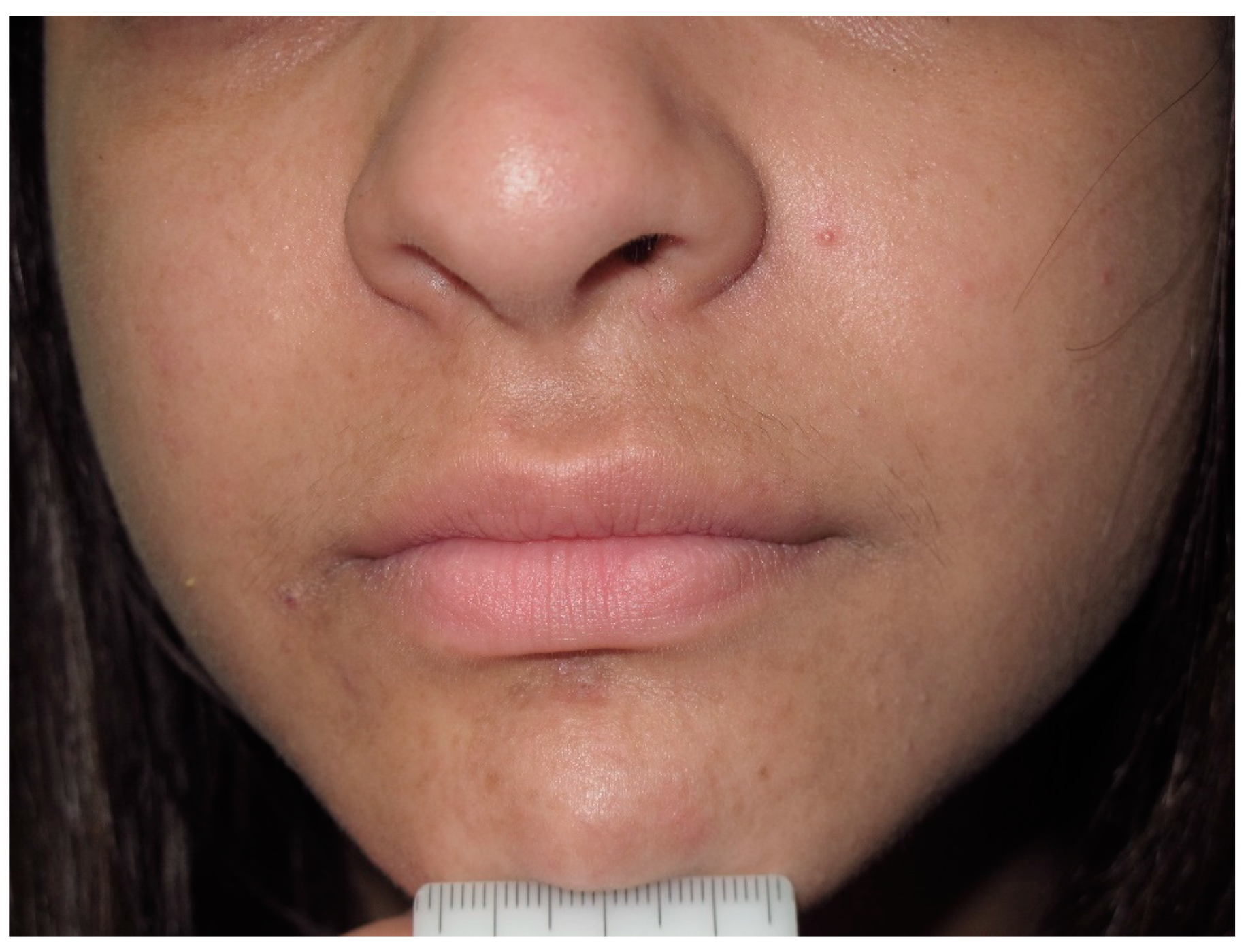
6. Lisch Nodules
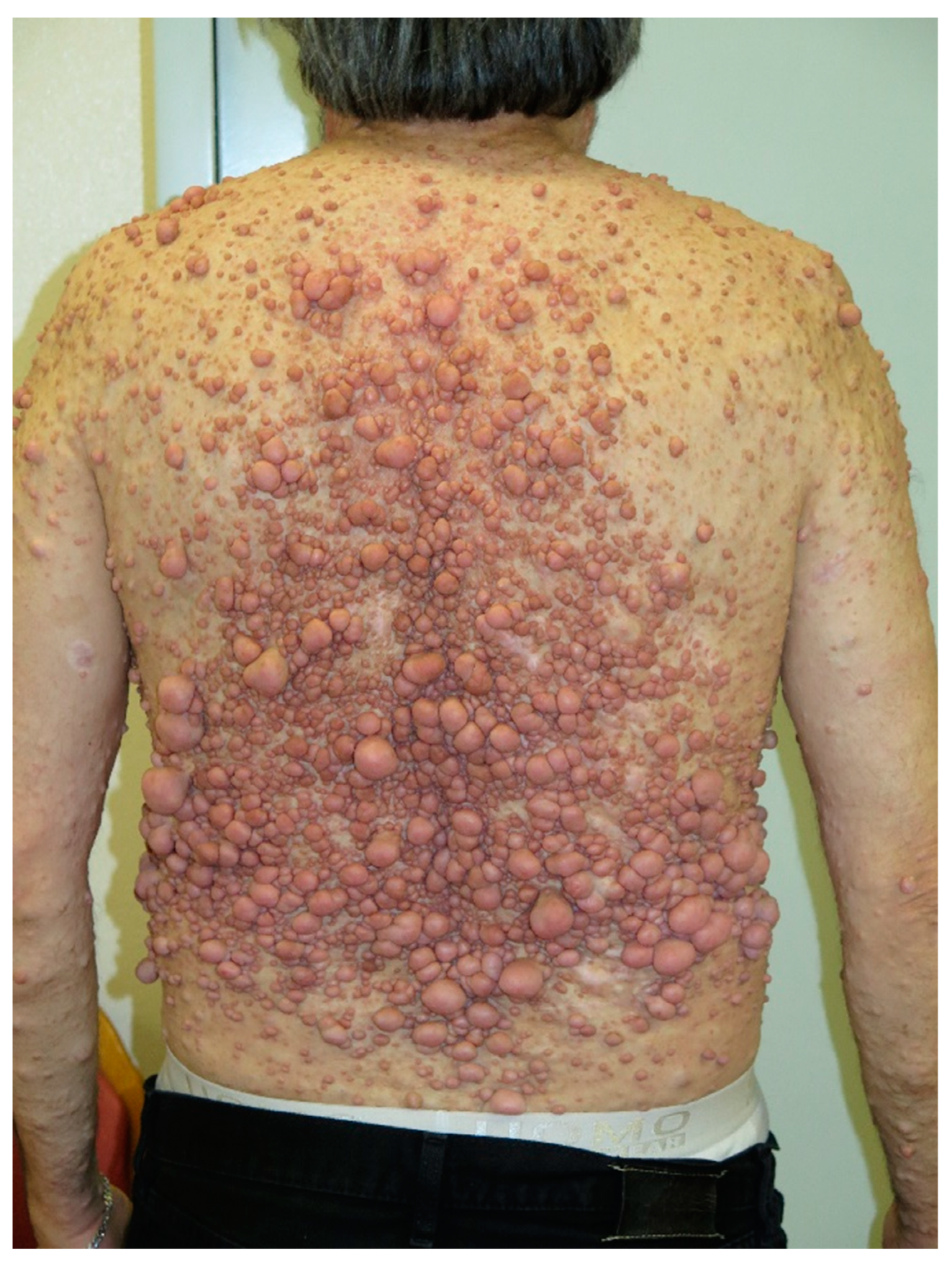
7. Neurofibromas
8. Malignant Peripheral Nerve Sheath Tumors
9. Optic Pathway Glioma
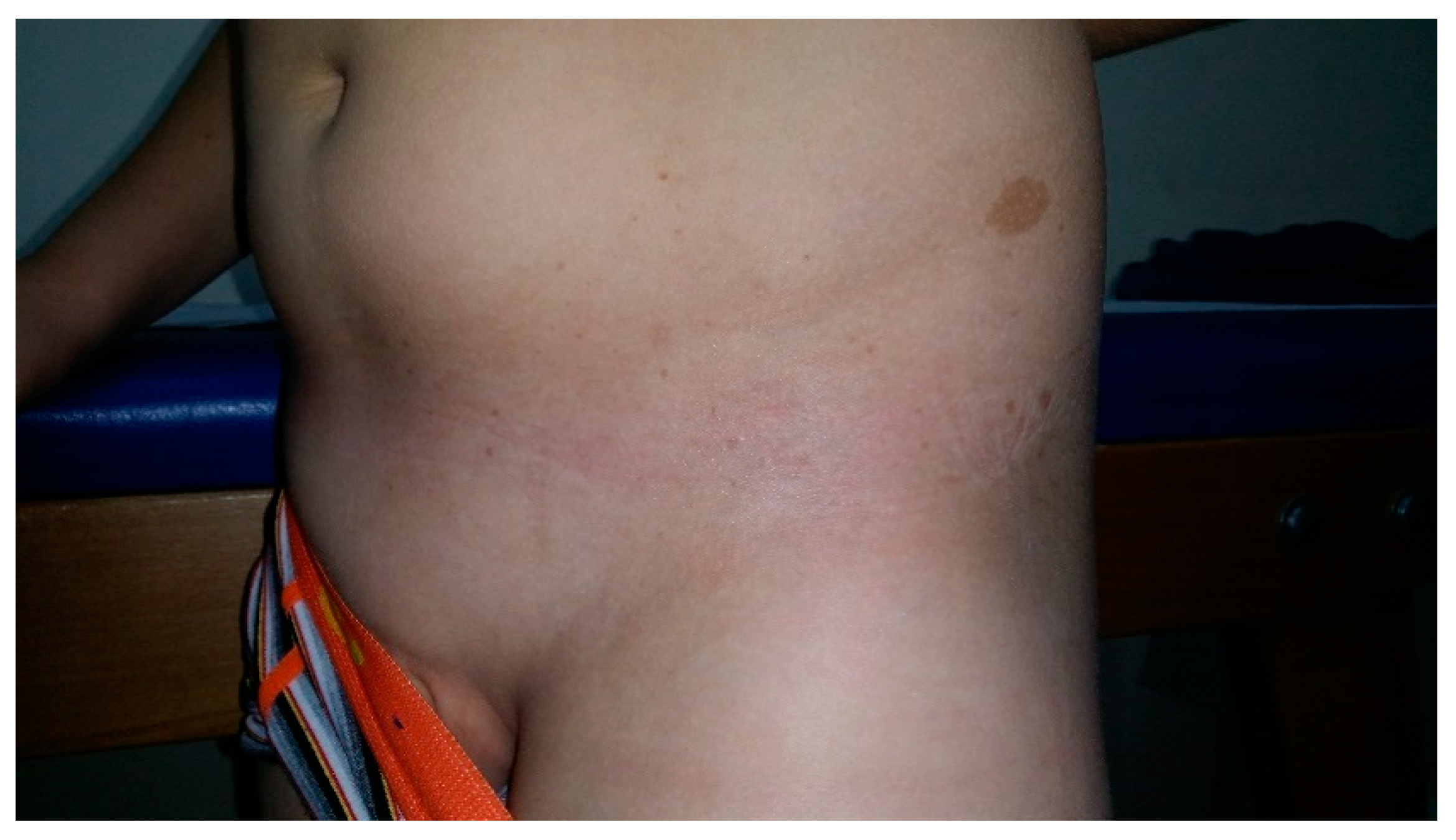
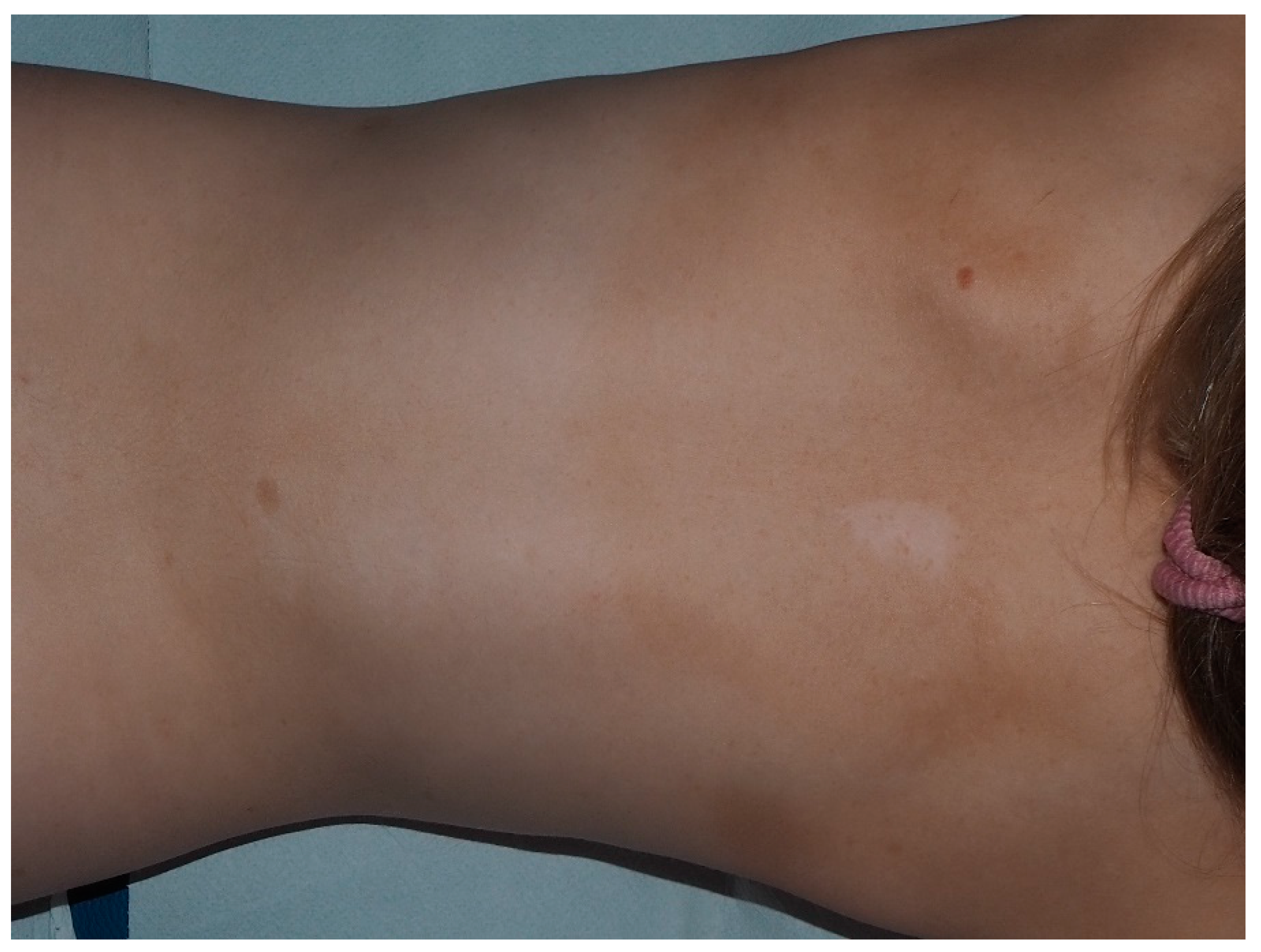
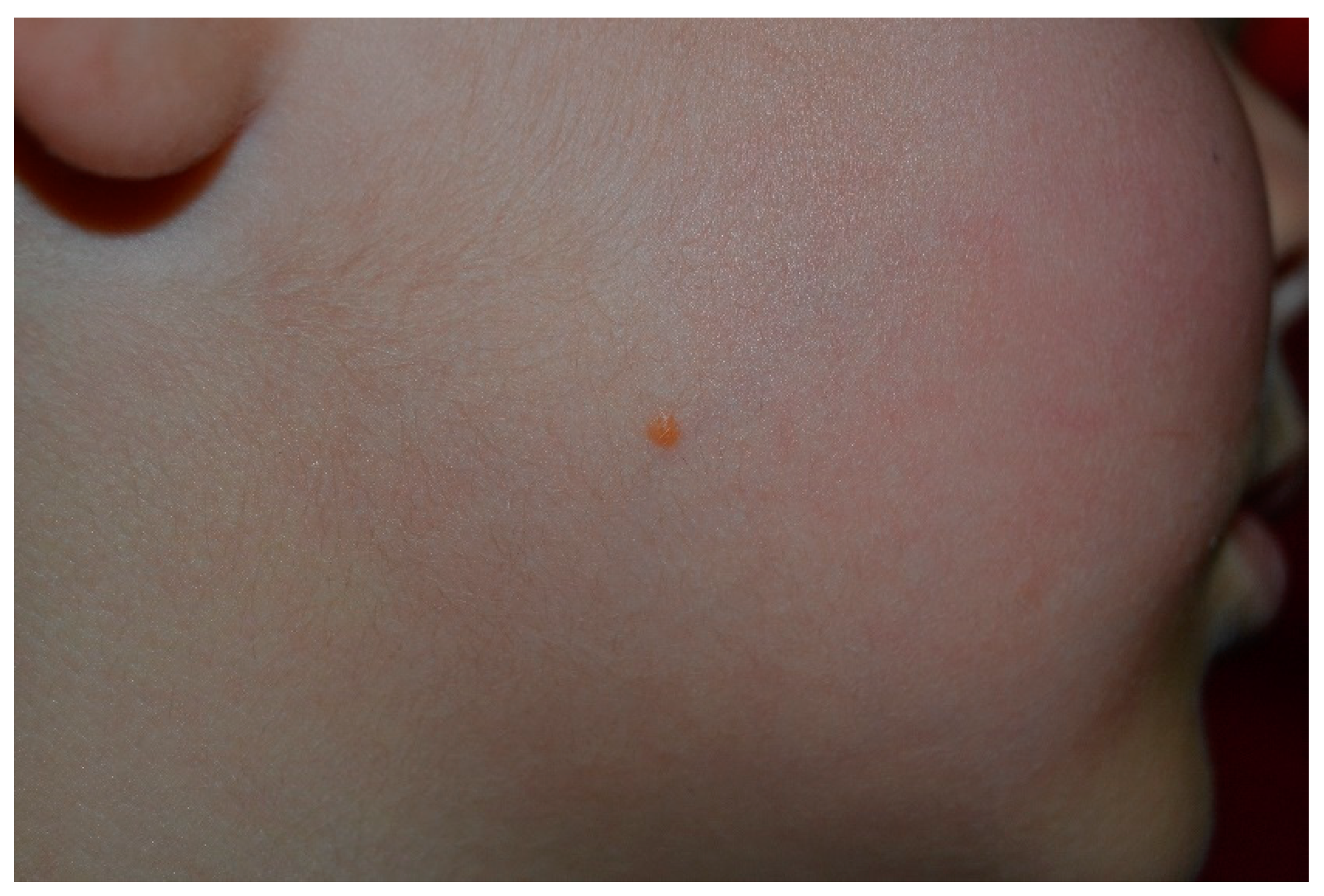
10. Nevus Anemicus
11. Juvenile Xanthogranuloma
12. Glomus Tumor
13. Melanoma
14. Other Neoplasms
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gutmann, D.H.; Wood, D.L.; Collins, F.S. Identification of the neurofibromatosis type 1 gene product. Proc. Natl. Acad. Sci. USA 1991, 88, 9658–9662. [Google Scholar] [CrossRef]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Prim. 2017, 3, nrdp20174. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J. Neurofibromatosis 1; Updated 2019; University of Washington: Washington, DC, USA, 1998. [Google Scholar]
- Trovó-Marqui, A.B.; Tajara, E.H. Neurofibromin: A general outlook. Clin. Genet. 2006, 70, 1–13. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health Consensus Development Conference Statement. Neurofibromatosis. Arch. Neurol. 1988, 45, 575–578. [Google Scholar] [CrossRef]
- Ars, E.; Kruyer, H.; Morell, M.; Pros, E.; Serra, E.; Ravella, A.; Estivill, X.; Lázaro, C. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J. Med. Genet. 2003, 40, e82. [Google Scholar] [CrossRef]
- Pros, E.; Gómez, C.; Martín, T.; Fábregas, P.; Serra, E.; Lázaro, C. Nature and mRNA effect of 282 differentNF1point mutations: Focus on splicing alterations. Hum. Mutat. 2008, 29, E173–E193. [Google Scholar] [CrossRef]
- Kang, E.; Kim, Y.-M.; Seo, G.H.; Oh, A.; Yoon, H.M.; Ra, Y.-S.; Kim, E.K.; Kim, H.; Heo, S.-H.; Kim, G.-H.; et al. Phenotype categorization of neurofibromatosis type I and correlation to NF1 mutation types. J. Hum. Genet. 2019, 65, 79–89. [Google Scholar] [CrossRef]
- Evans, D.G.; Bowers, N.; Burkitt-Wright, E.; Miles, E.; Garg, S.; Scott-Kitching, V.; Penman-Splitt, M.; Dobbie, A.; Howard, E.; Ealing, J.; et al. Comprehensive RNA analysis of the NF1 gene in classically affected NF1 affected individuals meeting NIH criteria has high sensitivity and mutation negative testing is reassuring in isolated cases with pigmentary features only. EBio Med. 2016, 7, 212–220. [Google Scholar] [CrossRef]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.-J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.; Atkin, J.; et al. High incidence of noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: Genotype–phenotype correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.-C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-phenotype correlation in NF1: Evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844–848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Theos, A.; Korf, B. Neurofibromatosis type 1. J. Am. Acad. Dermatol. 2009, 61, 401–407. [Google Scholar] [CrossRef]
- Ruggieri, M.; Huson, S.M. The clinical and diagnostic implications mosaicism in the neurofibromatoses. Neurology 2001, 56, 1433–1443. [Google Scholar] [CrossRef]
- Maertens, O.; De Schepper, S.; Vandesompele, J.; Brems, H.; Heyns, I.; Janssens, S.; Speleman, F.; Legius, E.; Messiaen, L. Molecular dissection of isolated disease features in mosaic neurofibromatosis Type 1. Am. J. Hum. Genet. 2007, 81, 243–251. [Google Scholar] [CrossRef]
- Shah, K.N. The diagnostic and clinical significance of café-au-lait macules. Pediatr. Clin. N. Am. 2010, 57, 1131–1153. [Google Scholar] [CrossRef]
- Nunley, K.S.; Gao, F.; Albers, A.C.; Bayliss, S.J.; Gutmann, D.H. Predictive value of café au lait macules at initial consultation in the diagnosis of neurofibromatosis Type 1. Arch. Dermatol. 2009, 145, 883–887. [Google Scholar] [CrossRef]
- Pemov, A.; Sung, H.; Hyland, P.L.; Sloan, J.L.; Ruppert, S.L.; Baldwin, A.M.; Boland, J.F.; Bass, S.E.; Lee, H.J.; Jones, K.M.; et al. Genetic modifiers of neurofibromatosis Type 1-associated café-au-lait macule count identified using multi-platform analysis. PLoS Genet. 2014, 10, e1004575. [Google Scholar] [CrossRef]
- Friedman, J.M.; Birch, P.H. Type 1 neurofibromatosis: A descriptive analysis of the disorder in 1728 patients. Am. J. Med. Genet. 1997, 70, 138–143. [Google Scholar] [CrossRef]
- Riccardi, V.M. Pathophysiology of neurofibromatosis. J. Am. Acad. Dermatol. 1980, 3, 157–166. [Google Scholar] [CrossRef]
- De Schepper, S.; Maertens, O.; Callens, T.; Naeyaert, J.-M.; Lambert, J.; Messiaen, L. Somatic mutation analysis in NF1 café au lait spots reveals two NF1 hits in the melanocytes. J. Investig. Dermatol. 2008, 128, 1050–1053. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Boucneau, J.; Haeghen, Y.V.; Messiaen, L.; Naeyaert, J.-M.; Lambert, J. Café-au-lait spots in neurofibromatosis type 1 and in healthy control individuals: Hyperpigmentation of a different kind? Arch. Dermatol. Res. 2006, 297, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Diwakar, G.; Zhang, D.; Jiang, S.; Hornyak, T.J. Neurofibromin as a regulator of melanocyte development and differentiation. J. Cell Sci. 2008, 121, 167–177. [Google Scholar] [CrossRef]
- Crowe, F.W. Axillary freckling as a diagnostic aid in neurofibromatosis. Ann. Intern. Med. 1964, 61, 1142–1143. [Google Scholar] [CrossRef]
- Obringer, A.C.; Meadows, A.T.; Zackai, E.H. The diagnosis of neurofibromatosis-1 in the child under the age of 6 years. Arch. Pediatr. Adolesc. Med. 1989, 143, 717. [Google Scholar] [CrossRef]
- Korf, B.R. Diagnostic outcome in children with multiple café au lait spots. Pediatrics 1992, 90, 924–927. [Google Scholar]
- Lubs, M.-L.E.; Bauer, M.S.; Formas, M.E.; Djokic, B. Lisch nodules in neurofibromatosis Type 1. N. Engl. J. Med. 1991, 324, 1264–1266. [Google Scholar] [CrossRef]
- Plotkin, S.R.; Bredella, M.A.; Cai, W.; Kassarjian, A.; Harris, G.J.; Esparza, S.; Merker, V.L.; Munn, L.L.; Muzikansky, A.; Askenazi, M.; et al. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS ONE 2012, 7, e35711. [Google Scholar] [CrossRef]
- Maertens, O.; Brems, H.; Vandesompele, J.; De Raedt, T.; Heyns, I.; Rosenbaum, T.; De Schepper, S.; De Paepe, A.; Mortier, G.; Janssens, S.; et al. ComprehensiveNF1 screening on cultured Schwann cells from neurofibromas. Hum. Mutat. 2006, 27, 1030–1040. [Google Scholar] [CrossRef]
- Feng-Chun, Y.; Ingram, D.A.; Chen, S.; Hingtgen, C.M.; Ratner, N.; Monk, K.R.; Clegg, T.; White, H.; Mead, L.; Wenning, M.J.; et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/– mast cells. J. Clin. Investig. 2003, 112, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Westerhof, W.; Konrad, K. Blue-red macules and pseudoatrophic macules. Arch. Dermatol. 1982, 118, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Chamseddin, B.H.; Le, L.Q. Management of cutaneous neurofibroma: Current therapy and future directions. Neuro-Oncol. Adv. 2019, 2 (Suppl. 1), i107–i116. [Google Scholar] [CrossRef] [PubMed]
- Ranibizumab for Neurofibromas Associated With Neurofibromatosis 1—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00657202 (accessed on 29 December 2020).
- Waggoner, D.J.; Towbin, J.; Gottesman, G.; Gutmann, D.H. Clinic-based study of plexiform neurofibromas in neurofibromatosis 1. Am. J. Med. Genet. 2000, 92, 132–135. [Google Scholar] [CrossRef]
- Mautner, V.-F.; Asuagbor, F.A.; Dombi, E.; Fünsterer, C.; Kluwe, L.; Wenzel, R.; Widemann, B.C.; Friedman, J.M. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro-oncology 2008, 10, 593–598. [Google Scholar] [CrossRef] [PubMed]
- James, W.D.; Elston, D.M.; Treat, J.R.; Rosenbach, M.A. Andrews’ Diseases of the Skin: Clinical Dermatology, 13th ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 550–553. [Google Scholar]
- Dombi, E.; Solomon, J.; Gillespie, A.J.; Fox, E.; Balis, F.M.; Patronas, N.; Korf, B.R.; Babovic-Vuksanovic, D.; Packer, R.J.; Belasco, J.; et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: Relationship to age and body weight. Neurology 2007, 68, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Prada, C.E.; Rangwala, F.A.; Martin, L.J.; Lovell, A.M.; Saal, H.M.; Schorry, E.K.; Hopkin, R.J. Pediatric plexiform neurofibromas: Impact on morbidity and mortality in neurofibromatosis Type 1. J. Pediatr. 2012, 160, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Waqar, M.; Huson, S.; Evans, D.G.; Ealing, J.; Karabatsou, K.; George, K.J.; Soh, C. C2 neurofibromas in neurofibromatosis type 1: Genetic and imaging characteristics. J. Neurosurg. Spine 2019, 30, 126–132. [Google Scholar] [CrossRef]
- Ruggieri, M.; Polizzi, A.; Spalice, A.; Salpietro, V.; Caltabiano, R.; D’Orazi, V.; Pavone, P.; Pirrone, C.; Magro, G.; Platania, N.; et al. The natural history of spinal neurofibromatosis: A critical review of clinical and genetic features. Clin. Genet. 2014, 87, 401–410. [Google Scholar] [CrossRef]
- Mauda-Havakuk, M.; Shofty, B.; Ben-Shachar, S.; Ben-Sira, L.; Constantini, S.; Bokstein, F. Spinal and paraspinal plexiform neurofibromas in patients with neurofibromatosis Type 1: A novel scoring system for radiological-clinical correlation. Am. J. Neuroradiol. 2017, 38, 1869–1875. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of selumetinib in neurofibromatosis Type 1–related plexiform neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; E Baser, M.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Dumas, V.; Martin-Garcia, N.; Falzone, M.C.; Voisin, M.C.; Wechsler, J.; Revuz, J.; Créange, A.; Levy, E.; Lantieri, L.; et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1: A clinicopathologic and molecular study of 17 patients. Arch. Dermatol. 2001, 137, 908–913. [Google Scholar] [PubMed]
- Coffin, C.M.; Cassity, J.; Viskochil, D.; Randall, R.L.; Albritton, K. Non-neurogenic sarcomas in four children and young adults with neurofibromatosis type 1. Am. J. Med. Genet. 2004, 40–43. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Granada, C.N.P.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- Farid, M.; Demicco, E.G.; A Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Cosper, P.F.; Dahiya, S.; Van Tine, B.A. Neoadjuvant ifosfamide and epirubicin in the treatment of malignant peripheral nerve sheath tumors. Sarcoma 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Sobczuk, P.; Teterycz, P.; Czarnecka, A.M.; Świtaj, T.; Kosela-Paterczyk, H.; Kozak, K.; Falkowski, S.; Rutkowski, P. Systemic treatment for advanced and metastatic malignant peripheral nerve sheath tumors—A sarcoma reference center experience. J. Clin. Med. 2020, 9, 3157. [Google Scholar] [CrossRef]
- Manji, G.A.; Van Tine, B.A.; Lee, S.M.; Raufi, A.; Patwardhan, P.; Blumberg, L.E.; Sender, N.; Wang, J.; Otap, D.; Singh-Kandah, S.V.; et al. Phase 1 combination therapy with pexidartinib (PEX) and sirolimus (S) to target tumor-associated macrophages in pigmented villonodular synovitis, malignant peripheral nerve sheath tumors, and other soft tissue sarcomas. J. Clin. Oncol. 2019, 37 (Suppl. 15), 11055. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Bohanes, P.; Bisig, B.; Missiaglia, E.; Tsantoulis, P.; Coukos, G.; Montemurro, M.; Homicsko, K.; Michielin, O. Deep response to anti-PD-1 therapy of metastatic neurofibromatosis type 1-associated malignant peripheral nerve sheath tumor with CD274/PD-L1 amplification. JCO Precis. Oncol. 2019, 1–6. [Google Scholar] [CrossRef]
- Prudner, B.C.; Ball, T.; Rathore, R.; Hirbe, A.C. Diagnosis and management of malignant peripheral nerve sheath tumors: Current practice and future perspectives. Neuro-Oncol. Adv. 2019, 2 (Suppl. 1), i40–i49. [Google Scholar] [CrossRef] [PubMed]
- Guillamo, J.; Créange, A.; Kalifa, C.; Grill, J.; Rodriguez, D.; Doz, F.; Barbarot, S.; Zerah, M.; Sanson, M.; Bastuji-Garin, S.; et al. Prognostic factors of CNS tumours in neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain 2002, 126, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Gerson, L.P.; Axelson, K.A.; Riccardi, V.M.; Whitford, R.P. Von Recklinghausen neurofibromatosis. Ophthalmology 1984, 91, 929–935. [Google Scholar] [CrossRef]
- Listernick, R.; Ferner, R.E.; Liu, G.T.; Gutmann, D.H. Optic pathway gliomas in neurofibromatosis-1: Controversies and recommendations. Ann. Neurol. 2007, 61, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Campen, C.J.; Gutmann, D.H. Optic pathway gliomas in neurofibromatosis type 1. J. Child Neurol. 2018, 33, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Martín, A.; García-Martínez, F.J.; Duat, A.; Lopez-Martin, I.; Noguera-Morel, L.; Torrelo, A. Nevus anemicus: A distinctive cutaneous finding in neurofibromatosis type 1. Pediatr. Dermatol. 2015, 32, 342–347. [Google Scholar] [CrossRef]
- Tadini, G.; Brena, M.; Pezzani, L.; Gelmetti, C.; Santagada, F.; Boldrini, M.P. Anemic nevus in neurofibromatosis type 1. Dermatology 2013, 226, 115–118. [Google Scholar] [CrossRef]
- Ferrari, F.; Masurel, A.; Olivier-Faivre, L.; Vabres, P. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol. 2014, 150, 42–46. [Google Scholar] [CrossRef]
- Marque, M.; Roubertie, A.; Jaussent, A.; Carneiro, M.; Meunier, L.; Guillot, B.; Pinson, L.; Pinson, S.; Bessis, D. Nevus anemicus in neurofibromatosis type 1: A potential new diagnostic criterion. J. Am. Acad. Dermatol. 2013, 69, 768–775. [Google Scholar] [CrossRef]
- Fenot, M.; Stalder, J.-F.; Barbarot, S. Juvenile xanthogranulomas are highly prevalent but transient in young children with neurofibromatosis type 1. J. Am. Acad. Dermatol. 2014, 71, 389–390. [Google Scholar] [CrossRef]
- Yoshimi, A.; Kojima, S.; Hirano, N. Juvenile myelomonocytic leukemia. Pediatr. Drugs 2010, 12, 11–21. [Google Scholar] [CrossRef]
- Zvulunov, A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia. Arch. Dermatol. 1995, 131, 904. [Google Scholar] [CrossRef]
- Cambiaghi, S.; Restano, L.; Caputo, R. Juvenile xanthogranuloma associated with neurofibromatosis 1: 14 patients without evidence of hematologic malignancies. Pediatr. Dermatol. 2004, 21, 97–101. [Google Scholar] [CrossRef]
- Liy-Wong, C.; Mohammed, J.; Carleton, A.; Pope, E.; Parkin, P.; Lara-Corrales, I. The relationship between neurofibromatosis type 1, juvenile xanthogranuloma, and malignancy: A retrospective case-control study. J. Am. Acad. Dermatol. 2017, 76, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Brems, H.; Park, C.; Maertens, O.; Pemov, A.; Messiaen, L.; Upadhyaya, M.; Claes, K.; Beert, E.; Peeters, K.; Mautner, V.; et al. Glomus tumors in neurofibromatosis type 1: Genetic, functional, and clinical evidence of a novel association. Cancer Res. 2009, 69, 7393–7401. [Google Scholar] [CrossRef]
- Harrison, B.; Sammer, D.M. Glomus tumors and neurofibromatosis. Plast. Reconstr. Surg.—Glob. Open 2014, 2, e214. [Google Scholar] [CrossRef]
- Okada, O.; Demitsu, T.; Manabe, M.; Yoneda, K. A case of multiple subungual glomus tumors associated with neurofibromatosis type 1. J. Dermatol. 1999, 26, 535–537. [Google Scholar] [CrossRef] [PubMed]
- De Smet, L.; Sciot, R.; Legius, E. Multifocal glomus tumours of the fingers in two patients with neurofibromatosis type 1. J. Med. Genet. 2002, 39, e45. [Google Scholar] [CrossRef]
- Zöller, M.E.T.; Rembeck, B.; Oden, A.; Samuelsson, M.; Angervall, L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer 1997, 79, 2125–2131. [Google Scholar]
- Rauen, K.A.; Huson, S.M.; Burkitt-Wright, E.; Evans, D.G.; Farschtschi, S.; Ferner, R.E.; Gutmann, D.H.; Hanemann, C.O.; Kerr, B.; Legius, E.; et al. Recent developments in neurofibromatoses and RASopathies: Management, diagnosis and current and future therapeutic avenues. Am. J. Med. Genet. Part A 2014, 167, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Seminog, O.O.; Goldacre, M. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: Population-based record-linkage study. Br. J. Cancer 2013, 108, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Guillot, B.; Dalac, S.; Delaunay, M.; Baccard, M.; Chevrant-Breton, J.; Dereure, O.; Machet, L.; Sassolas, B.; Zeller, J.; Bernard, P.; et al. Cutaneous malignant melanoma and neurofibromatosis type 1. Melanoma Res. 2004, 14, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Kiuru, M.; Scott, S.N.; Arcila, M.; Halpern, A.C.; Hollmann, T.; Berger, M.F.; Busam, K.J. NF1 mutations are common in desmoplastic melanoma. Am. J. Surg. Pathol. 2015, 39, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, T.J.; Plesec, T.P.; Singh, A.D. Desmoplastic melanoma of the eyelid and conjunctival melanoma in neurofibromatosis type 1: A clinical pathological correlation. Surv. Ophthalmol. 2015, 60, 72–77. [Google Scholar] [CrossRef]
- Zafar, W.; Chaucer, B.; Davalos, F.; Beenish, S.; Chevenon, M.; Nfonoyim, J. Neurofibromatosis type 1 with a pheochromocytoma: A rare Presentation of Von Recklinghausen disease. J. Endocrinol. Metab. 2015, 5, 309–311. [Google Scholar] [CrossRef]
- Crucis, A.; Richer, W.; Brugières, L.; Bergeron, C.; Marie-Cardine, A.; Stephan, J.-L.; Girard, P.; Corradini, N.; Munzer, M.; Lacour, B.; et al. Rhabdomyosarcomas in children with neurofibromatosis type I: A national historical cohort. Pediatr. Blood Cancer 2015, 62, 1733–1738. [Google Scholar] [CrossRef]
- Salvi, P.F.; Lorenzon, L.; Caterino, S.; Antolino, L.; Antonelli, M.S.; Balducci, G. Gastrointestinal stromal tumors associated with neurofibromatosis 1: A single centre experience and systematic review of the literature including 252 cases. Int. J. Surg. Oncol. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cafe-au-lait Macules | Light to dark brown-pigmented, oval or round skin patches with regular, well-demarcated margin |
| Freckles | Small, clustered pigmented lentigines |
| Neurofibroma | Dome—shaped, pink to brown, pedunculated or sessile cutaneous tumors |
| Plexiform Neurofibroma | Thickened, slightly raised and pigmeted firm masses located along the nerve; “Bag of worms” appearance; hypertrichosis |
| Nevus Anemicus | Hypopigmented, pale, well-defined skin patch |
| Juvenile Xantogranuloma | Yellowish-pink dome-shaped granulomatous papules and nodules |
| Glomus Tumor | Small reddish-blue papulonodule often located at the subungual area |
| Melanoma | Unevenly pigmented unusual looking macule |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozarslan, B.; Russo, T.; Argenziano, G.; Santoro, C.; Piccolo, V. Cutaneous Findings in Neurofibromatosis Type 1. Cancers 2021, 13, 463. https://doi.org/10.3390/cancers13030463
Ozarslan B, Russo T, Argenziano G, Santoro C, Piccolo V. Cutaneous Findings in Neurofibromatosis Type 1. Cancers. 2021; 13(3):463. https://doi.org/10.3390/cancers13030463
Chicago/Turabian StyleOzarslan, Bengisu, Teresa Russo, Giuseppe Argenziano, Claudia Santoro, and Vincenzo Piccolo. 2021. "Cutaneous Findings in Neurofibromatosis Type 1" Cancers 13, no. 3: 463. https://doi.org/10.3390/cancers13030463
APA StyleOzarslan, B., Russo, T., Argenziano, G., Santoro, C., & Piccolo, V. (2021). Cutaneous Findings in Neurofibromatosis Type 1. Cancers, 13(3), 463. https://doi.org/10.3390/cancers13030463