
Small Molecule-Based Prodrug Targeting Prostate Specific Membrane Antigen for the Treatment of Prostate Cancer

 , , ,
, , , 
Simple Summary
Abstract
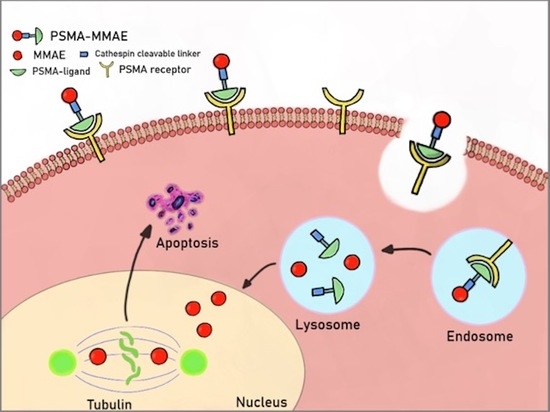
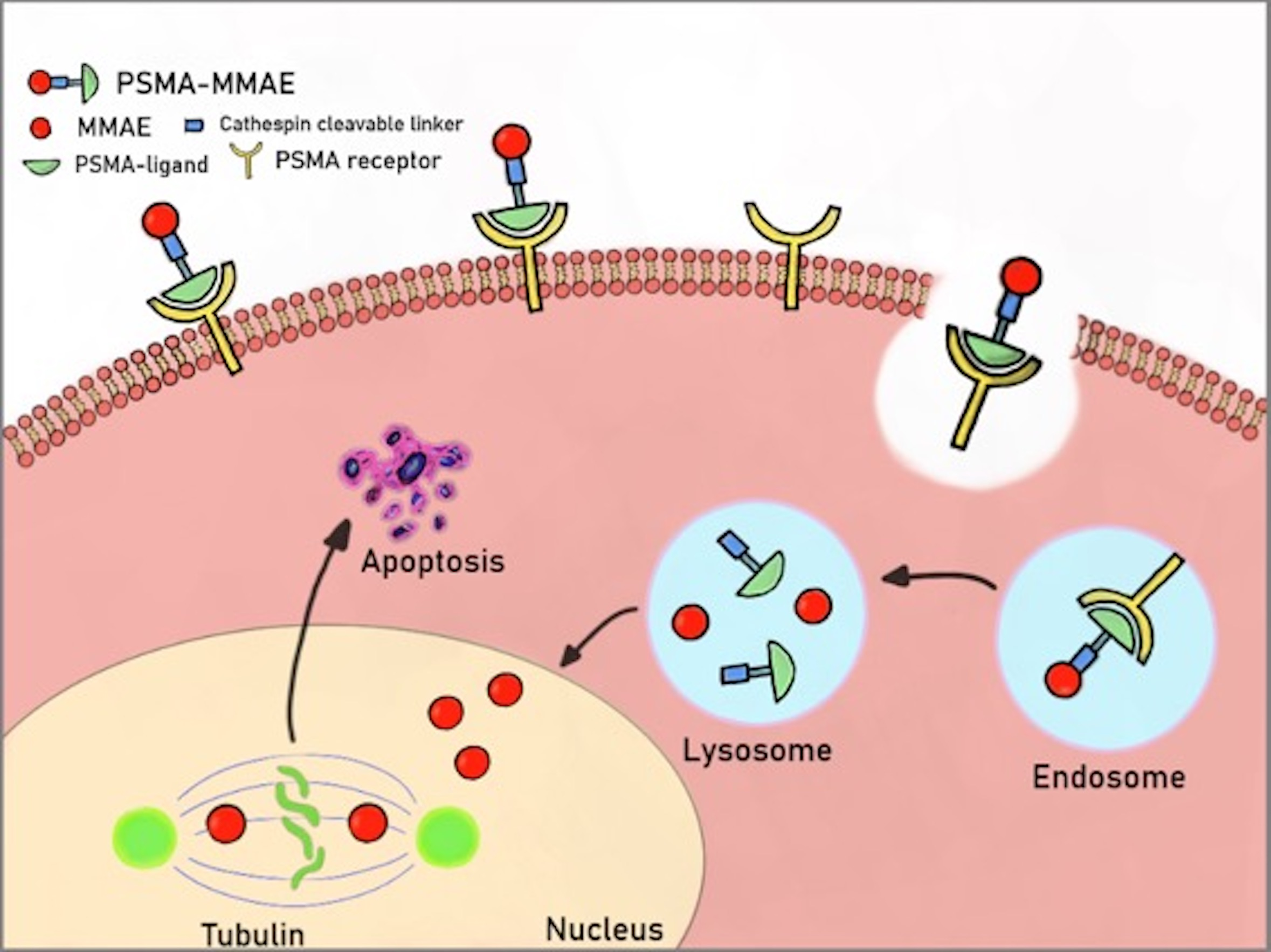
1. Introduction
2. Results
2.1. A Prodrug Strategy Is Crucial for Antitumor Activity
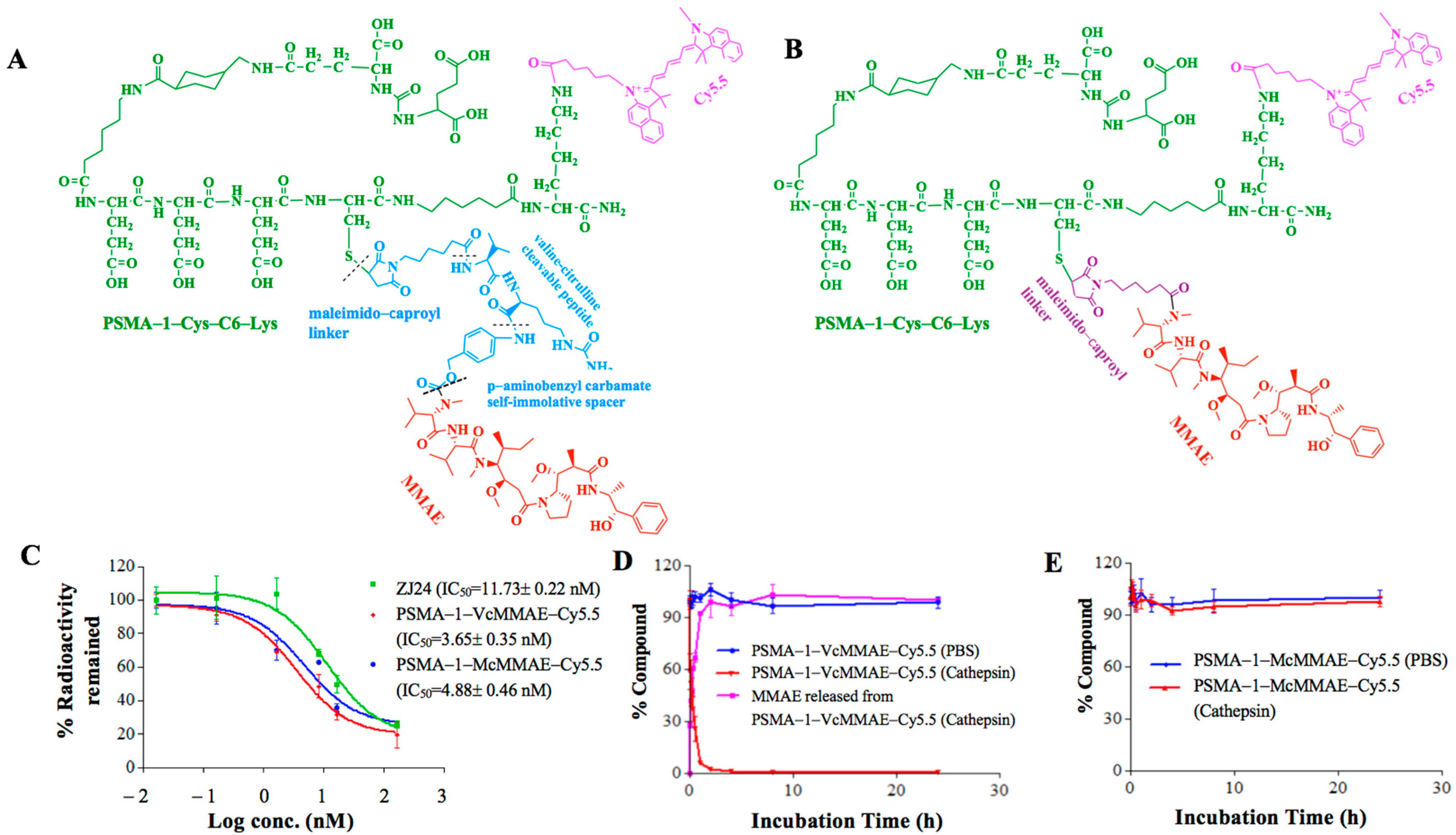
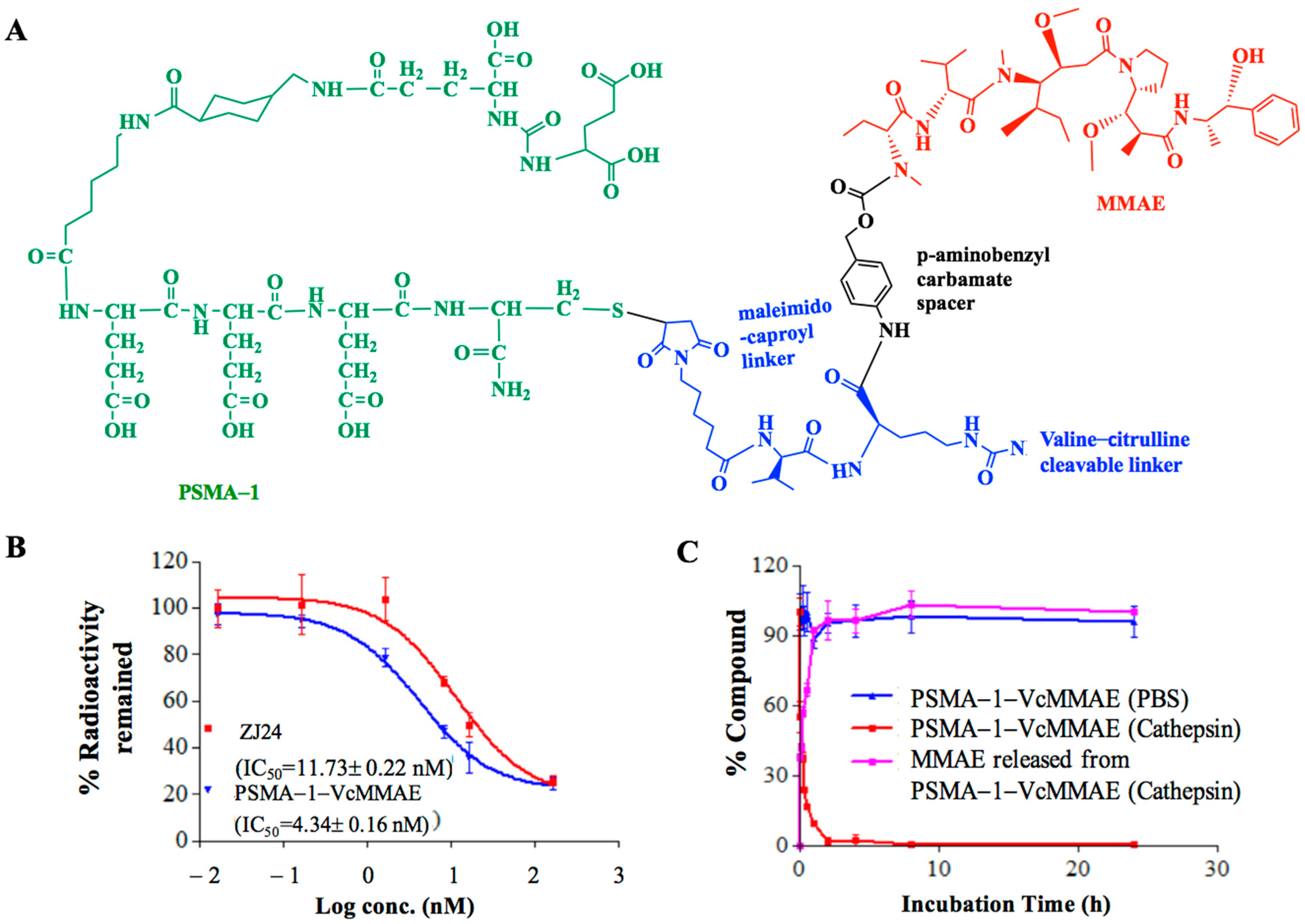
2.1.1. Binding Affinity of PSMA-Targeted MMAE-Cy5.5 Conjugates
2.1.2. Cathepsin Cleavage of PSMA-Targeted MMAE-Cy5.5 Conjugates
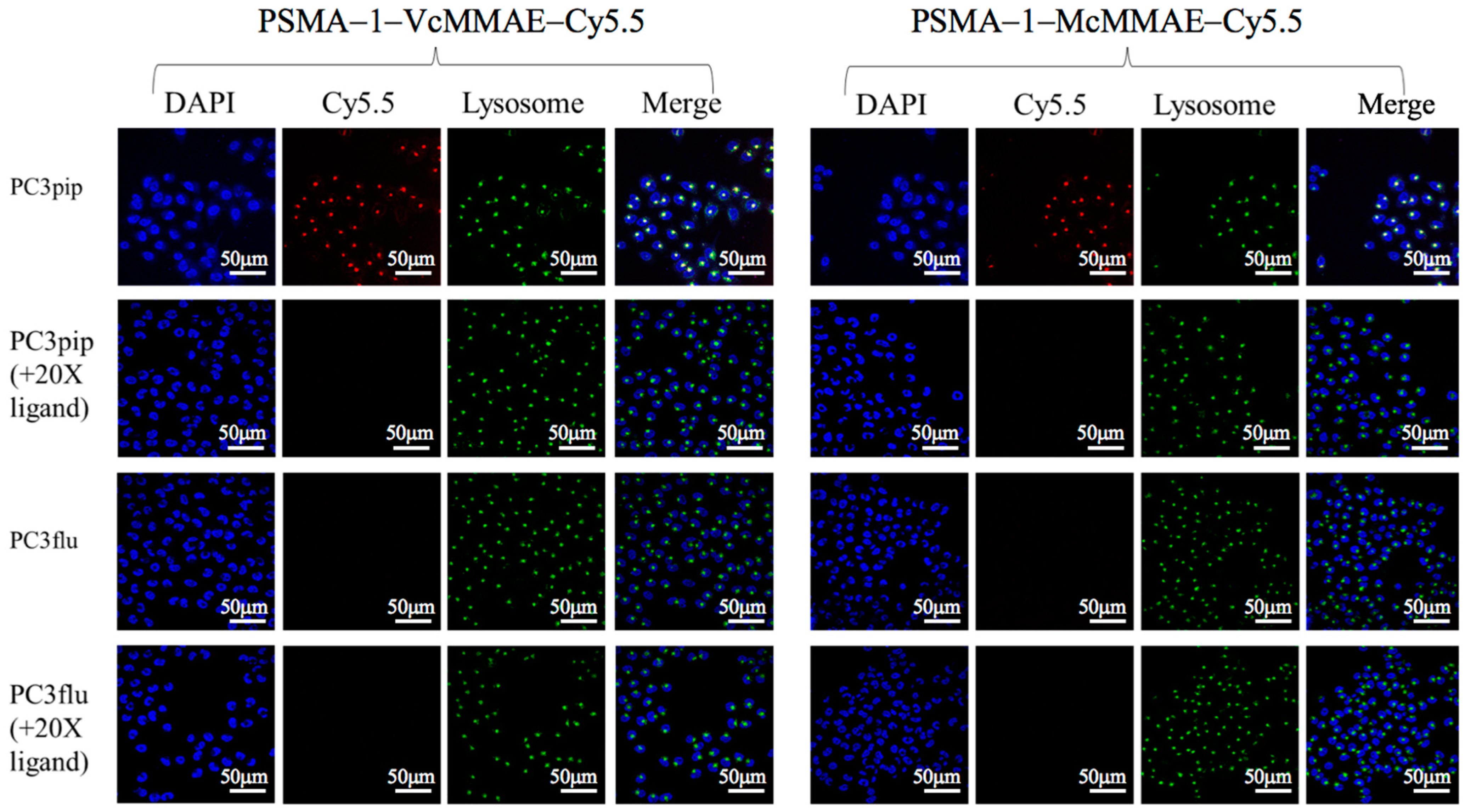
2.1.3. Cellular Uptake Studies
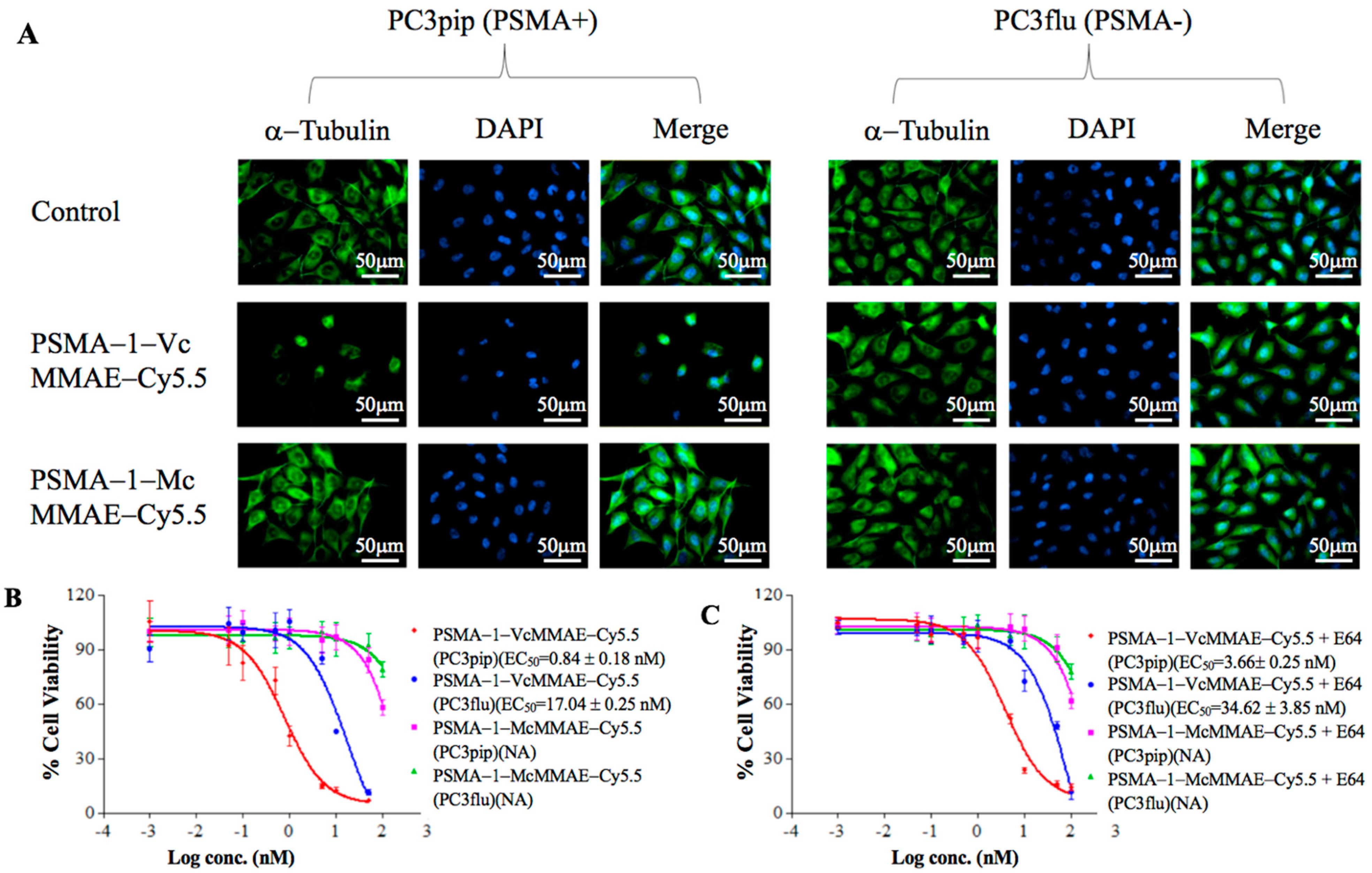
2.1.4. Disruption of α-Tubulin
2.1.5. In Vitro Cytotoxicity of PSMA-Targeted MMAE-Cy5.5 Conjugates
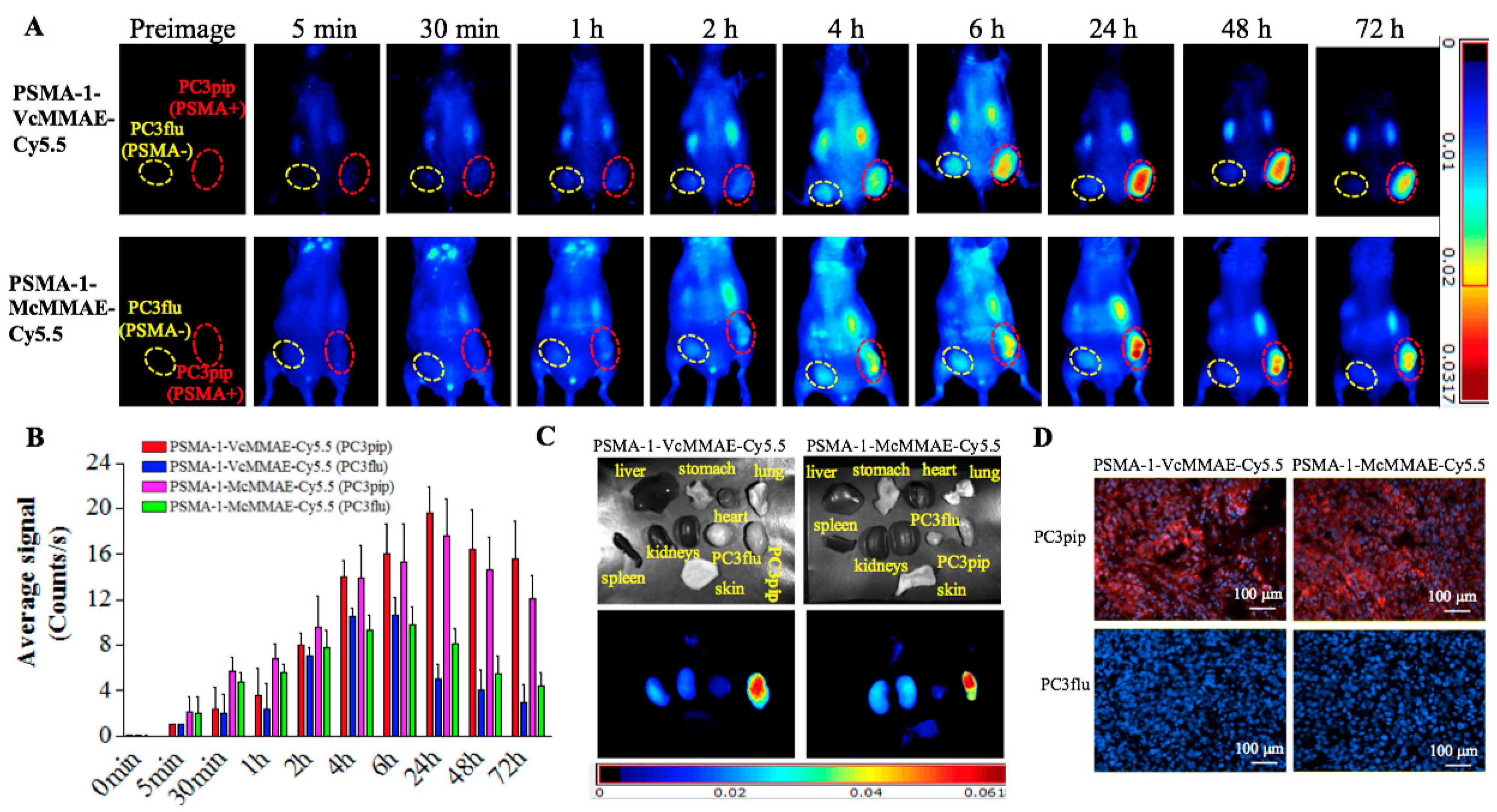
2.1.6. In Vivo Fluorescence Imaging of PSMA-Targeted MMAE-Cy5.5 Conjugates
2.1.7. In Vivo Antitumor Activity of PSMA-Targeted MMAE-Cy5.5 Conjugates
2.2. Efficacy of PSMA-1-VcMMAE
2.2.1. In Vitro Characterization of PSMA-1-VcMMAE
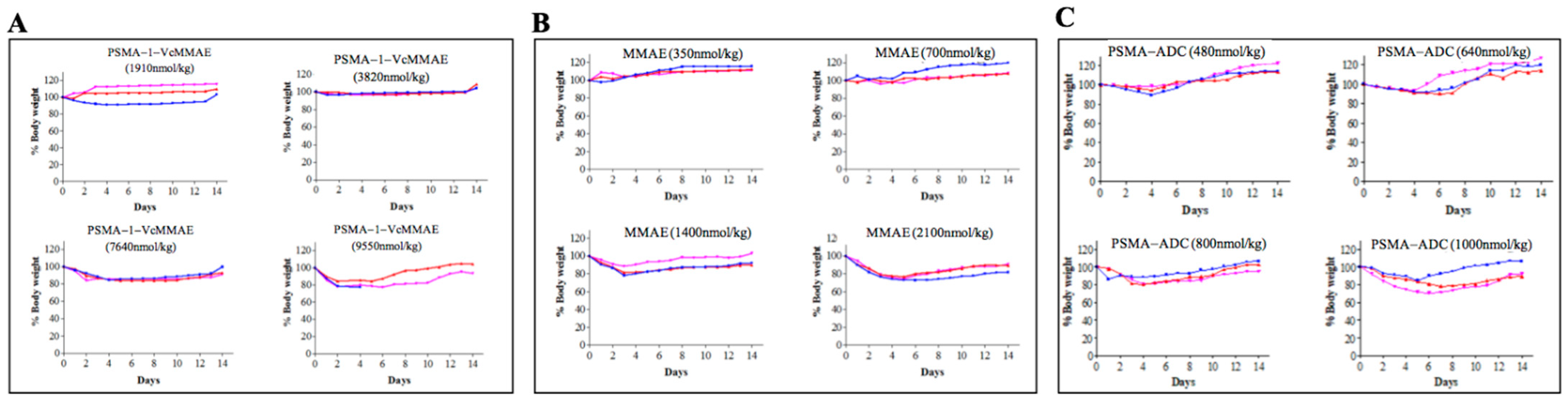
2.2.2. Maximum Tolerated Dose of PSMA-1-VcMMAE
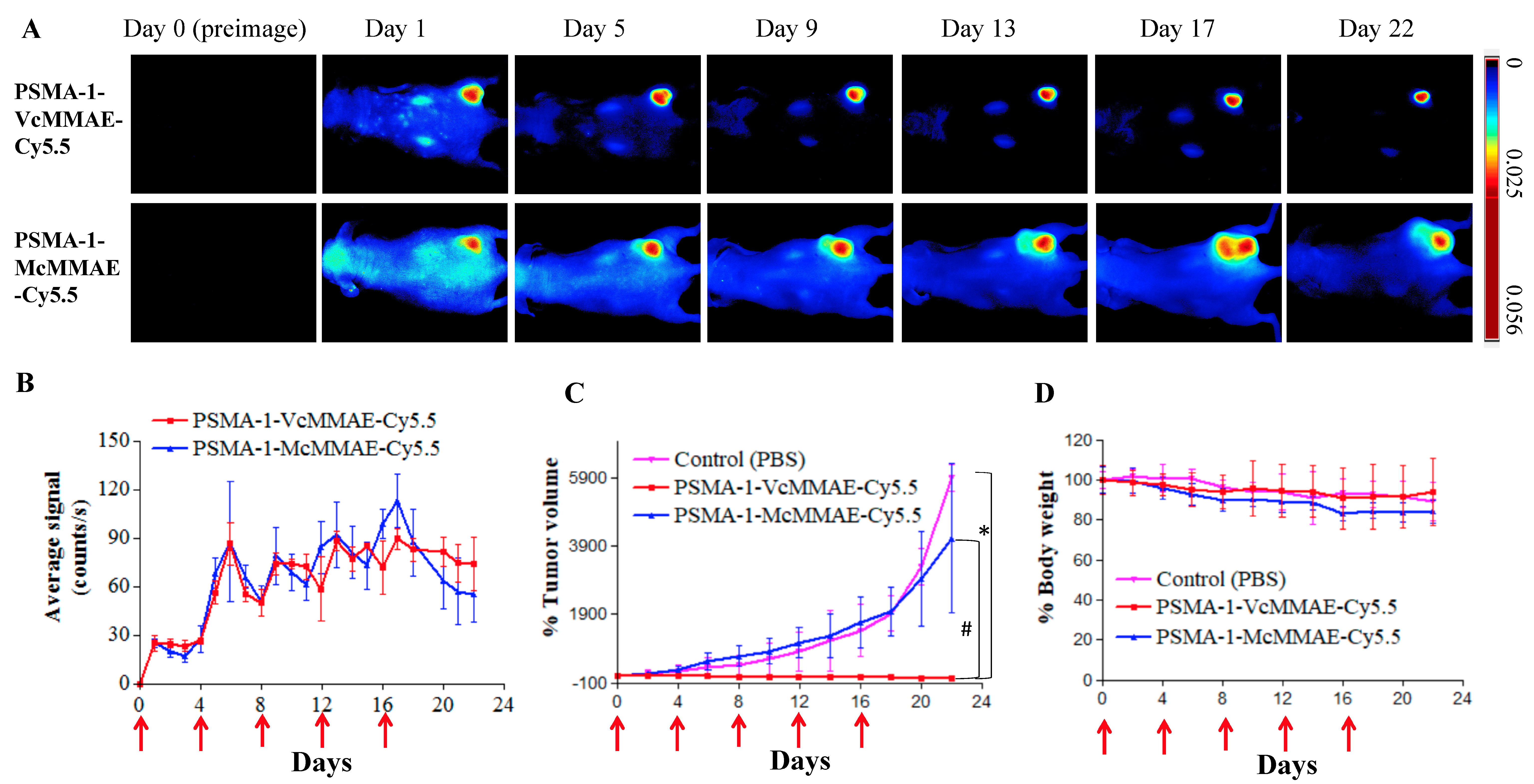
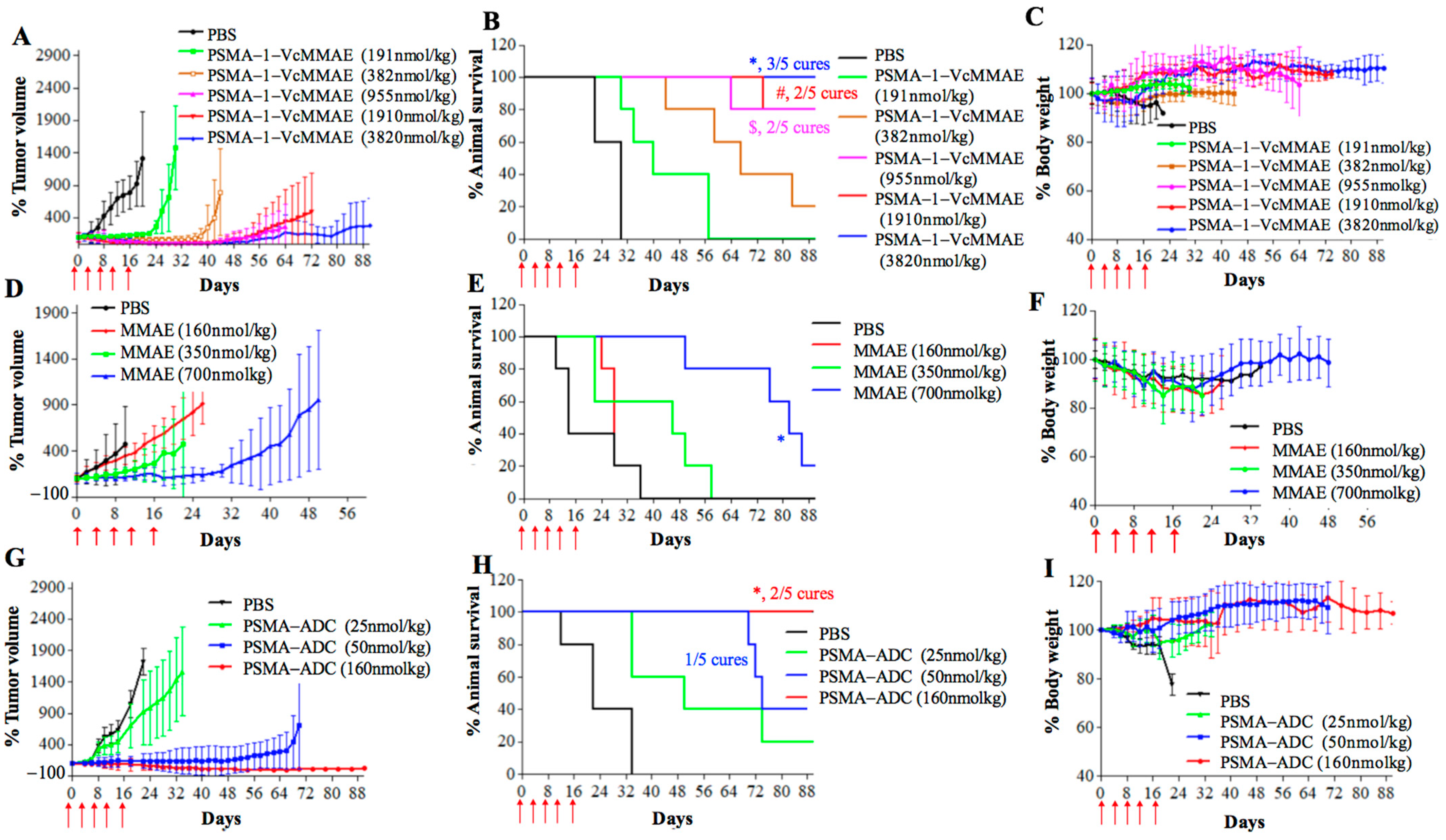
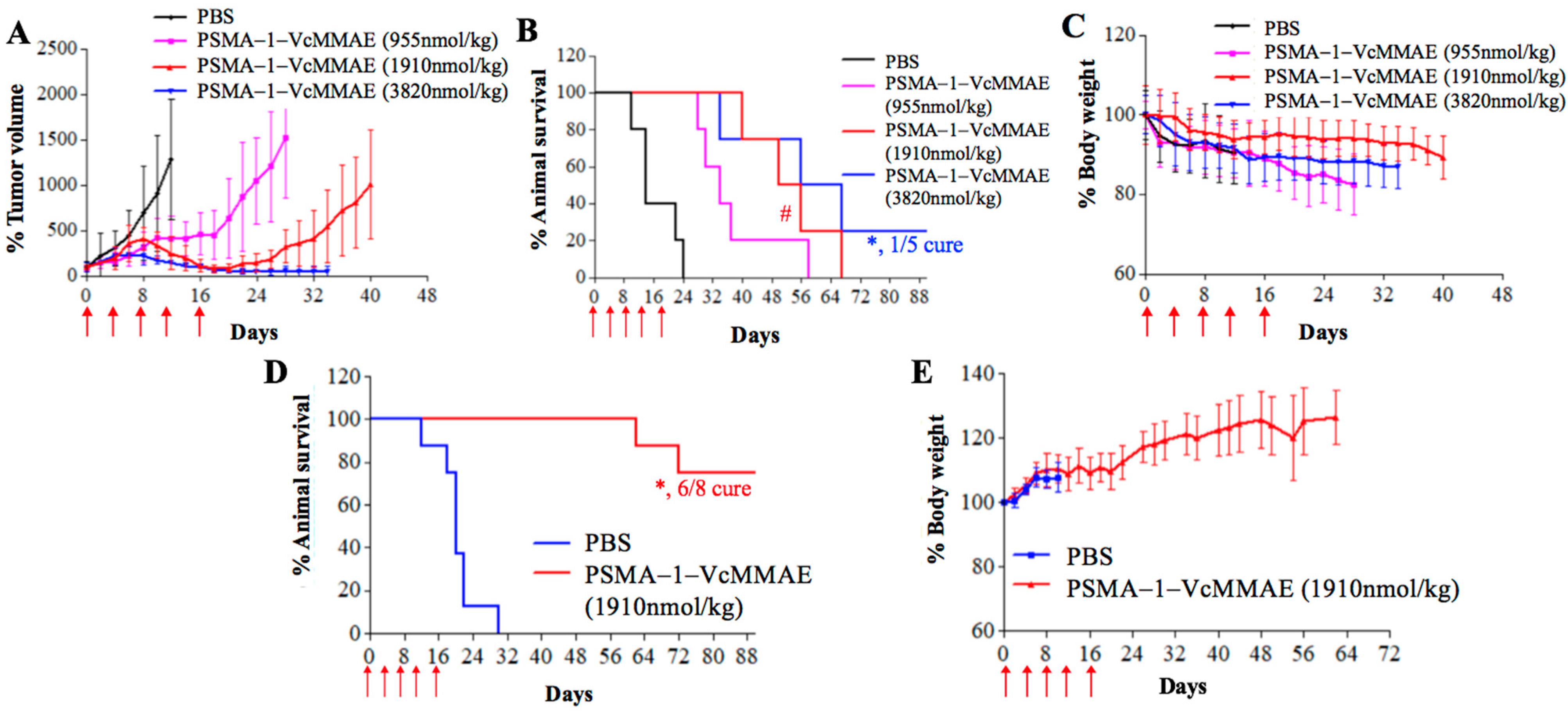
2.2.3. In Vivo Antitumor Activity of PSMA-1-VcMMAE
3. Discussion
4. Materials and Methods
4.1. General
4.2. Synthesis of PSMA-Targeting MMAE Conjugates
4.2.1. Synthesis of PSMA-1-VcMMAE
4.2.2. Synthesis of PSMA-1-VcMMAE-Cy5.5
4.2.3. Synthesis of PSMA-1-McMMAE-Cy5.5
4.3. Cell Culture
4.4. Competitive Binding Assay
4.5. Enzymatic Cleavage of PSMA-1-MMAE-Cy5.5 by Cathepsin B
4.6. In Vitro Cellular Uptake Studies
4.7. Immunofluorescence Analysis of Alpha-Tubulin
4.8. In Vitro Cytotoxicity Assay
4.9. In Vivo NIR Imaging Studies
4.10. Determination of Maximum Tolerated Dose (MTD)
4.11. Heterotopic Survival Study
4.12. Orthotopic Survival Study
4.13. Metastatic Survival Study
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Eisenberger, M. Advances in the Treatment of Metastatic Prostate Cancer. Mayo Clin. Proc. 2015, 90, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Isaacs, J.T. A history of prostate cancer treatment. Nat. Rev. Cancer 2002, 2, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Dagher, R.; Li, N.; Abraham, S.; Rahman, A.; Sridhara, R.; Pazdur, R. Approval summary: Docetaxel in combination with prednisone for the treatment of androgen-independent hormone-refractory prostate cancer. Clin. Cancer Res. 2004, 10, 8147–8151. [Google Scholar] [CrossRef]
- Gulley, J.; Dahut, W.L. Chemotherapy for prostate cancer: Finally an advance! Am. J. Ther. 2004, 11, 288–294. [Google Scholar] [CrossRef]
- DeVita, V.T.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- de Goeij, B.E.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Martin, M.; Symmans, W.F.; Jung, K.H.; Huang, C.-S.; Thompson, A.M.; Harbeck, N.; Valero, V.; Stroyakovskiy, D.; Wildiers, H.; et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 115–126. [Google Scholar] [CrossRef]
- Horwitz, S.M.; O’Connor, O.A.; Pro, B.; Illidge, T.; Fanale, M.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019, 393, 229–240. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-targeted chemotherapy with CMC-544: A CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjug. Chem. 2002, 13, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.-S.; Tice, D.A.; Soria, J.-C. Antibody–Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019, 25, 5441–5448. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Triguero, M.; Yi, J.-H.; Dere, R.; Qiu, Z.J.; Lei, C.; Li, Y.; Mahood, C.; Wang, B.; Leipold, D.; Poon, K.A.; et al. Immunogenicity assays for antibody–drug conjugates: Case study with ado-trastuzumab emtansine. Bioanalysis 2013, 5, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Borsi, L.; Balza, E.; Bestagno, M.; Castellani, P.; Carnemolla, B.; Biro, A.; Leprini, A.; Sepulveda, J.; Burrone, O.; Neri, D.; et al. Selective targeting of tumoral vasculature: Comparison of different formats of an antibody (L19) to the ED-B domain of fibronectin. Int. J. Cancer 2002, 102, 75–85. [Google Scholar] [CrossRef]
- Krall, N.; Pretto, F.; Decurtins, W.; Bernardes, G.J.L.; Supuran, C.T.; Neri, D. A Small-Molecule Drug Conjugate for the Treatment of Carbonic Anhydrase IX Expressing Tumors. Angew. Chem. Int. Ed. 2014, 53, 4231–4235. [Google Scholar] [CrossRef]
- Vlahov, I.R.; Leamon, C.P. Engineering Folate–Drug Conjugates to Target Cancer: From Chemistry to Clinic. Bioconjugate Chem. 2012, 23, 1357–1369. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Corso, A.D.; Widmayer, F.; Neri, D. Chemically Defined Antibody– and Small Molecule–Drug Conjugates for in Vivo Tumor Targeting Applications: A Comparative Analysis. J. Am. Chem. Soc. 2018, 140, 1617–1621. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discov. 2015, 14, 203–219. [Google Scholar] [CrossRef]
- Israeli, R.S.; Powell, C.T.; Corr, J.G.; Fair, W.R.; Heston, W.D. Expression of the prostate-specific membrane antigen. Cancer Res. 1994, 54, 1807–1811. [Google Scholar]
- Wright, G.L.; Grob, B.M.; Haley, C.; Grossman, K.; Newhall, K.; Petrylak, D.; Troyer, J.; Konchuba, A.; Schellhammer, P.F.; Moriarty, R. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology 1996, 48, 326–334. [Google Scholar] [CrossRef]
- Bostwick, D.G.; Pacelli, A.; Blute, M.; Roche, P.; Murphy, G.P. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: A study of 184 cases. Cancer 1998, 82, 2256–2261. [Google Scholar] [CrossRef]
- Mannweiler, S.; Amersdorfer, P.; Trajanoski, S.; Terrett, J.A.; King, D.; Mehes, G. Heterogeneity of Prostate-Specific Membrane Antigen (PSMA) Expression in Prostate Carcinoma with Distant Metastasis. Pathol. Oncol. Res. 2009, 15, 167–172. [Google Scholar] [CrossRef]
- Ross, J.S.; E Sheehan, C.; Fisher, H.A.G.; Kaufman, R.P.; Kaur, P.; Gray, K.; Webb, I.; Gray, G.S.; Mosher, R.; Kallakury, B.V.S. Correlation of primary tumor prostate-specific membrane antigen expression with disease recurrence in prostate cancer. Clin. Cancer Res. 2003, 9, 6357–6362. [Google Scholar] [PubMed]
- Mitsiades, C.S.; Lembessis, P.; Sourla, A.; Milathianakis, C. Molecular staging by RT-PCR analysis for PSA and PSMA in peripheral blood and bone marrow samples is an independent predictor of time to biochemical failure following radical prostatectomy for clinically localized prostate cancer. Clin. Exp. Metastasis 2004, 21, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rajasekaran, A.K.; Moy, P.; Xia, Y.; Kim, S.; Navarro, V.; Rahmati, R.; Bander, N.H. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998, 58, 4055–4060. [Google Scholar]
- Galsky, M.D.; Eisenberger, M.; Moore-Cooper, S.; Kelly, W.K.; Slovin, S.F.; DeLaCruz, A.; Lee, Y.; Webb, I.J.; Scher, H.I. Phase I Trial of the Prostate-Specific Membrane Antigen–Directed Immunoconjugate MLN2704 in Patients with Progressive Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2008, 26, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Hopf, C.E.; Malewicz, A.D.; Donovan, G.P.; Senter, P.D.; Goeckeler, W.F.; Maddon, P.J.; Olson, W.C. Potent Antitumor Activity of an Auristatin-Conjugated, Fully Human Monoclonal Antibody to Prostate-Specific Membrane Antigen. Clin. Cancer Res. 2006, 12, 2591–2596. [Google Scholar] [CrossRef]
- Eder, M.; Neels, O.C.; Müller, M.; Bauder-Wüst, U.; Remde, Y.; Schäfer, M.; Hennrich, U.; Eisenhut, M.; Afshar-Oromieh, A.; Haberkorn, U.; et al. Novel Preclinical and Radiopharmaceutical Aspects of [68Ga]Ga-PSMA-HBED-CC: A New PET Tracer for Imaging of Prostate Cancer. Pharmaceuticals 2014, 7, 779–796. [Google Scholar] [CrossRef]
- Cho, S.Y.; Gage, K.L.; Mease, R.C.; Senthamizhchelvan, S.; Holt, D.P.; Jeffrey-Kwanisai, A.; Endres, C.J.; Dannals, R.F.; Sgouros, G.; Lodge, M.; et al. Biodistribution, Tumor Detection, and Radiation Dosimetry of 18F-DCFBC, a Low-Molecular-Weight Inhibitor of Prostate-Specific Membrane Antigen, in Patients with Metastatic Prostate Cancer. J. Nucl. Med. 2012, 53, 1883–1891. [Google Scholar] [CrossRef]
- Vallabhajosula, S.; Nikolopoulou, A.; Babich, J.W.; Osborne, J.R.; Tagawa, S.T.; Lipai, I.; Solnes, L.; Maresca, K.P.; Armor, T.; Joyal, J.L.; et al. 99mTc-labeled small-molecule inhibitors of prostate-specific membrane antigen: Pharmacokinetics and biodistribution studies in healthy subjects and patients with metastatic prostate cancer. J. Nucl. Med. 2014, 55, 1791–1798. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Hetzheim, H.; Kratochwil, C.; Benešová, M.; Eder, M.; Neels, O.C.; Eisenhut, M.; Kübler, W.; Holland-Letz, T.; Giesel, F.L.; et al. The novel theranostic PSMA-ligand PSMA-617 in the diagnosis of prostate cancer by PET/CT: Biodistribution in humans, radiation dosimetry and first evaluation of tumor lesions. J. Nucl. Med. 2015, 56, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Mena, E.; Rowe, S.P.; Plyku, D.; Nidal, R.; Eisenberger, M.A.; Antonarakis, E.S.; Fan, H.; Dannals, R.F.; Chen, Y.; et al. Initial Evaluation of [(18)F]DCFPyL for Prostate-Specific Membrane Antigen (PSMA)-Targeted PET Imaging of Prostate Cancer. Mol. Imaging Biol. 2015, 17, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Weineisen, M.; Schottelius, M.; Šimeček, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-Labeled PSMA I&T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, K.; Ahmadzadehfar, H.; Seifert, R.; Boegemann, M. [177 Lu]-PSMA-617 radionuclide therapy in patients with metastatic castration-resistant prostate cancer. Lancet Oncol. 2018, 19, e371. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-Targeted -Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef]
- Sathekge, M.M.; Knoesen, O.; Meckel, M.; Modiselle, M.; Vorster, M.; Marx, S. 213Bi-PSMA-617 targeted alpha-radionuclide therapy in metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1099–1100. [Google Scholar] [CrossRef]
- Kularatne, S.A.; Wang, K.; Santhapuram, H.-K.R.; Low, P.S. Prostate-Specific Membrane Antigen Targeted Imaging and Therapy of Prostate Cancer Using a PSMA Inhibitor as a Homing Ligand. Mol. Pharm. 2009, 6, 780–789. [Google Scholar] [CrossRef]
- Lv, Q.; Yang, J.; Zhang, R.; Yang, Z.; Yang, Z.; Han, X.; Xu, Y.; He, Z. Prostate-Specific Membrane Antigen Targeted Therapy of Prostate Cancer Using a DUPA–Paclitaxel Conjugate. Mol. Pharm. 2018, 15, 1842–1852. [Google Scholar] [CrossRef]
- Roy, J.; Nguyen, T.X.; Kanduluru, A.K.; Venkatesh, C.; Lv, W.; Reddy, P.V.N.; Low, P.S.; Cushman, M. DUPA Conjugation of a Cytotoxic Indenoisoquinoline Topoisomerase I Inhibitor for Selective Prostate Cancer Cell Targeting. J. Med. Chem. 2015, 58, 3094–3103. [Google Scholar] [CrossRef]
- Leamon, C.P.; Reddy, J.A.; Bloomfield, A.; Dorton, R.; Nelson, M.; Vetzel, M.; Kleindl, P.; Hahn, S.; Wang, K.; Vlahov, I.R. Prostate-Specific Membrane Antigen-Specific Antitumor Activity of a Self-Immolative Tubulysin Conjugate. Bioconjugate Chem. 2019, 30, 1805–1813. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Nan, F.; Conti, P.; Zhang, J.; Ramadan, E.; Bzdega, T.; Wroblewska, B.; Neale, J.H.; Pshenichkin, S.; Wroblewski, J.T. Design of Remarkably Simple, Yet Potent Urea-Based Inhibitors of Glutamate Carboxypeptidase II (NAALADase). J. Med. Chem. 2001, 44, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Wang, X.; Zhang, Y.; Doke, A.; DiFilippo, F.P.; Heston, W.D. Improving the biodistribution of PSMA-targeting tracers with a highly negatively charged linker. Prostate 2014, 74, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, S.S.; Heston, W.D.; Guo, H.; Wang, B.-C.; Basilion, J.P. Development of Targeted Near-Infrared Imaging Agents for Prostate Cancer. Mol. Cancer Ther. 2014, 13, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsui, B.; Ramamurthy, G.; Zhang, P.; Meyers, J.; Kenney, M.E.; Kiechle, J.; Ponsky, L.; Basilion, J.P. Theranostic Agents for Photodynamic Therapy of Prostate Cancer by Targeting Prostate-Specific Membrane Antigen. Mol. Cancer Ther. 2016, 15, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Mangadlao, J.D.; Wang, X.; McCleese, C.; Escamilla, M.; Ramamurthy, G.; Wang, Z.; Govande, M.; Basilion, J.P.; Burda, C. Prostate-Specific Membrane Antigen Targeted Gold Nanoparticles for Theranostics of Prostate Cancer. ACS Nano 2018, 12, 3714–3725. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Wang, X.; Zeng, S.; Ramamurthy, G.; Burda, C.; Basilion, J.P. Targeted Gold Nanocluster-Enhanced Radiotherapy of Prostate Cancer. Small 2019, 15, e1900968. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Wang, X.; Zeng, S.; Ramamurthy, G.; Burda, C.; Basilion, J. Prostate-specific membrane antigen targeted gold nanoparticles for prostate cancer radiotherapy: Does size matter for targeted particles? Chem. Sci. 2019, 10, 8119–8128. [Google Scholar] [CrossRef]
- Sheng, X.; Yan, X.; Wang, L.; Shi, Y.; Yao, X.; Luo, H.; Shi, B.; Liu, J.; He, Z.; Yu, G.; et al. Open-label, Multicenter, Phase II Study of RC48-ADC, a HER2-Targeting Antibody-Drug Conjugate, in Patients with Locally Advanced or Metastatic Urothelial Carcinoma. Clin. Cancer Res. 2021, 21, 43–51. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; Deblanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Huang, C.T.; Guo, X.; Barinka, C.; Lupold, S.E.; Pomper, M.G.; Gabrielson, K.; Raman, V.; Artemov, D.; Hapuarachchige, S. Development of 5D3-DM1: A Novel Anti-Prostate-Specific Membrane Antigen Antibody-Drug Conjugate for PSMA-Positive Prostate Cancer Therapy. Mol. Pharm. 2020, 17, 3392–3402. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Tibbitts, J.; Holden, S.N.; Sliwkowski, M.X.; Lewis Phillips, G.D. Trastuzumab emtansine (T-DM1): A novel agent for targeting HER2+ breast cancer. Clin. Breast Cancer 2011, 11, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Morris, C.Q. Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135. [Google Scholar] [CrossRef]
- Cunningham, D.; Parajuli, K.R.; Zhang, C.; Wang, G.; Mei, J.; Zhang, Q.; Liu, S.; You, Z. Monomethyl Auristatin E Phosphate Inhibits Human Prostate Cancer Growth. Prostate 2016, 76, 1420–1430. [Google Scholar] [CrossRef]
- Matsumoto, K.; Mizoue, K.; Kitamura, K.; Tse, W.-C.; Huber, C.P.; Ishida, T. Structural basis of inhibition of cysteine proteases by E-64 and its derivatives. Pept. Sci. 1999, 51, 99–107. [Google Scholar] [CrossRef]
- Wang, X.; Ma, D.; Olson, W.C.; Heston, W.D. In Vitro and In Vivo Responses of Advanced Prostate Tumors to PSMA ADC, an Auristatin-Conjugated Antibody to Prostate-Specific Membrane Antigen. Mol. Cancer Ther. 2011, 10, 1728–1739. [Google Scholar] [CrossRef]
- De Groot, F.M.; Damen, E.W.; Scheeren, H.W. Anticancer prodrugs for application in monotherapy: Targeting hypoxia, tumor-associated enzymes, and receptors. Curr. Med. Chem. 2001, 8, 1093–1122. [Google Scholar] [CrossRef]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef]
- Dal Pozzo, A.; Esposito, E.; Ni, M.; Muzi, L.; Pisano, C.; Bucci, F.; Vesci, L.; Castorina, M.; Penco, S. Conjugates of a novel 7-substituted camptothecin with RGD-peptides as alpha(v)beta(3) integrin ligands: An approach to tumor-targeted therapy. Bioconjug. Chem. 2010, 21, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Crisp, J.L.; Savariar, E.N.; Glasgow, H.L.; Ellies, L.G.; Whitney, M.A.; Tsien, R.Y. Dual targeting of integrin alphavbeta3 and matrix metalloproteinase-2 for optical imaging of tumors and chemotherapeutic delivery. Mol. Cancer Ther. 2014, 13, 1514–1525. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs 2016, 8, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Caculitan, N.G.; Chuh, J.D.C.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q.; et al. Cathepsin B Is Dispensable for Cellular Processing of Cathepsin B-Cleavable Antibody–Drug Conjugates. Cancer Res. 2017, 77, 7027–7037. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Fan, S.-Y.; Xiao, D.; Xie, F.; Zhao, L.; Zhong, W.; Zhong, W. Antibody-Drug Conjugate Using Ionized Cys-Linker-MMAE as the Potent Payload Shows Optimal Therapeutic Safety. Cancers 2020, 12, 744. [Google Scholar] [CrossRef] [PubMed]
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.O.; Sussman, D.; Prota, A.E. Structural Basis of Microtubule Destabilization by Potent Auristatin Anti-Mitotics. PLoS ONE 2016, 11, e0160890. [Google Scholar] [CrossRef]
- Autio, K.A.; Dreicer, R.; Anderson, J.; Garcia, J.A.; Alva, A.; Hart, L.L.; Milowsky, M.I.; Posadas, E.M.; Ryan, C.J.; Graf, R.P.; et al. Safety and Efficacy of BIND-014, a Docetaxel Nanoparticle Targeting Prostate-Specific Membrane Antigen for Patients With Metastatic Castration-Resistant Prostate Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 1344–1351. [Google Scholar] [CrossRef]
- Sathekge, M.; Bruchertseifer, F.; Knoesen, O.; Reyneke, F.; Lawal, I.; Lengana, T.; Davis, C.; Mahapane, J.; Corbett, C.; Vorster, M.; et al. (225)Ac-PSMA-617 in chemotherapy-naive patients with advanced prostate cancer: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2018, 48, 129–138. [Google Scholar]
- Kratochwil, C.; Haberkorn, U.; Giesel, F.L. 225Ac-PSMA-617 for Therapy of Prostate Cancer. Semin. Nucl. Med. 2020, 50, 133–140. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Bronzel, M.; Apostolidis, C.; Weichert, W.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Targeted α-Therapy of Metastatic Castration-Resistant Prostate Cancer with225Ac-PSMA-617: Dosimetry Estimate and Empiric Dose Finding. J. Nucl. Med. 2017, 58, 1624–1631. [Google Scholar] [CrossRef]
- Taïeb, D.; Foletti, J.-M.; Bardiès, M.; Rocchi, P.; Hicks, R.J.; Haberkorn, U. PSMA-Targeted Radionuclide Therapy and Salivary Gland Toxicity: Why Does It Matter? J. Nucl. Med. 2018, 59, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-Labile Dipeptide Linkers for Lysosomal Release of Doxorubicin from Internalizing Immunoconjugates: Model Studies of Enzymatic Drug Release and Antigen-Specific In Vitro Anticancer Activity. Bioconjugate Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Park, S.I.; Kim, S.J.; McCauley, L.K.; Gallick, G.E. Pre-clinical mouse models of human prostate cancer and their utility in drug discovery. Curr. Protoc. Pharmacol. 2010, 51, 14–15. [Google Scholar]
- Wu, T.T.; Sikes, R.A.; Cui, Q.; Thalmann, G.N.; Kao, C.; Murphy, C.F.; Yang, H.; Zhau, H.E.; Balian, G.; Chung, L.W.K. Establishing human prostate cancer cell xenografts in bone: Induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int. J. Cancer 1998, 77, 887–894. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | PSMA-1-VcMMAE (EC50, nM) | PSMA-1-VcMMAE + PSMA-1 (EC50, nM) | PSMA-1-VcMMAE + E64 (EC50, nM) | MMAE (EC50, nM) | PSMA-ADC (EC50, nM) |
|---|---|---|---|---|---|
| PC3pip | 4.64 ± 0.83 | 241.1 ± 8.6 | 31.09 ± 2.11 | 0.24 ± 0.05 | 0.063 ± 0.003 |
| PC3flu | 221.7 ± 2.8 | 261.3 ± 5.6 | 384.9 ± 5.3 | 0.21 ± 0.04 | 241.8 ± 9.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Shirke, A.; Walker, E.; Sun, R.; Ramamurthy, G.; Wang, J.; Shan, L.; Mangadlao, J.; Dong, Z.; Li, J.; et al. Small Molecule-Based Prodrug Targeting Prostate Specific Membrane Antigen for the Treatment of Prostate Cancer. Cancers 2021, 13, 417. https://doi.org/10.3390/cancers13030417
Wang X, Shirke A, Walker E, Sun R, Ramamurthy G, Wang J, Shan L, Mangadlao J, Dong Z, Li J, et al. Small Molecule-Based Prodrug Targeting Prostate Specific Membrane Antigen for the Treatment of Prostate Cancer. Cancers. 2021; 13(3):417. https://doi.org/10.3390/cancers13030417
Chicago/Turabian StyleWang, Xinning, Aditi Shirke, Ethan Walker, Rongcan Sun, Gopolakrishnan Ramamurthy, Jing Wang, Lingpeng Shan, Joey Mangadlao, Zhipeng Dong, Jing Li, and et al. 2021. "Small Molecule-Based Prodrug Targeting Prostate Specific Membrane Antigen for the Treatment of Prostate Cancer" Cancers 13, no. 3: 417. https://doi.org/10.3390/cancers13030417
APA StyleWang, X., Shirke, A., Walker, E., Sun, R., Ramamurthy, G., Wang, J., Shan, L., Mangadlao, J., Dong, Z., Li, J., Wang, Z., Schluchter, M., Luo, D., Wang, Y., Stauffer, S., Brady-Kalnay, S., Hoimes, C., Lee, Z., & Basilion, J. P. (2021). Small Molecule-Based Prodrug Targeting Prostate Specific Membrane Antigen for the Treatment of Prostate Cancer. Cancers, 13(3), 417. https://doi.org/10.3390/cancers13030417